
Abstract
Cardiomyopathy developed in mice deficient for α-kinase 3 (ALPK3), a nuclear kinase previously implicated in the differentiation of cardiomyocytes. Alpk3–/– mice were produced according to normal Mendelian ratios and appeared normal except for a nonprogressive cardiomyopathy that had features of both hypertrophic and dilated forms of cardiomyopathy. Cardiac hypertrophy in Alpk3–/– mice was characterized by increased thickness of both left and right ventricular (LV and RV) walls and by markedly increased heart weight and increased heart weight/body weight and heart weight/tibia length ratios. Magnetic resonance imaging studies confirmed the increased thickness in both septal and LV free walls at end-diastole, although there was no significant change in LV wall thickness at end-systole. Myocardial hypertrophy was the predominant feature in Alpk3–/– mice, but several changes more typically associated with dilated cardiomyopathy included a marked increase in end-diastolic and end-systolic LV volume, as well as reduced cardiac output, stroke volume, and ejection fractions, suggesting LV chamber dilation. Magnetic resonance imaging showed a 50% reduction in both septal and free wall LV contractility in Alpk3–/– mice. Interstitial fibrosis and inflammation were notably absent in Alpk3–/– mice; however, light and electron microscopy revealed altered cardiomyocyte architecture, characterized by reduced numbers of abnormal intercalated discs being associated with mild disarray of myofibrils. These lesions could account for the impaired contractility of the myofibrillar apparatus and contribute to the pathogenesis of cardiomyopathy in Alpk3 –/– mice.
Primary cardiomyopathies are myocardial disorders that result in reduced contractile function and deficient systolic and/or diastolic ventricular function. The primary cardiomyopathies are classified by the World Health Organization as (1) hypertrophic cardiomyopathy, (2) dilated cardiomyopathy, (3) restrictive cardiomyopathy, and (4) arrhythmogenic right ventricular (RV) dysplasia–cardiomyopathy. 33 Increased evidence suggests that cardiac remodeling can be a primary cause of the progression of heart failure, 24 and hypertrophic and dilated cardiomyopathies are 2 major clinical conditions that present with ventricular remodeling. Although hypertrophic and dilated cardiomyopathies are clinically recognized as distinct diseases, a progression from hypertrophic cardiomyopathy to ventricular dilation with systolic and diastolic dysfunction has been shown, suggesting that these 2 types of cardiomyopathy may represent different stages within a pathological spectrum. 14
Hypertrophic cardiomyopathy is the most common inherited cardiac disorder, 25 with a prevalence of 1:500 in the young. 26 Hypertrophic cardiomyopathy is associated with normal or enhanced systolic function but impaired cardiac relaxation due to thickened, fibrotic ventricular walls. Hypertrophic cardiomyopathy is characterized by thickening of the ventricular myocardium, with little or no increase in cardiac chamber volume, and is typified by enlarged disorganized cardiomyocytes, with extensive myofibrillar disarray and fibrosis. In contrast, dilated cardiomyopathy is characterized by ventricular chamber dilation with normal or reduced thickness of the ventricular wall and impaired systolic function. 8 Dilated cardiomyopathy has an estimated prevalence in the United States of 36.5 per 100 000 people 27 and an annual incidence in children of 0.57 cases per 100 000 per year. 36
Although monogenic disorders appear to account for only a small proportion of heart failure in humans, insights into cardiac development and function provided by specific gene mutations are likely to be relevant to more common causes of heart failure. 29 The identification of genetic mutations responsible for the development of cardiomyopathy has advanced understanding of the molecular mechanisms involved in the pathogenesis of heart failure and may assist in developing diagnostics and treatments that permit effective interventions at early stages of disease. It is possible that classification of cardiac diseases in the future may be based on identification of genetic mutations that affect heart morphogenesis, myocyte survival, biomechanical stress responses, cardiac contractility, and electrical conduction instead of the currently used clinical criteria. 8 In this regard, knockout mouse models have proven to be powerful tools for defining the functional role of novel genes in cardiovascular diseases.
To identify pivotal genes for cardiac development and function, we applied high-throughput phenotypic analysis to 4650 independent lines of knockout mice generated by homologous recombination or gene trapping. We have identified novel genes that positively and negatively regulate cardiac function in vivo and report here the development of a nonprogressive dilated cardiomyopathy with hypertrophy in α-kinase 3 (ALPK3)–deficient mice. The ALPK3 enzyme is encoded by Alpk3, which is the mouse ortholog of myocytic induction/differentiation originator or muscle α-kinase (Midori, also known as MAK, AW319487, KIAA1330, mKIAA1330, and D330016D04). ALPK3 was first identified by using differential display technique in P19CL6 embryonal carcinoma cells 16 and belongs to the α-kinase family, which represents a class of protein kinases that are structurally and evolutionarily unrelated to conventional eukaryotic protein kinases. 11 The 6 different α-kinases identified thus far in mammals appear to have a wide range of functions. 11 For example, the kinase eEF-2 phosphorylates and inactivates elongation factor-2 and thus may regulate the global rate of protein synthesis. 11 In contrast, TRPM6 and 7 both phosphorylate the assembly domain of myosin IIA, IIB, and IIC. 9
In mouse embryos, Alpk3 is expressed only in the cardiac crescent and developing heart and appears to play a critical role in cardiomyocyte differentiation. 16 Overexpression of Alpk3 in P19CL6 cells promotes their differentiation into cardiomyocytes, whereas overexpression of antisense Alpk3 in P19CL6 cells negatively affects differentiation, suggesting that ALPK3 may act as a transcriptional regulator of cardiomyocyte differentiation. 16 It is known that transcriptional regulators that control cardiomyocyte differentiation are essential for early cardiac development and that deletion of these types of genes often results in cardiac malformations and embryonic lethality. For example, the transcription factors myocyte enhancer factor-2 (MEF2) are involved in the development of cardiac hypertrophy, 37 and targeted disruption of the MEF2C specifically results in defective cardiac myogenesis and RV development. 23 However, the function of the Alpk3 gene in vivo has not been reported previously. In this study, detailed histological and ultrastructural analysis of hearts from Alpk3–/– mice revealed cytoarchitectural defects that could account for the deficient myocardial contractility and cardiomyopathy in these Alpk3 null mice.
