
Abstract
Catecholamine-induced cardiomyopathy (CCM) is an entity associated with increased levels of catecholamines causing subendocardial and papillary muscle cardiomyocyte degeneration and necrosis. In 2020, 49 autopsies from early rabbit deaths in a colony used for medical device biocompatibility studies were submitted for microscopic examination. Of the 49 rabbits, 26 had histologic changes consistent with CCM. No common stressor for CCM was determined in affected rabbits. Animals were generally male, were 12–16-wk-old, and were found dead or had bloating, lethargy, and/or diarrhea. Those observed with clinical signs were euthanized and autopsied per the organization’s standard operating procedures. Heart lesions consisted of various degrees of apical subendocardial myocardial degeneration and necrosis. Common non-cardiac lesions included pulmonary congestion and edema, hepatic congestion and centrilobular hepatocellular degeneration, and/or variable intestinal submucosal edema.
Several well-recognized cardiomyopathies are associated with supraphysiologic catecholamine release in humans and other mammalian species. Perhaps the best characterized of these entities in veterinary medicine are the cardiomyopathies seen with functional pheochromocytomas and the “capture” cardiomyopathies associated with stress.11,33 Here I present the cardiac changes consistent with catecholamine cardiomyopathy (CCM) in laboratory rabbits and briefly review the pathophysiology associated with CCM.
Stress cardiomyopathy was first described in rabbits in 1973 further to investigating models for idiopathic endomyocardiopathy in humans. 37 By creating stress in rabbits using intermittent crowding, changes consistent with heart failure were found at autopsy. The microscopic heart lesions that were correlated with heart failure in these rabbits included myocytolysis, interstitial edema, and increased amounts of interstitial acid mucopolysaccharides. Longer-term survivors also had coagulative myocardial necrosis, myocardial fibrosis, endocardial fibroelastosis, and basophilic cardiomyocyte degeneration. A key distinguishing feature of these changes was the consistent apical and subendocardial distribution of lesions. Lesions and progression of this disease were similar to the human idiopathic endomyocardiopathy that was being modeled.11,25,37
Long before this work with rabbits, 37 a paper was published on voodoo death, which recounted anecdotal experiences of human death from fright.6,33 “Voodoo death” was defined as sudden unexplained death resulting from a voodoo curse. These deaths were hypothesized to have a physiologic basis in activation of the sympathetic or sympathicoadrenal divisions of the nervous system. This hypothesis forms the basis for much of the current understanding linking emotions or fear to some forms of sudden, unexplained cardiac death.1,5–7,9–13,27,32
Here I provide comprehensive histologic findings in rabbits with CCM, briefly discuss the pathophysiology of rabbit CCM, clarify distinguishing characteristics of CCM from other rabbit heart diseases, and provide observations on the incidence of CCM in laboratory New Zealand White rabbits.
Materials and methods
Findings in this paper are based on observations of diagnostic autopsies from the rabbit colony of a medical device contract research organization. The decision to conduct autopsies and collect tissues was based on U.S. Food and Drug Administration (FDA) guidelines for early-death animals. The research facility that houses the rabbits is certified to ISO 9001:2015, and is accredited to ISO/IEC 17025:2017; continued compliance is maintained with all relevant regulatory and statutory requirements including but not limited to ISO 13485:2016, 21CFR211, and 21CFR820. The facility has full accreditation by the Association for Assessment and Accreditation of Laboratory Animal Care and follows all USDA regulations. The procedures for all rabbits used in studies were approved by the organization’s Institutional Animal Care and Use Committee.
Rabbits were single-housed in standard stainless-steel cages (40 × 61 × 61 cm) in a room with other rabbits. Animals were provided with a 12:12-h light:dark cycle; room temperature was maintained at 17–21°C; and relative humidity was 30–70%. Animals had access at all times to water distributed by an automatic watering system. The water valves were tested once daily for patency. Each rabbit was fed 240 g of rabbit chow (Hi-Fiber Diet 5326; Purina Mills) and one handful of timothy hay (Kaytee Products) daily. Collection pans were cleaned daily with water.