Methods
Generation of Alpk3 Mutant Mice
Methods for gene trapping in ES cells, identification of trapped genes using Omnibank Sequence Tags (OSTs), characterization of retroviral gene-trap vector insertion site, and reverse transcription–polymerase chain reaction (RT-PCR) analysis of knockout and wild-type transcripts are published.41,42 This gene trap mutation of Alpk3 (designated as Alpk3Gt(OST149512)Lex in MGI) was generated in strain 129S5/SvEvBrd-derived embryonic stem cells using clone OST149512 from Lexicon’s OmniBank library.1,41 OST149512 was selected for microinjection based on sequence similarity between the 3′ RACE tag obtained from this clone and the Alpk3 gene (accession NM_054085). Prior to injection, the mutagenicity of this clone was assessed by determining whether the gene-trapping vector had inserted intragenically. The precise location of insertion was determined by amplification and sequencing of the vector/genomic junction site using inverse genomic PCR with primers complementary to the gene trap vector. 15 The targeted ES cell clone was microinjected into C57BL/6J-Tyrc–Brd/Lex (albino) blastocysts to generate chimeric animals that were bred to C57BL/6J-Tyrc–Brd/Lex (albino) females, and the resulting heterozygous offspring were interbred to produce homozygous Alpk3-deficient mice. Mutant F2 mice used in phenotyping studies were produced by intercrossing the F1 heterozygous (–/+) mice. Gene disruption in vivo was confirmed by a direct analysis of gene expression using quantitative real-time polymerase chain reaction (qRT-PCR). Total RNA was isolated from skeletal muscle and heart tissue of wild-type and homozygous mutant animals using TRIzol (Invitrogen, Carlsbad, CA), and reverse transcription was performed using a High Capacity cDNA RT Kit (Applied Biosystems, Foster City, CA) and random hexamer primers, according to the manufacturer’s instructions. Gene expression of Alpk3 was assessed by relative quantitation of Alpk3 gene expression, normalized to the 18S endogenous control, in wild-type and mutant tissues using the TaqMan Gene Expression Assay Mm00475369_m1 (Applied Biosystems). Individual mouse genotypes were determined on DNA isolated from tail biopsy samples by qPCR for the NEO gene present in the gene trapping vector (VICTR 48, accession EU676804) as described previously. 15
Mouse Husbandry
Mice were housed in microisolator cages within a barrier facility at 24°C on a fixed 12-hour light and 12-hour dark cycle and were provided ad libitum acidified water and Purina rodent chow No. 5001 (Purina, St Louis, MO). Procedures involving animals were conducted in conformity with the Institutional Animal Care and Use Committee guidelines that are in compliance with the state and federal laws and the standards outlined in the Guide for the Care and Use of Laboratory Animals (National Research Council, 1996). Quarterly sentinel surveillance showed no evidence of pathogenic rodent viruses or Mycoplasma or Helicobacter spp. in the Lexicon Pharmaceutical source colonies. Unless indicated otherwise, 12-week-old male mice were used in experiments described in this report.
Blood Pressure, Electrocardiogram, and Heart Rate
Blood pressure was measured using a tail-cuff system (Visitech Systems, Apex, NC) with an average of 4 days of readings taken to determine the systolic blood pressure. Conscious and unrestrained electrocardiographic (ECG) tracings were collected by implantable radio telemetry devices (TA10ETA-F20, Data Sciences Int, St Paul, MN) as described previously. 17 Twenty-four-hour heart rate was analyzed 3 days after the implantation of the telemetry device, and raw ECG waveforms from all mice were individually scanned for arrhythmias.
Cardiac Magnetic Resonance Imaging
In vivo cardiac function was determined by magnetic resonance imaging (MRI) analysis on a 7-T horizontal-bore PharmaScan MRI scanner (Bruker, Billerica, MA) using a standard 300-MHz, 38-mm transmit/receive resonator. Mice were anesthetized with 1.5–2.5% (volume) isoflurane via a nose cone. A fast, low-angle shot (FLASH) gradient echo cine sequence gated to both the respiratory movement and the cardiac cycle was used to acquire 10 phases of heart images per cardiac cycle. Both long- and short-axis images of the heart were acquired. Typically, 8 contiguous 1-mm-thick short-axis slices were used to cover the heart from apex to base. For quantitative assessment of myocardial mass and the global function, borders of the left ventricular (LV) epicardium and the LV cavity on each short-axis slice were manually delineated at the end-diastolic and/or the end-systolic phases. LV mass was determined as the total LV volume at the end-diastole, multiplied by the myocardial specific gravity (1.05 g/cm3). Stroke volume (SV) was calculated as LV cavity volume at end-diastole (LVEDV) minus that at end-systole (LVESV), and ejection fraction (EF) was given as SV divided by LVEDV. Cardiac output (CO) was determined by multiplying SV by the average heart rate. The index of each physiological parameter was calculated by normalizing individual parameters with corresponding body weight.
Wall Thickness and Contractility
Short-axis MRI images at end-diastole and end-systole were analyzed for ventricular wall thickness using image analysis software Image-Pro (MediaCybernetics, Bethesda, MD). Cross-sectional area, epicardial and endocardial length of LV free wall, and septal wall were measured. Average wall thickness was calculated with the formula Wall Thickness = 2 * [Wall Area/(Epi-Length + Endo-Length)]. Corresponding contractility of LV (both free wall and septal) was calculated with the formula Contractility (%) = 100 * (Wall Thicknesssystole – Wall Thicknessdiastole)/Wall Thicknessdiastole.
Dobutamine Challenge
Male mice were anesthetized with 2.5% isoflurane. Body temperature of anesthetized animals was maintained at 37°C using a water circulating heating pad. A pressure catheter (1.4F, Millar Instruments, Winston-Salem, NC) connected to a pressure module (Indus Instruments, Houston, TX) was cannulated into the right carotid artery and gently advanced to left ventricle. Baseline LV pressure and heart rate were monitored. Dobutamine (3 μg/kg) was administered via intraperitoneal injection after the baseline LV pressure and heart rate were stabilized for at least 5 minutes. The LV pressure was recorded every 30 seconds for 5 minutes following dobutamine injection, and LV pressure changes in 30 seconds were calculated.
Gross Morphology of Cardiovascular System
Animals were euthanatized via inhalation of lethal levels of isoflurane. Thoracic and abdominal cavities were opened and large blood vessels and heart were examined for any grossly apparent malformations. Hearts were excised and dried on filter paper, and heart weights were measured using an analytical balance. Tibia length was measured using a caliper. Cardiac hypertrophy was expressed as heart weight/body weight ratio (HW/BW) and heart weight/tibia length ratio (HW/TL).
Pathology
Immediately after euthanasia, 14 Alpk3 null mice (7 males and 7 females) with equivalent numbers of wild-type littermates were fixed by cardiac perfusion with 10% neutral buffered formalin. Standard tissues examined included heart, skeletal muscle, aorta, lung, kidney, renal lymph node, trachea, thyroid gland, parathyroid gland, mediastinal lymph node, adrenal gland, pituitary gland, thymus, salivary glands, cervical lymph node, esophagus, stomach, pancreas, duodenum, jejunum, ileum, cecum, colon, rectum, mesenteric lymph node, liver, gallbladder, spleen, brain, spinal cord, eyes, Harderian gland, urinary bladder, uterus, ovaries, fallopian tube, skin, mammary gland, bone, bone marrow, adipose tissue, blood, teeth, prostate gland, testes, epididymis, seminal vesicle, vas deferens, urethral glands, inguinal lymph node, inner ear, middle ear, and nasal turbinates. Tissues were collected and immersed in 10% neutral buffered formalin for an additional 48 hours except for the eyes, which were removed and fixed by immersion in Davidson’s fixative (Poly Scientific, Bay Shore, NY) overnight at room temperature. All tissues were embedded in paraffin, sectioned at 4 μm, and mounted on positively charged glass slides (Superfrost Plus, Fisher Scientific, Pittsburgh, PA) and stained with hematoxylin and eosin (HE) for histopathologic examination. For immunohistochemistry, 4-μm sections were first deparaffinized in xylene and then rehydrated through graded alcohols to phosphate-buffered saline (PBS); endogenous peroxidase activity was blocked by incubation in 3.0% hydrogen peroxide in PBS for 5 minutes. Nonspecific binding of primary antibody was blocked by 20-minute pretreatment with 5% normal goat serum, and negative controls were obtained by omission of the primary antibody. The primary antibody, rabbit anti-desmin antibody (No. 32362, Abcam Inc, Cambridge, MA), was diluted 1:500 in PBS and applied for 1 hour. After rinsing, sections were incubated for 1 hour in the biotinylated goat anti-rabbit immunoglobulin G secondary antibody (Vector Laboratories, Burlingame, CA) diluted 1:400 in PBS. Bound antibodies were detected by an avidin–biotin enzyme complex method with 3,3′-diaminobenzidine as chromogenic substrate following the manufacturer’s instructions (Elite ABC Kit, Vector Laboratories, Burlingame, CA). Sections were counterstained with Mayer’s hematoxylin.