Autopsies were performed by trained technical staff. Tissues submitted for microscopic evaluation were part of a standard tissue set (liver, spleen, kidneys, adrenals, submandibular and mesenteric lymph nodes, thymus, heart, lungs, cerebrum, cerebellum, brain stem, thalamus, skeletal muscle, skin, and femur with bone marrow). Collection of gastrointestinal (GI) tract was not performed routinely. Rabbits either died spontaneously or were euthanized because of diarrhea, bloating, anorexia, and/or respiratory distress. These rabbits were either stock animals or were part of biocompatibility studies evaluating biologic responses to various medical devices. Routine histopathology consisted of paraffin embedding and microtome sectioning at 3–5 µm followed by staining with H&E. I received resulting slides and brief notes regarding clinical signs (if any) and autopsy findings (if recognized).
Results
Over 1 y (2020), 49 rabbits of the > 8,000 rabbits used annually by this facility underwent an autopsy as prescribed by company policy and FDA guidance. Of those 49, 26 had histologic changes consistent with CCM, indicating a relatively high incidence of CCM in the population of rabbits undergoing a pathology evaluation following death or euthanasia for various reasons. Each submission of tissue for evaluation was accompanied by a form providing limited history and macroscopic findings (Table 1).
Information provided on submission of 12–16-wk-old laboratory New Zealand White rabbits for autopsy.
BHF = biventricular heart failure based on changes consistent with both LHF and RHF; CCM = catecholamine cardiomyopathy based on degeneration and/or necrosis localized to the apical subendocardial myocardium; F = female; implant = animals implanted with a test article or control article to assess local reactivity to the article; LHF = left heart failure based on pulmonary congestion and edema with alveolar erythrophagocytosis; M = male; pyrogen = animals administered test article extracts and evaluated for febrile response or reaction; RHF = right heart failure based on centrilobular congestion and hepatocellular degeneration and/or necrosis; stock = animals either awaiting inclusion on a future study or off a previous non-terminal study.
Test procedures and handling of the affected rabbits varied; a common history of testing or handling was not established. Affected rabbits were 12–16-wk-old (~2.5 kg); generally, rabbits were at the testing facility for 2–3 wk before developing clinical signs or dying. More specific weights and ages were not available.
Most rabbits with CCM were male. Although the approximate ratio of males to females in acute studies at this facility was 2:1, a markedly greater proportion of CCM rabbits were male (> 8:1 male:female incidence).
As expected, rabbits with CCM often died suddenly or had signs of heart failure. Many rabbits diagnosed with CCM also had antemortem signs of illness related to the GI system, including diarrhea, bloating, and inappetence or anorexia.
Systemic lesions associated with right heart failure (RHF) dominated those associated with left heart failure (LHF) or biventricular heart failure (BHF). The right-sided dominance was correlated with the GI signs frequently seen in affected animals.