Electron Microscopy
For ultrastructural studies, deep anesthesia was induced in mice by administering Avertin (1.25% solution 2,2,2, tribromoethanol in 2-methyl-2-butanol in PBS) at 0.2 ml/10 g body weight given intraperitoneally. Anesthetized mice were then perfused with 3.5% glutaraldehyde in 0.1 M phosphate buffer. Sections of heart were incubated in 2% osmium tetroxide, stained with 2% uranyl acetate, dehydrated in graded ethanols, and embedded in Spurr’s resin. Semithin sections were stained with 1% Toluidine blue. Ultrathin sections were obtained and collected on 200 mesh grids, poststained with 4% methanolic uranyl acetate and Venable’s lead citrate, and examined with a transmission electron microscope.
Results
Production of Alpk3–/– Mice
Inverse genomic PCR of DNA isolated from OST149512 confirmed that the retroviral gene-trap vector inserted into the intron between the coding exons 2 and 3 of Alpk3 (Fig. 1a). At 3 weeks of age, offspring of founder parents were present in an approximately 1:2:1 Mendelian ratio (126 Wild type [WT], 255 Heterozygous [HET], 103 Homozygous [HOM], χ 2 = 3.58, significance = 0.16696018), indicating the absence of significant embryonic lethality. Homozygous mutant mice were viable and exhibited similar growth curve with their wild-type littermates (data not shown). Both male and female Alpk3 null mice were fertile, and no other grossly or histologically apparent abnormalities were noted in heterozygous and homozygous mutant mice. Analysis of tissues by RT-qPCR demonstrated that the α-kinase 3 transcripts were absent in the heart and the skeletal muscle of the Alpk3 null mice (Fig. 1b). In agreement with previous reports, 16 the strongest gene expression in adult wild-type was observed in heart and skeletal muscle (data not shown).
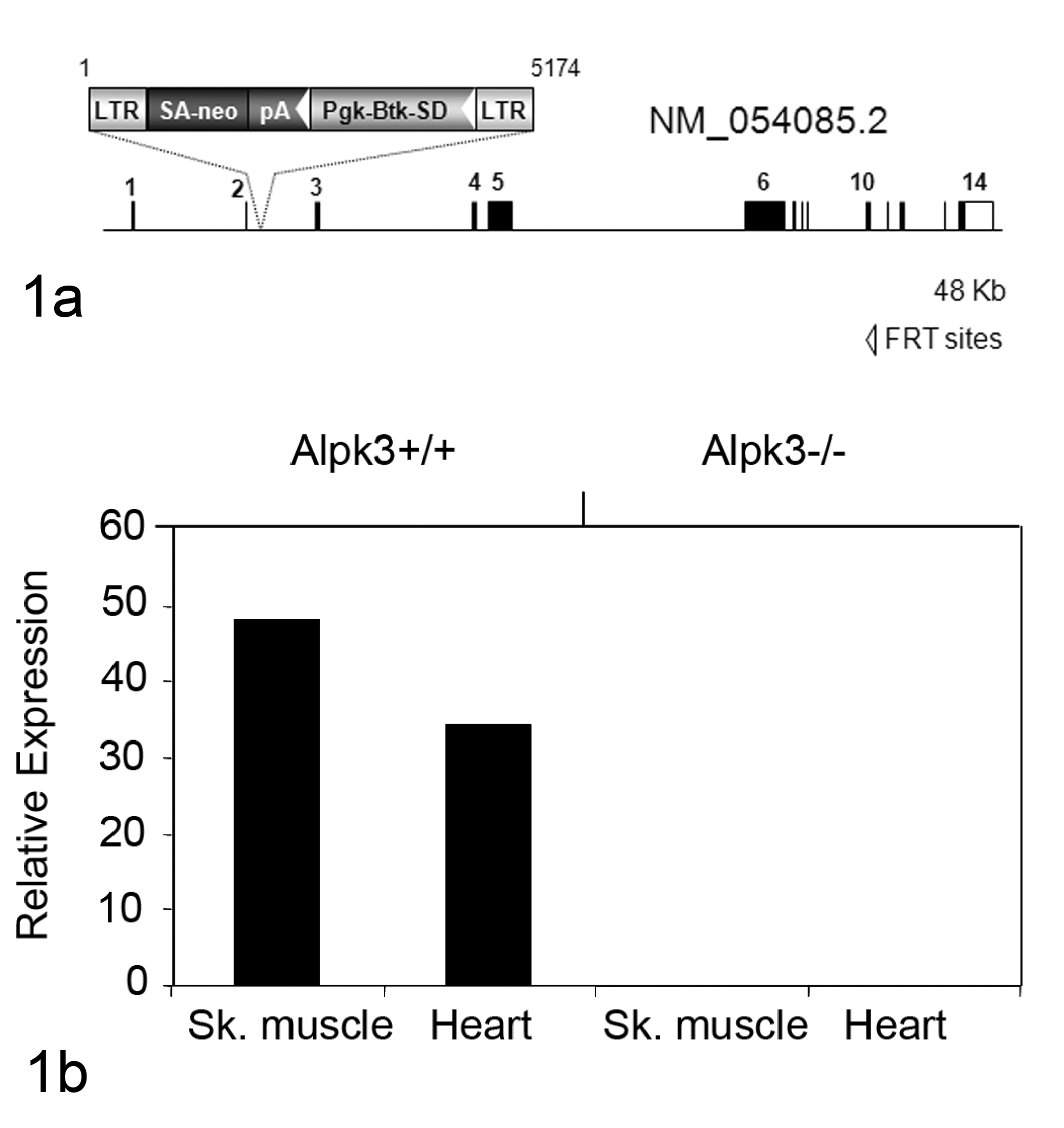
Disruption of the Alpk3 gene by gene trapping. (a) Illustration of the mutant locus identified in OmniBank ES cell clone OST149512. The VICTR48 gene trap vector insertion occurred intragenically within intron 2 of the Alpk3 gene on mouse chromosome 7. Btk-SD, Bruton’s tyrosine kinase splice donor sequence; LTR, viral long terminal repeat; neo, neomycin phosphotransferase gene; pA, polyadenylation sequence; Pgk, phosphoglycerate kinase-1 promoter; SA, splice acceptor sequence. (b) RT-qPCR expression analysis of the Alpk3 transcript in adult tissues. Endogenous Alpk3 transcript was detected in skeletal muscle and heart of wild-type (+/+) mice. No endogenous Alpk3 transcript was detected in homozygous (–/–) tissues. Template input was normalized using an 18S ribosomal RNA endogenous control assay; values are shown in arbitrary units.
Alpk3–/– Mice Showed Decreased System Blood Pressure at 12 Weeks of Age
Despite their grossly normal appearance and behavior, 12-week-old male Alpk3–/– mice showed a significant reduction in systolic blood pressure in the initial phenotypic screen (Fig. 2a). These Alpk3–/– mice exhibited normal heart rates (Fig. 2b) and had no obvious conductance abnormalities or arrhythmia determined by telemetric ECG analysis (data not shown).
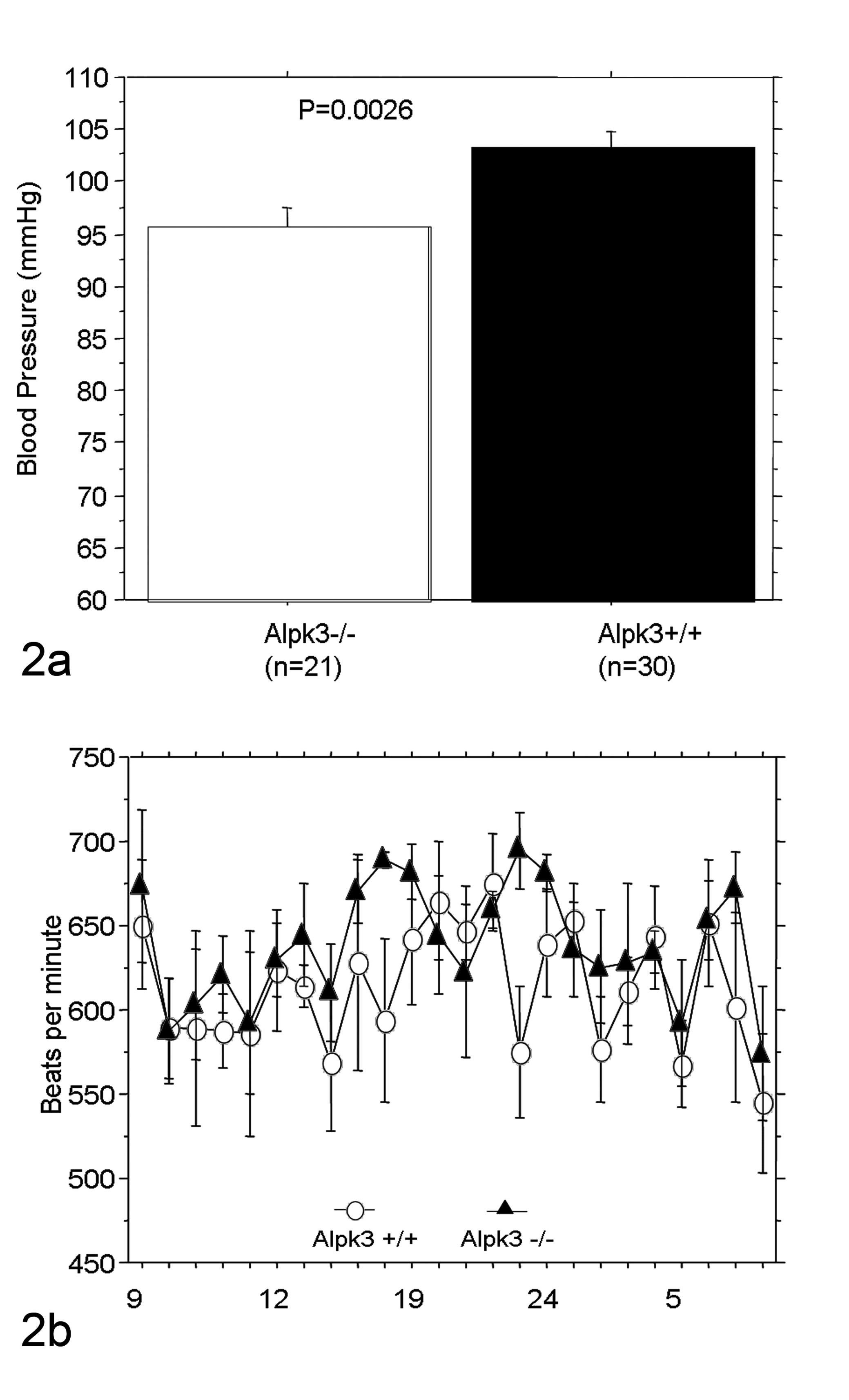
Measurements of systolic blood pressure and heart rate in Alpk3–/– mice. (a) Systolic blood pressure measured by tail cuff methods was significantly decreased in Alpk3–/– mice. The values represent mean systolic blood pressure +/− standard error of the mean (SEM). Data were analyzed by Student’s 2-tailed t-test. (b) There was no significant difference between heart rates of Alpk3+/+ and Alpk3–/– mice in the 24 -hour heart rate monitored by a telemetric system (n = 5 in each group).
Alpk3–/– Mice Developed Spontaneous Cardiomyopathy
The most striking phenotype in Alpk3–/– mice was an enlargement of the heart that became evident during the first month of postnatal life. To further characterize the cardiovascular function in Alpk3–/– mice, a cohort of 3-month-old male homozygous mice and their wild-type littermates were examined by functional cardiac MRI. The Alpk3–/– mice exhibited reduced CO, SV, and EF (Table 1). Furthermore, they exhibited a marked increase in end-diastolic and end-systolic volumes of left ventricle, suggesting LV chamber dilation. Alpk3–/– mice showed an increased thickness in both septal and LV free walls at end-diastole (Fig. 3a), but there was no significant change in wall thickness at end-systole (Fig. 3b). No spontaneous deaths were observed in either male or female Alpk3–/– mice up to 12 months of age, despite the presence of heart enlargement. To assess any progression of heart disease in Alpk3–/– mice, we measured cardiac function by MRI in 3- and 9-month-old male mice. We found that Alpk3–/– mice at 3 and 9 months of age showed similarly reduced EFs compared with their wild-type littermates at both time points (67% at 2 months of age, n = 8; and 68% at 9 months of age, n = 8) indicating that EFs did not deteriorate over time.
Alpk3 –/– Mice Exhibit Compromised Cardiac Function
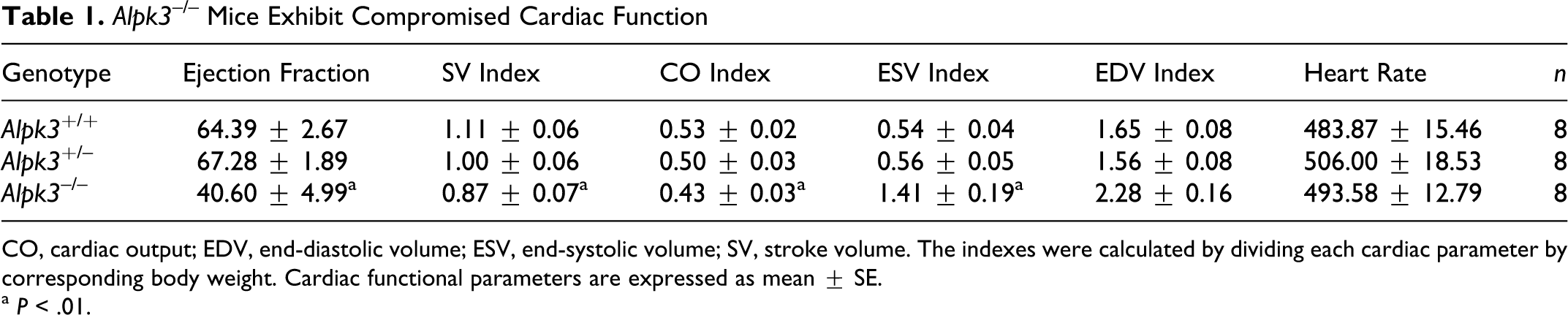
CO, cardiac output; EDV, end-diastolic volume; ESV, end-systolic volume; SV, stroke volume. The indexes were calculated by dividing each cardiac parameter by corresponding body weight. Cardiac functional parameters are expressed as mean ± SE.
a P < .01.