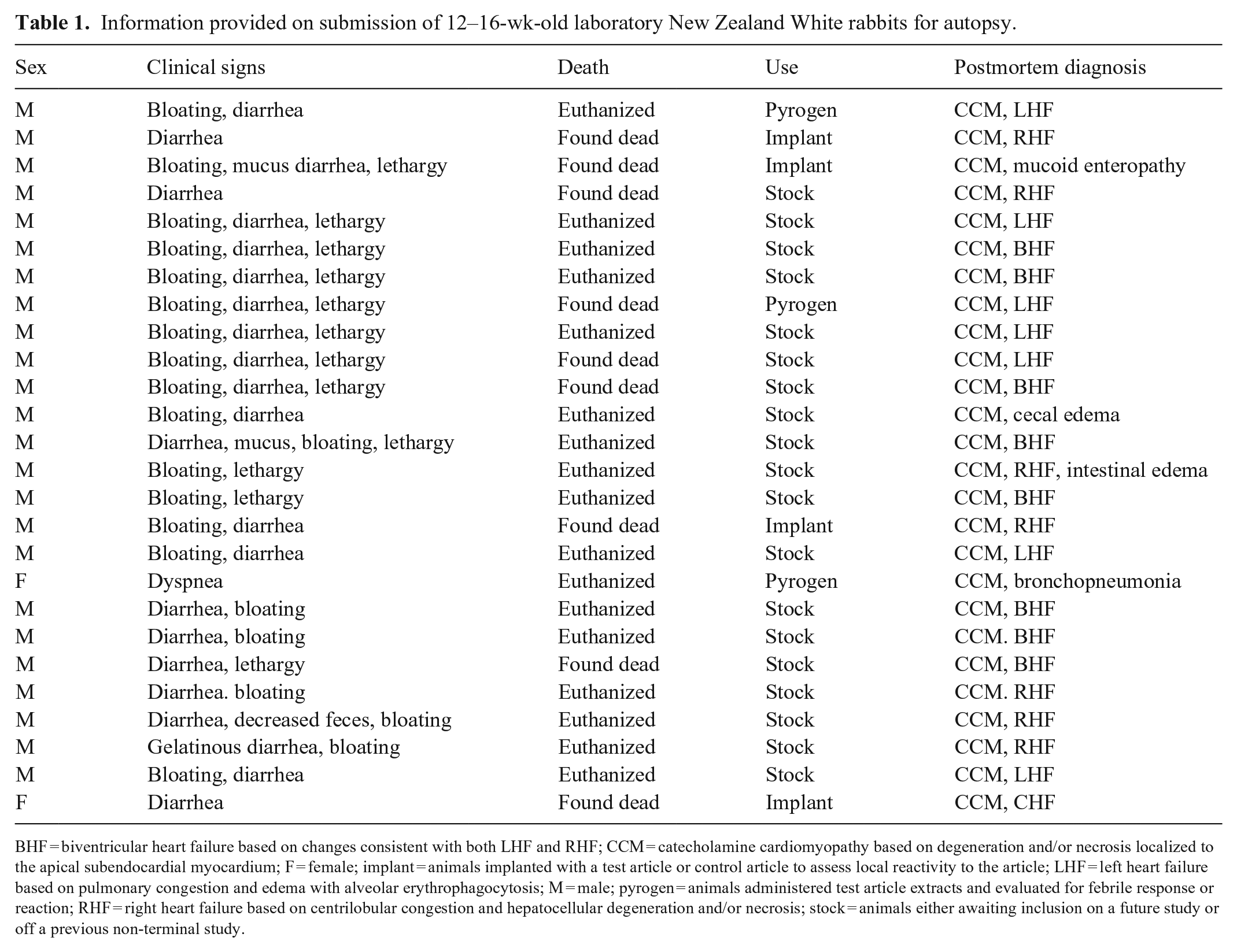
Cardiac histologic findings consistent with CCM varied in degree and duration. Acute cardiac lesions were myocardial degeneration, necrosis, and loss, with endomysial edema and mucopolysaccharide deposition (Fig. 1A). As would be expected with cardiomyocyte necrosis, subacute and chronic lesions were attended by infiltrates of histiocytes with lesser numbers of heterophils (Fig. 1B). Additionally, in rabbits in chronic heart failure that survived for a longer time than rabbits with only inflammatory cell reactions, dystrophic mineralization of cardiac lesions was seen as well as fibrosis (Fig. 1C). Lesions were consistently subendocardial and concentrated in the apical myocardium (Fig. 1). Histologically, no marked preference was seen for left ventricular free wall, right ventricular free wall, or interventricular septum.
Cardiomyocyte degeneration, necrosis, and loss in the heart of a laboratory New Zealand White rabbit.
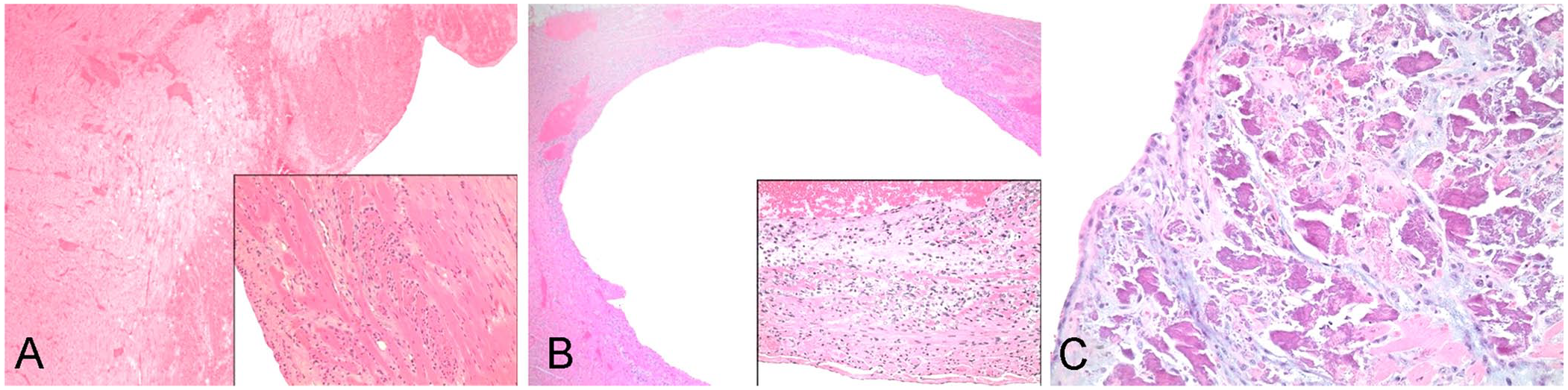
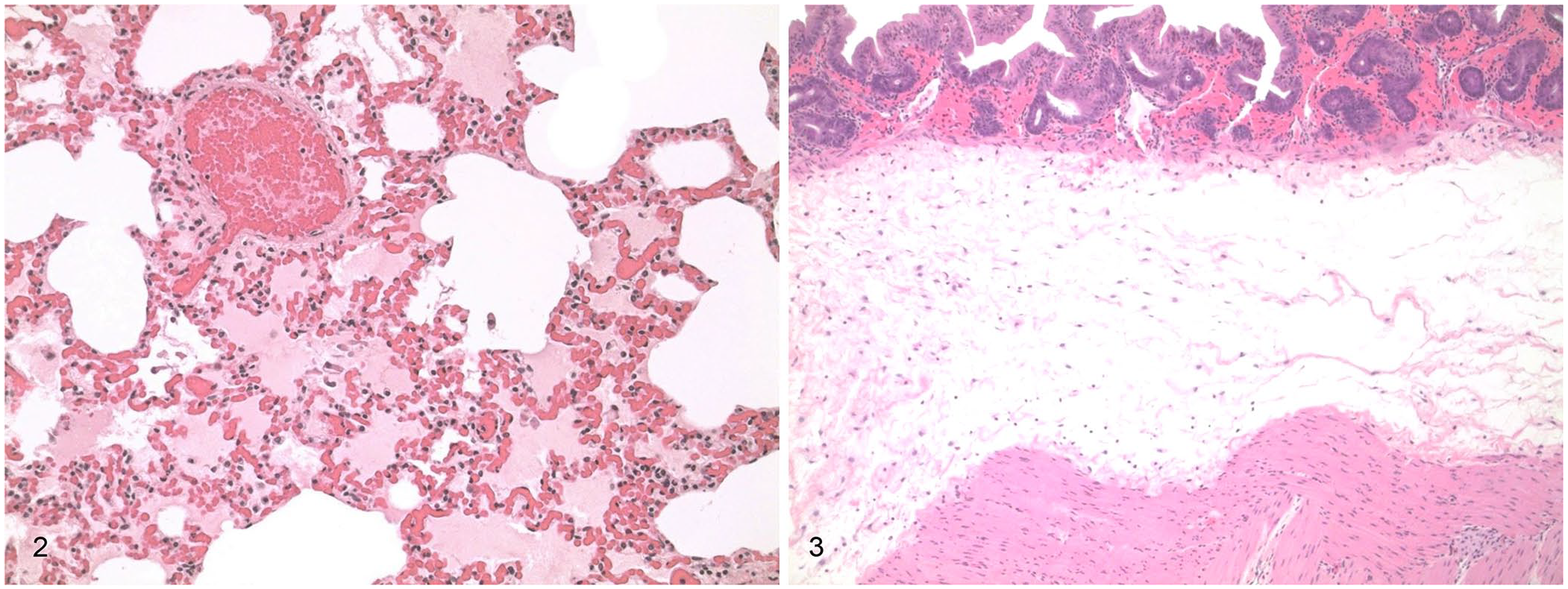
Lesions supportive of LHF and BHF were pulmonary congestion, edema, and hemorrhage (Fig. 2). In animals with RHF and BHF, chronic passive hepatic congestion was consistent; additionally, congestion and edema affecting the GI submucosa (Fig. 3) and mesenteric lymph nodes were common. Tissue sets of only 6 of the CCM animals included intestines, and 5 of the 6 had small intestinal submucosal edema.
Marked pulmonary congestion and edema in a rabbit, consistent with left heart failure. H&E.
Discussion
The histologic changes in 26 of 49 rabbits are consistent with changes known to be associated with CCM, 37 but did not include the broader subset of rabbit inflammatory cardiac lesions included in a 2002 publication. 30 These observations, despite limitations regarding the lack of in-life data and observations, are consistent with classic findings in CCM along with other findings that are worthy of further study, such as the predominance of males and of GI signs in CCM-affected rabbits.
The GI signs of diarrhea, bloating, and inappetence or anorexia were presumed to be the result of RHF. Although a cause-and-effect relationship between the GI changes and RHF was not definitive, the GI changes in cases of CCM were not characteristic of other GI illnesses typically seen in rabbits. Shiga toxin–producing Escherichia coli is one of the few primary intestinal diseases that could manifest with submucosal edema, but the localization in these cases (pan-intestinal) would be atypical for Shiga toxin, which usually is localized in the large intestine. In many of the animals, sections of the GI tract were not included in tissues evaluated microscopically; of the 6 examined, 5 had submucosal edema. Although listed as a GI sign, bloating in many of the animals may have been a consequence of aerophagia in dyspneic animals (husbandry personnel were not trained to detect rabbit dyspnea). The low diagnostic incidence of this syndrome and abbreviated history and signs made further determination of definitive factors associated with the disease problematic. No definitive dietary or husbandry changes have been recognized to date in this colony.