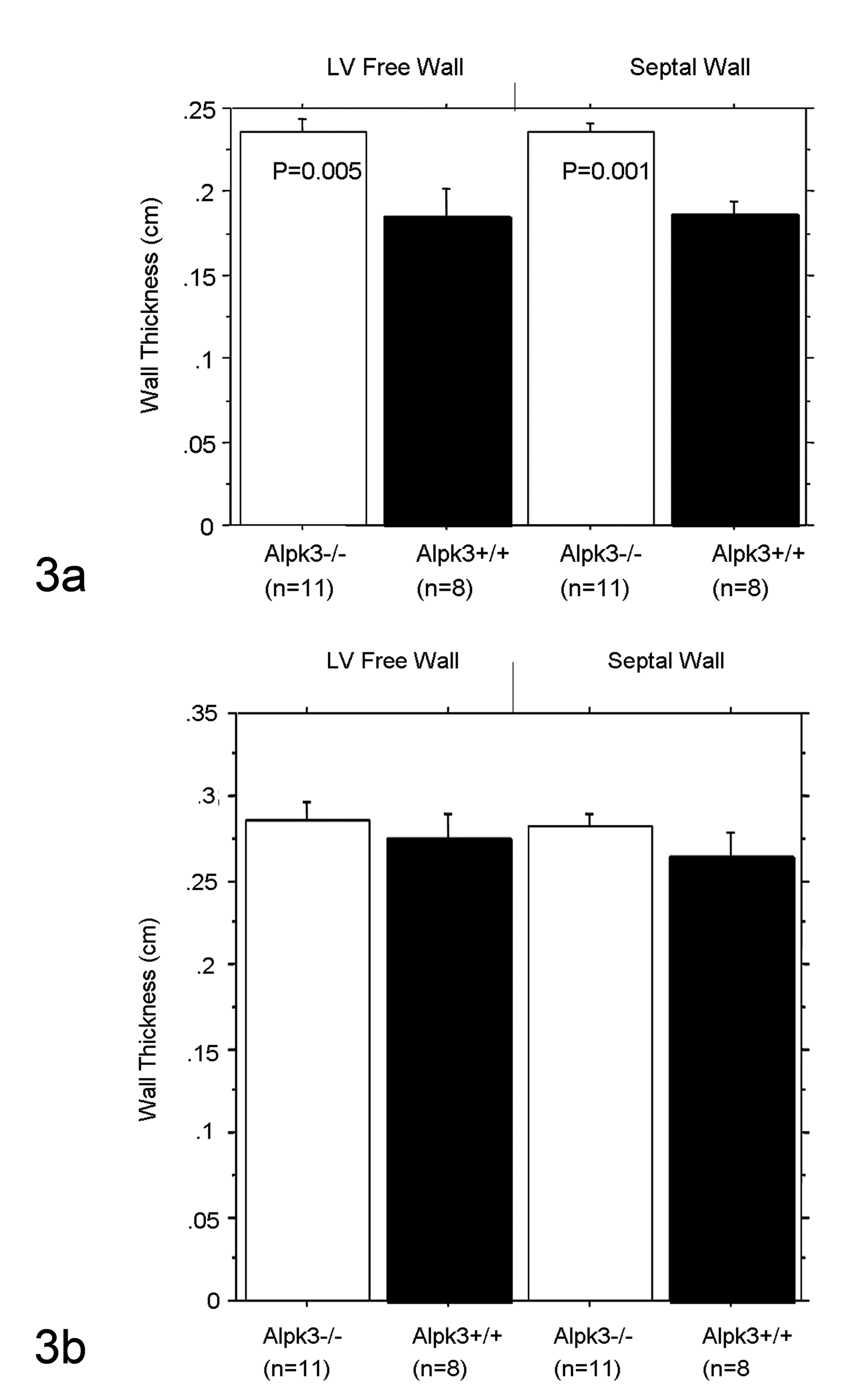
Increased ventricular wall thickness in Alpk3–/– mice. Short-axis ventricular images at end-diastole and end-systole were acquired using MRI imaging system and analyzed using the software Image-Pro (Media Cybernetics, Bethesda, MD). The LV free and septal walls in Alpk3–/– mice were significantly thicker compared with those of their littermates at end-diastole (a), whereas no significant difference was detected at end-systole (b). These data were analyzed by analysis of variance followed by post hoc test Bonferroni/Dunn.
Impaired Myocardial Contractility in Alpk3 –/– Mice at Baseline and Following Dobutamine Challenge
The contractility of LV wall was assessed using 2 different approaches. First, we analyzed LV contractility by MRI. The LV contractility in naive Alpk3–/– mice was reduced more than 50% in both septal and LV free walls (Fig. 4a). We then measured LV pressures using intravascular transducers and found that baseline LV pressures in Alpk3–/– mice were markedly elevated in comparison with Alpk3+/+ animals (Fig. 4b). In the first 5 minutes following dobutamine stimulation, heart rate increased gradually in both homozygous and wild-type mice (100 and 60 beats/min, respectively), with LV pressure in WT mice increasing in conjunction with dobutamine-induced increased heart rates. In contrast, LV pressure in Alpk3–/– mice did not increase in response to dobutamine treatment (Fig. 4c).
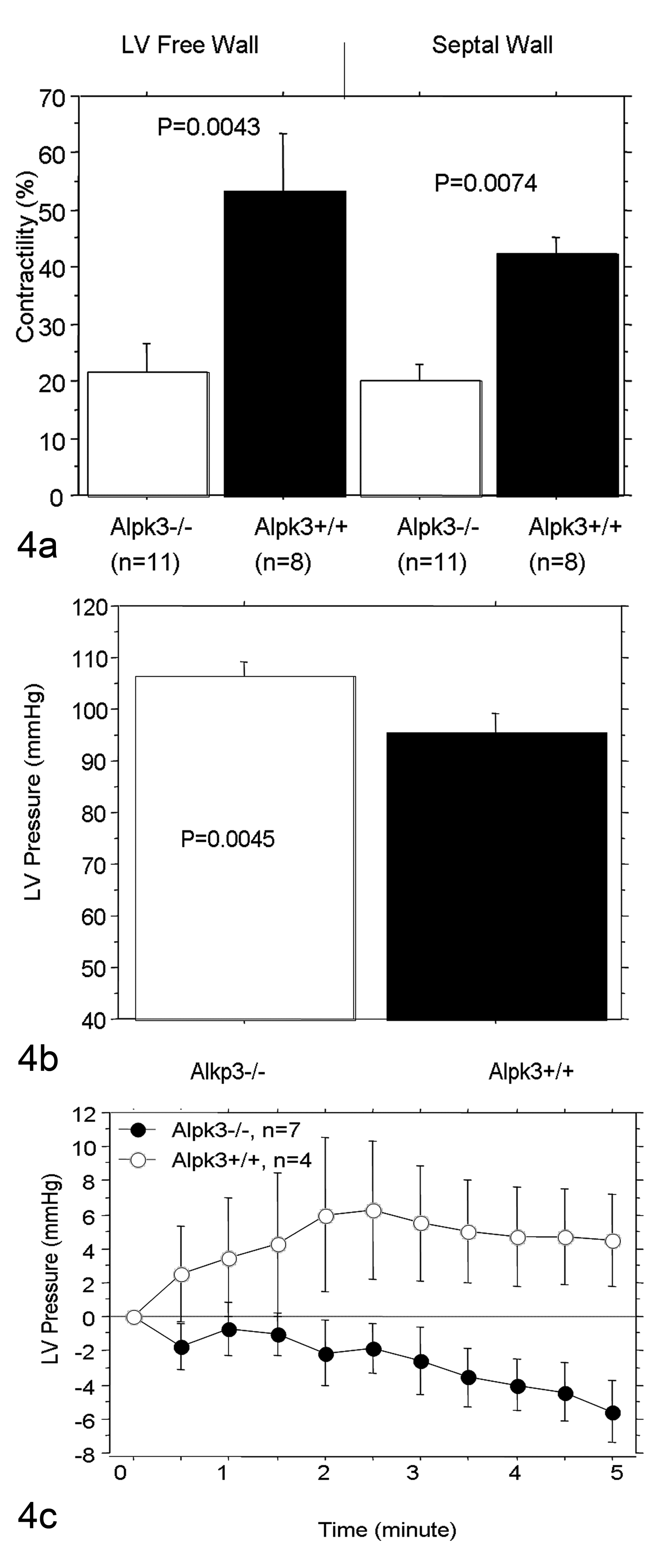
Decreased left ventricular contractility in Alpk3–/– mice. Decreased left ventricular contractility was detected in Alpk3–/– mice by functional MRI analysis indicated by wall thickness change during contraction (a) and by hemodynamic analysis indicated by baseline LV pressure (b) and LV pressure changes during dobutamine challenge (c). These data were analyzed by analysis of variance followed by post hoc test Bonferroni/Dunn and t-test in (a) and (b), respectively. The values represent mean LV pressure +/− standard error.
Pathology Findings in Alpk3–/– Mice
The hearts of Alpk3–/– mice were grossly enlarged in comparison with wild-type littermates, and this enlargement was characterized by thickened walls of all 4 chambers and the interventricular septum. There was no overt malformation detected either grossly or histologically in the large elastic vessels, which included the aorta, pulmonary, carotid, and femoral arteries in Alpk3–/– mice. The Alpk3–/– mice had increased absolute heart weight and increased heart weight/body weight (Fig. 5a) and heart weight/tibia length ratios (Fig. 5b). The measured cross-sections of hearts from Alpk3–/– mice exhibited increased thickness of LV and RV walls compared with those of wild-type littermates, confirming the increased wall thickness detected by earlier MRI studies.
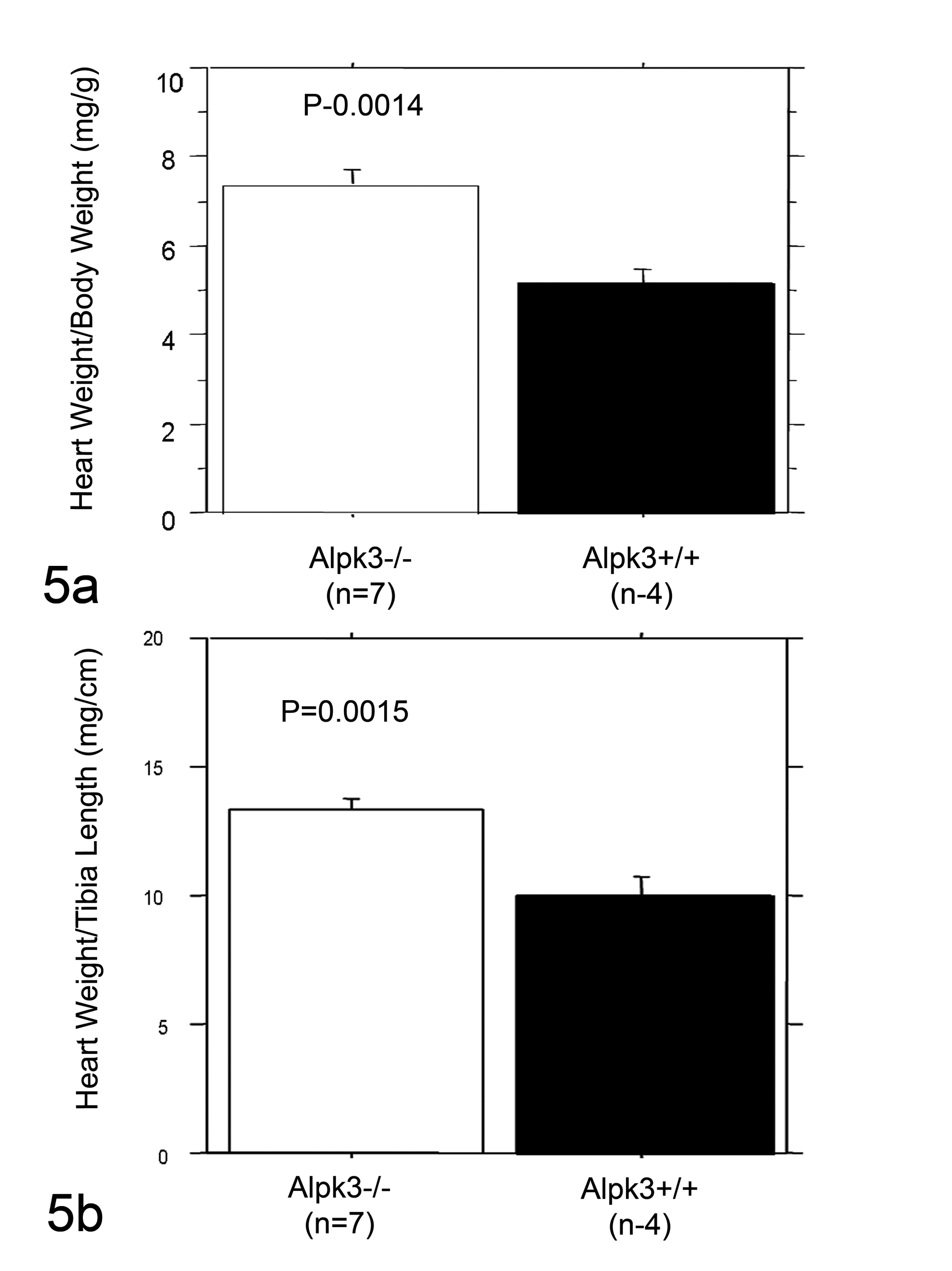
Increased heart size in Alpk3 –/– mice. Heart size was assessed by measuring heart weight/body weight and heart weight/tibia length ratios. Alpk3–/– mice exhibited increased heart size at 24 weeks of age evidenced by the increased heart weight/body weight (a) and heart weight/tibia length (b) ratios. These data were analyzed by the Student’s t-test.
Histologically, the myofibrils of cardiomyocytes in wild-type mice were generally oriented parallel to each other and the long axis of cardiac muscle cells (Figs. 6, 8). This parallel arrangement of contractile myofibrils maximizes the effective mechanical work output by cardiomyocytes. In addition, the intercellular junctions (intercalated disks) connecting individual cardiomyocytes were numerous and well-defined. Cardiomyocyte nuclei in wild-type hearts were generally elongate with blunted ends and located in the center of the cells.