CCM in humans can be precipitated by acute emotional stress, acute intracranial events (including subarachnoid and intracranial hemorrhage), head trauma, ischemic stroke, acute medical illnesses (including sepsis), surgical procedures, pheochromocytoma, and administration of exogenous catecholaminergic agents (methylxanthines, amphetamines, cocaine, epinephrine).1,5,7–9 CCM in humans has histologic findings similar to those in rabbits and is also associated with a variety of kinetic alterations. Alterations may include apical and mid-ventricular ventricular dysfunction, isolated mid-ventricular and basal ventricular dysfunction, global hypokinesis, and other wall motion abnormalities without a coronary artery distribution pattern. 4 Dynamic changes in rabbits with classic spontaneous CCM have not been assessed with echocardiography or other imaging modalities. Echocardiograms of a rabbit model of takotsubo cardiomyopathy, a reversible stress cardiomyopathy in people, have been investigated but it is unclear how applicable those findings are to spontaneous CCM in rabbits. 34
Diagnosis of CCM is based on characteristic apical subendomyocardial microscopic lesions and extracardiac changes compatible with heart failure. Random background cardiac changes seen in laboratory rabbits 30 can be differentiated from CCM based on microanatomic localization and characteristic changes seen with CCM. In CCM, myocardial histologic lesions are variable cardiomyocyte degeneration, necrosis, and loss. Lesions may include histiocytes and lesser numbers of heterophils infiltrating endomysium that have been expanded by mucopolysaccharides, fibrin, edema, and fibrous connective tissue. A key diagnostic feature of CCM is that lesions in rabbits and people are consistently localized to the subendocardial apex and papillary muscles. The characteristic apical and subendocardial histologic lesions of CCM are localized to the area of the heart with the highest concentration of catecholamine receptors and the area of highest metabolic activity (papillary muscles).5,6,11,14,23,24 Rabbits receiving high-dose catecholamine infusions have a similar stereotypical cellular reaction with infiltration by mononuclear cells, macrophages, and heterophils.13,30 In both people and rabbits, 38 cardiomyocyte changes within lesions include intense cytoplasmic eosinophilia with loss of cross-striations (hyalinization) designated as coagulative myocytolysis, myofibrillar degeneration, or contraction band necrosis. With light microscopy, lesions may not be readily apparent in all sections of the heart, and serial sections may be required to rigorously exclude its presence. The findings on ultrastructural examination are invariably more widespread, often involving nearly every muscle cell, even when the light microscopic appearance is unimpressive. 29
In non-spontaneously fatal lesions, 4 of the animals had mineralization with multifocal subendocardial predisposition. Intense rapid mineralization makes it likely that the subcellular mechanisms that underlie the development of coagulative myocytolysis involve calcium entry. Ionized calcium plays a major role in the progression of CCM. In normal hearts, stimulation of beta receptors by catecholamines results in increased intracellular cyclic adenosine monophosphate (cAMP) by stimulating adenylate cyclase. cAMP increases intracellular protein kinases. These kinases phosphorylate phospholamban, which results in increased ingress of calcium into the sarcoplasmic reticulum, which in turn results in an increased cell relaxation rate. Catecholamines also directly cause membrane phosphorylation, which further contributes to ingress of calcium and inhibits the troponin–tropomyosin system, resulting in the inhibition of excitation–contraction coupling. Excitation–contraction coupling can also be the consequence of impaired calcium transport in the sarcoplasmic reticulum.7,17
Catecholamines and their oxidation products also may have an acute direct toxic effect on the myocardium. Longer-term elevation of catecholamines leads to downregulation of beta-adrenergic receptors thereby inducing decreased cardiomyocyte function and decreasing the contractile units.10,20,21,31 Catecholamine-mediated myocardial stunning due to direct toxicity is possible through a variety of mechanisms. 23 Additionally, catecholamine-induced vasoconstriction results in compromised blood flow through small arterioles, which would also be expected to exacerbate cardiomyocyte hypoxic injury. 7 Myocardial damage itself may then be a source of oxygen free radicals, which cause further direct cardiomyocyte injury.7,18,20,23,35,38