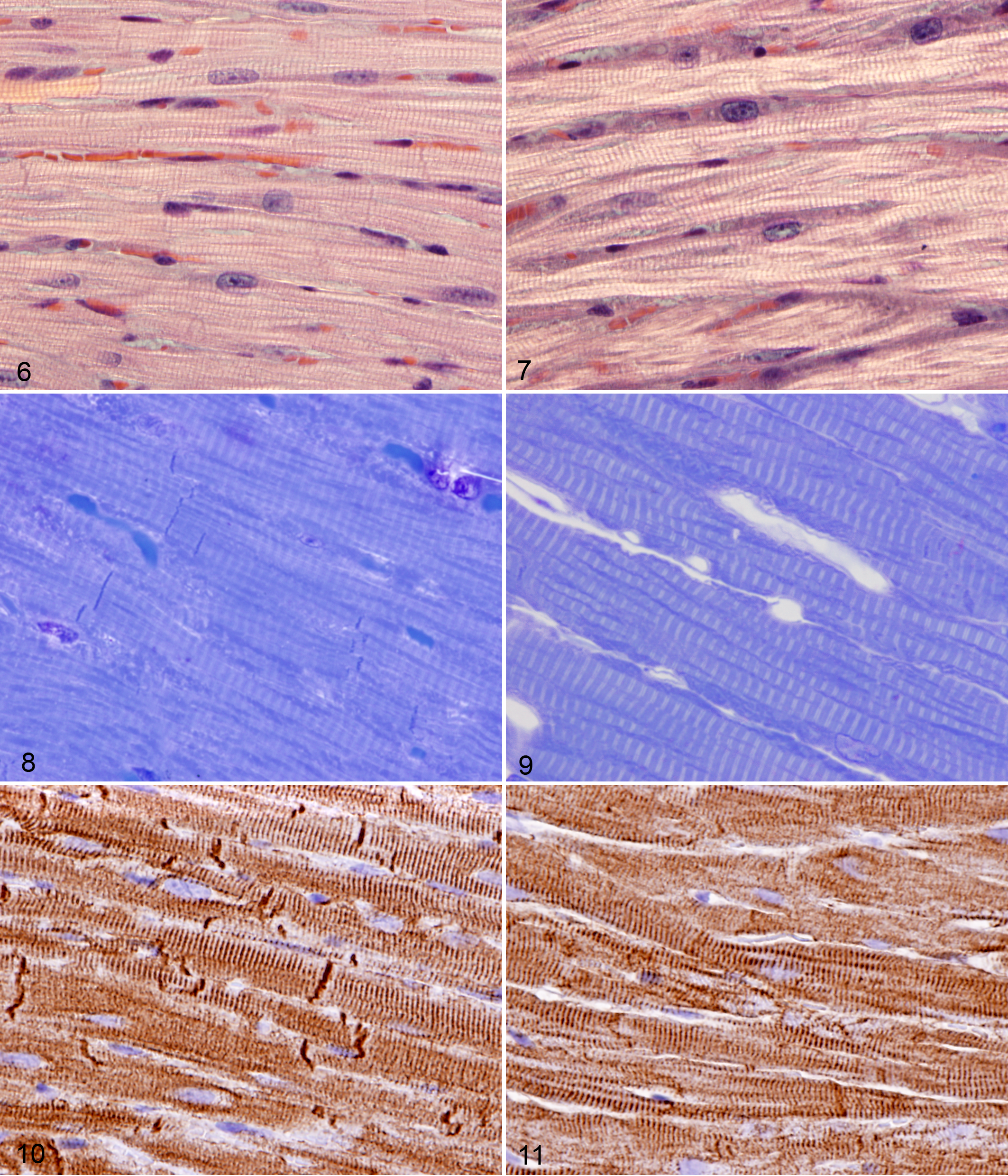
In contrast, cardiomyocytes in Alpk3 –/– mice appeared to be slightly thicker in most areas of the heart muscle than those in corresponding areas in wild-type littermates, with changes evident in the myofibrils, intercalated discs, and nuclei. The most consistent difference from normal was that instead of being arrayed in tight parallel bundles, myofibrils in Alpk3–/– mice had a looser arrangement characterized by increased waviness, overlapping, and crossing of myofibrils within cardiomyocytes (Figs. 7, 9). Multifocally, increased space appeared to be present between individual myofibrils. Another notable difference was that intercalated discs were markedly reduced in number and often had an indistinct and fragmented appearance. In addition, cell nuclei in Alpk3–/– cardiomyocytes were often enlarged, rounded, or lobulated and frequently contained large nucleoli. No lesions were noted in other tissues examined.
Desmin immunostaining accentuated the striated pattern of cardiomyocytes due to labeling of the Z-bands in sarcomeres and highlighted the reduced number and abnormal appearance of intercalated discs, due to very prominent expression of desmin at these locations. In longitudinal segments of myocardium in wild-type mice, relatively large numbers of intercalated discs appeared as sharply defined dense transverse lines, with occasional steps at irregular intervals (Fig. 10). In contrast, intercalated discs in Alpk3–/– mice were sparse in number and tended to be broad, jagged, and less distinct when stained by desmin immunohistochemistry (Fig. 11).
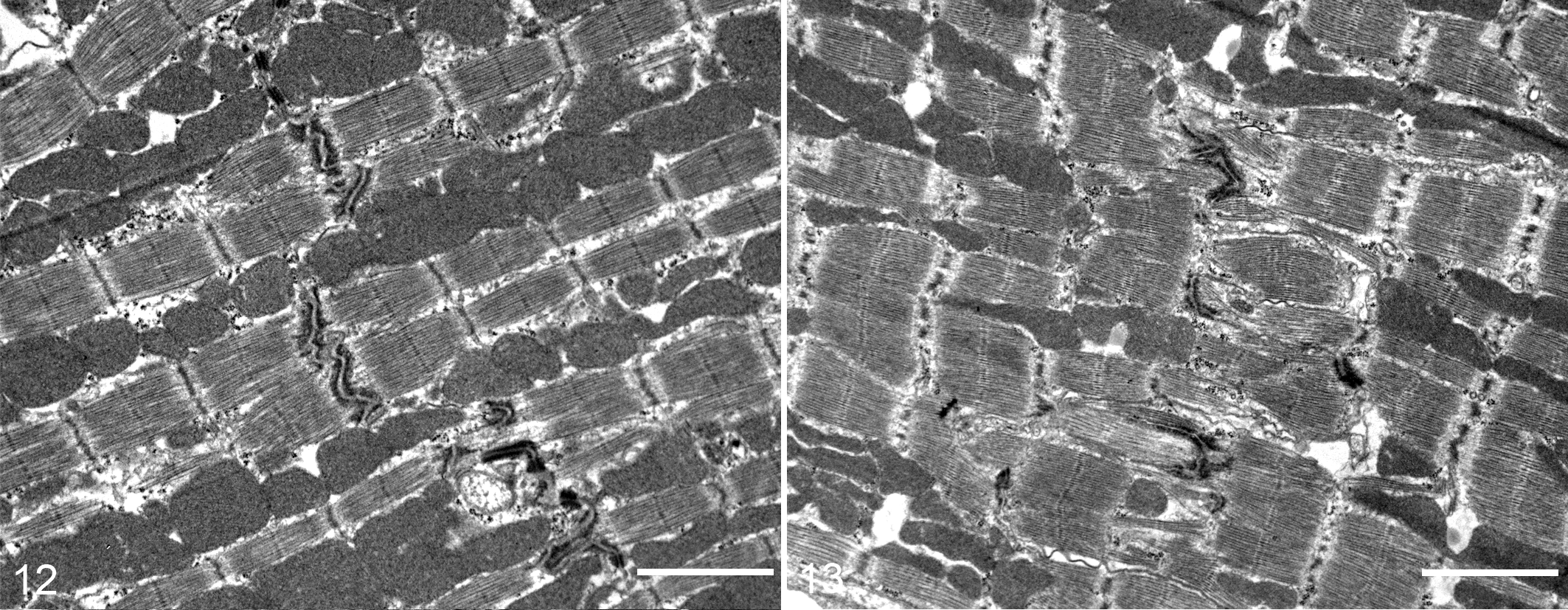
Electron microscopy findings confirmed the altered cytoarchitecture of myofibrils and intercalated discs in Alpk3–/– mice, with changes in cardiomyocytes noted in all regions of the heart in Alpk3–/– mice. In wild-type cardiomyocytes, there was an orderly parallel alignment of myofibrils, which were usually separated by tightly packed rows of mitochondria (Fig. 12). In contrast, the markedly reduced numbers of intercalated discs associated with a diffuse mild disarray of more loosely arranged cardiac myofibrils were evident in Alpk3–/– mice myocardium (Fig. 13). Where present, the intercalated discs in Alpk3–/– mice were less electron-dense and well-defined than those observed in normal control myocardium. Often, the intercalated discs in Alpk3–/– mice were highly convoluted and sometimes fragmented. Other milder ultrastructural changes in the myocardium of Alpk3–/– mice were characterized by a mild increase in nonmyofibrillar and extracellular space, but no increases in interstitial fibrosis or inflammation were observed.
Discussion
Gross cardiac enlargement with mural thickening typically associated with hypertrophic cardiomyopathy was present in Alpk3–/– mice. The development of myocardial hypertrophy in Alpk3–/– mice was confirmed by MRI, gross examination, heart and body weight measurements, histopathology, and ultrastructural analysis. The increased ratios of the total heart weight and the heart weight to body weight strongly supported the presence of cardiac hypertrophy, which is a constant feature of heart failure in humans. Primary cardiac hypertrophy was indicated by the absence of overt malformations in heart valves or large vessels, including aorta, pulmonary, carotid, and femoral arteries, in Alpk3–/– mice. The myocardial hypertrophy in Alpk3–/– mice was characterized by increased thickness of LV and RV walls compared with that of the wild-type mice and by the markedly increased absolute heart weight and increased heart weight/body weight and heart weight/tibia length ratios. MRI studies confirmed an increased thickness in both septal and LV free walls at end-diastole, but there was no significant change in wall thickness at end-systole.
Several characteristic histopathological lesions of hypertrophic cardiomyopathy were conspicuously absent in these mice. The absence of interstitial fibrosis in Alpk3–/– mice is particularly significant. Generally, hallmarks of pathologic cardiac hypertrophy include widespread increased interstitial fibrosis and inflammation, whereas myocardial collagen concentration remains unchanged in adaptive hypertrophy of the heart. The accumulation of fibrillar collagen in pathologic cardiac hypertrophy is a major determinant of the increased stiffness and reduced contractility in the heart in hypertrophic cardiomyopathy. Although myocardial hypertrophy was the predominant feature in Alpk3 –/– mice, several significant findings were present that are more typically associated with dilated cardiomyopathy. Most important, these included the marked increase in end-diastolic and end-systolic volumes of left ventricle, suggesting LV chamber dilation. We also demonstrated reduced CO, SV, and EF in Alpk3–/– mice; MRI findings showed a 50% reduction in both septal and free wall LV contractility in naive Alpk3–/– mice, and baseline LV pressure in Alpk3–/– mice was significantly elevated compared with Alpk3+/+ littermates. Significantly, although dobutamine stimulation increased heart rates in both homozygous and wild-type mice, there was not a corresponding increase in LV pressure in response to dobutamine treatment in Alpk3–/– mice. Although these findings are supportive of dilated cardiomyopathy, it is important to note that Alpk3–/– mice did not develop the thinning of ventricular walls that is a hallmark characteristic of dilated cardiomyopathy.
We believe that the subtle intercalated disc and myofibrillar defects present in Alpk3–/– mice would likely reduce the contractile efficiency of cardiomyocytes and contribute to the development of myocardial hypertrophy in these animals. Thus we propose that Alpk3 should be included with other genes and pathways already known to be involved in cardiac morphogenesis and in the pathogenesis of congenital heart disease.5,6 Despite their importance, the molecular basis of most cardiomyopathies is still unknown.2,28 Many of the single-gene mutations responsible for cardiomyopathy affect proteins that are involved in the generation or transmission of contractile forces or otherwise affect metabolism, calcium homeostasis, or transcriptional control in cardiomyocytes. 29 For example, studies using embryonic stem cells suggested that desmin could be critical for the differentiation of skeletal and cardiac myocytes, 39 but desmin-deficient mice initially appeared to develop normally. 22 However, detailed examination revealed the development of skeletal myopathy and dilated cardiomyopathy in desmin-deficient mice.22,35 Electron microscopic analysis of these mice suggested that desmin helps maintain the integrity of the sarcolemma and myofibrils. 35 As a result of these studies in desmin-deficient mice, specific desmin mutations were identified in human patients with phenotypes similar to those observed in the mouse model.10,21 Mutations in components of the sarcomere and Z-disc have been linked predominantly to hypertrophic cardiomyopathy, 19 whereas changes in the membranous and cytoskeletal structures are more often associated with dilated cardiomyopathy, suggesting that defective transduction of contractile force is an important mechanism underlying this disorder. 18 Nevertheless, approximately 10% of cases of familial dilated cardiomyopathy involve mutations in sarcomere protein genes, 18 demonstrating an overlap in disease genes responsible for hypertrophic and dilated cardiomyopathy. 7 The cytoskeletal proteins associated with some forms of dilated cardiomyopahty (such as dystrophin, sarcoglycan, metavinculin, desmin, and laminin) are involved in the anchorage or lateral integration of myofibrils. 31 For example, dystrophin and other components of the dystroglycan complex connect myofilaments to the membrane and extracellular matrix, and it is believed that destabilization of the dystroglycan complex leads to membrane fragility and loss of membrane integrity that culminates in the degeneration of skeletal muscle and cardiomyocytes. 20 Other genetic causes of dilated cardiomyopathy involve mutations in components of sarcomeres, Z-discs, nuclear membranes, ion channels, titin-N2B, and intracellular Ca2+ modulating systems (see review by Kimura 19 ).
In addition to diffuse hypertrophy, we noted a marked reduction in the numbers of well-defined intercalated discs in histological sections and confirmed this finding with immunohistochemistry and electron microscopy. The relatively mild cytoarchitectural lesions affecting myofibrils and intercalated discs that we observed in Alpk3-deficient mice have not been previously reported. Intercalated discs are important in the mechanical coupling and chemical communication between cardiomyocytes, 4 and our findings strongly suggest that ALPK3 is involved in the formation of these highly organized structures. Intercalated discs play a central role in establishing the proper alignment of myofibrils and in maintaining the overall structure and mechanical efficiency of cardiomyocytes, and it is known that abnormalities in cardiomyocyte cytoarchitecture can impair efficient energy transfer and contractile force production. 40
The loss of myofibril organization and disruption of intercalated disc structure are common features in many models of cardiomyopathy, and in recent years mutations affecting the structure and composition of the intercalated discs have been recognized in many types of dilated cardiomyopathy.12,13,31 For example, myofibrillar disarray and intercalated disc abnormalities are hallmark features of cardiomyopathy in muscle LIM protein (MLP)-deficient mice.3,12 In those mice, altered expression levels of cytoskeletal proteins (either the lack of MLP or an increased expression of tropomodulin) lead to alterations in intercalated disks and impaired myofibrillar contractility. 12 Similarly, mutations in metavinculin disrupt the connection between intercalated discs and actin filaments, which impedes transmission of contractile forces at the thin filament-intercalated disc interface and represents the most likely underlying mechanism responsible for the development of dilated cardiomyopathy in metavinculin-deficient mice.30,38 Disruption of intercalated disc structures can also have lethal effects; the adherens-type junctions in intercalated discs contain N-cadherin, which has an important role in myofibrillogenesis, 34 and mice lacking the N-cadherin protein die in utero with severe structural defects of the myocardium. 32
In conclusion, we have discovered a role for ALPK3 in establishing normal structure and function of the myocardium. The markedly reduced numbers of apparently defective intercalated discs resulting in diffuse mild myofibrillar disarray in Alpk3 null mice suggest that this gene is involved in the assembly of intercalated discs and thus in maintaining the tight parallel arrangement of adjacent myofibrils. Taken together, our data suggest that defective intercalated disc formation in Alpk3-deficient hearts is the most likely cause of the mild myofibrillar disarray present in these mice. It is likely that myofibril disarray impairs normal contractility of cardiomyocytes and that the inability to generate sufficient muscle tension is the precipitating cause of cardiac insufficiency and cardiomyopathy in these mice.
Footnotes
Acknowledgements
We thank Brent Sharp and Angela Harris for their assistance in measuring cardiac function, David R. Powell for helpful discussions, Ryan Vance for necropsy assistance, and Kathy Henze and Mary Thiel for histology support.
The authors declared that they had no conflicts of interest with respect to their authorship or the publication of this article.
The authors declared that they received no financial support for their research and/or authorship of this article.