Cardiac lesions in CCM are not prevented by adrenalectomy, suggesting that lesions are caused by the direct neural connection rather than the blood-borne route of sympathetic hyperactivity. Clinical and pharmacologic data support the concept that myocardial necrosis is caused by catecholamine toxicity and that catecholamines released directly into the heart via neural connections are more toxic than those that reach the heart via the bloodstream. Nevertheless, the 2 routes may be additive and, during an autonomic storm, both neurologic and adrenal inputs are increased.4,9,12,15,29 Thus, histologic lesions may reflect the effects of large volumes of norepinephrine released into the myocardium from sympathetic nerve terminals. Myocardial necrosis is greatest near the nerve terminals in the endocardium and is progressively less severe in muscle cells near the epicardium, providing additional evidence that catecholamine toxicity produces lesions seen in CCM. The secondary cardiomyopathy associated with functional pheochromocytomas manifests with similar changes and has a pathophysiology similar to other types of CCM; the primary difference is that humoral catecholamines may play a much greater role than local release from regional nerve terminals.4,9,16,22,23,26,28,36,38
Arrhythmias associated with CCM and resulting in sudden death can also contribute to rates of mortality. Because cardiomyocyte degeneration is predominantly subendocardial, lesions may also affect the cardiac conducting system, predisposing to cardiac arrhythmias. This predisposition combined with the propensity of catecholamines themselves to produce arrhythmias may raise risks of sudden death.5,10,14,16,17,21,23 –25,38
Rabbits diagnosed with CCM were 12–16-wk-old in my study. Several publications suggest that younger rabbits may be less susceptible. There are fewer sympathetic nerves and lower epinephrine stores in fetal and newborn rabbits than in adults. Ingrowths of large preterminal nerve trunks enclose catecholamine-containing terminal varicosities to form the autonomic ground plexus in maturing rabbits; therefore, basic neural networks and pathologic concentrations of catecholamines may not be present prior to maturation.2,3 Interestingly, young rabbits are also less sensitive to the alpha-2 receptor agonist, dexmedetomidine, compared to adult animals. 2 Among the other cardiac changes occurring during maturation, expression of the cardiac Na+-Ca2+ exchanger is high at birth and declines rapidly to adult levels by ~21 d in rabbits, and this decline is regulated by the thyroid hormone. Given the role of disruption of calcium homeostasis in the pathogenesis of CCM, loss of this exchanger may also be contributory in the age group affected. 3 Additionally, changes in excitation–contraction coupling that occur during maturation are also governed by the ability to regulate intracytoplasmic and sarcoplasmic calcium levels 19 ; hence, it is not surprising that susceptibility to CCM may be greatest at a specific developmental period in cardiac maturation.
In this colony of laboratory rabbits, CCM appears to be a relatively uncommon cause of morbidity and mortality based on a very limited sampling. Nevertheless, the overall incidence of fatal CCM is enough to suggest several general observations. Generally, affected rabbits appear to succumb to the effects of CCM relatively rapidly or show a variety of acute morbidities (respiratory distress, diarrhea, boating, and/or anorexia) necessitating euthanasia. GI signs are common and may be associated with RHF. Despite the low number of documented CCM animals in this colony, the overall incidence may be greater, particularly in those animals that survive the acute manifestations of CCM. Not all animals are autopsied, and many animals in studies are primarily evaluated for study-specific changes rather than overall postmortem findings.
We were unable to link any specific handling, husbandry, or experimental manipulations to rabbits that developed CCM. The study facility has a variety of background stressors that exist for all rabbits (noise, cage movement, handling, examination). The manipulations for affected rabbits were heterogeneous and did not support any trends or associated interventions.
Given the rapid onset of signs and the low incidence of this syndrome, the overall impact on subacute and chronic studies was considered minimal. At this facility, incidental cardiac lesions are rare (lesions unassociated with morbidity or premature mortality) in those animals examined. To date, lesions characteristic of CCM have not been recognized in subclinical animals (but most are not evaluated for these lesions). Specific stressors were not identified, but rabbits were generally 12–16-wk-old males.
Footnotes
Acknowledgements
My thanks to the veterinary professional and paraprofessional staff at NAMSA for in-life care and for conducting the autopsy prosection, specimen collection, and histologic processing for my study. Thanks also to Dr. Francisco Alejandro Uzal for helping prepare images, and to reviewers as well as Dr. Grant Maxie for valuable feedback on the manuscript.
Declaration of conflicting interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.