
Abstract
The pharmaceutical industry and regulatory agency toxicology testing paradigms in the United States currently appear successful, in part because of the continuously increasing life expectancy and the declining age-adjusted cancer rates in the United States. Although drugs likely have a minimal impact on the population statistics for cancer rates, pharmaceutical pathologists and toxicologists must focus on the individual risk for pharmaceutical carcinogenesis. As our understanding of carcinogenesis increases exponentially, and after hundreds if not thousands of rodent cancer tests, significant improvement in the precision of human pharmaceutical carcinogenesis hazard identification should now be possible and would enable a reduction in the substantial false-negative and false positive-rates reported herein. The appropriate use of acute, subchronic, chronic, and special toxicology tests to identify the major associated cancer risk factors, specifically, hormonal modulation, immunosuppression, genetic toxicity, and chronic toxicity, can be recognized through this review of pharmaceutical carcinogens. Significant opportunities exist for improving the effectiveness and efficiency of the current cancer risk assessment paradigm.
To provide context for a multinational initiative to explore opportunities to improve the effectiveness and efficiency of the pharmaceutical cancer hazard identification process, we briefly review here the historical foundations for testing and the success of the testing paradigm on a population basis in a United States-centric fashion. An outline of the current regulatory testing paradigm then sets the stage for a review of the labels of currently marketed pharmaceuticals and a comparison of the animal cancer responses with the human experience. After decades of experience and hundreds of lifetime rodent pharmaceutical cancer tests, this opportunity to take stock of the successes and failures of the existing paradigm forms the cornerstone for process change that may have the potential to improve the prediction of human cancer risk.
Supplementary material (including results of every drug reviewed from the Physicians’ Desk Reference [PDR]) for this article is available at http://vet.sagepub.com/supplemental.
Historical Context
Although xenobiotic-induced carcinogenesis was recognized as early as the 1700s, in the United States the modern regulatory basis for controlling exposure to carcinogens in food, drugs, and cosmetics was established with passage of the Food, Drug, and Cosmetic Act of 1938. 12 Two tragedies—scores of deaths from the administration of an adulterant, diethylene glycol, in an elixir of sulfanilamide and multiple occurrences in women of blindness induced by corrosive eye cosmetics—were driving forces leading to this Act. Public recognition of and concerns about carcinogenic hazard are also apparent in the same time frame and are reflected in the political response manifested through the creation of the National Cancer Institute (NCI) in 1937. Although carcinogenicity testing of pharmaceuticals in the United States can be traced back to the 1930s, when diethylstilbestrol was tested in mice, 6 public pressure for regular testing and regulatory control of pharmaceutical cancer risk was not really felt until the late 1950s and early 1960s. The political response to this public awareness and concern was reflected, in part, in the Delaney Clause, a food additive amendment established in 1958 that banned the use of food additives shown to cause cancer in animals; and by the initiation of the NCI bioassay program in 1961, 42 which was transformed into the National Toxicology Program (NTP) as part of the National Institute of Environmental Health Sciences (NIEHS) in 1978. Further public awareness and political responsiveness was driven by the publication, in 1962, of Rachel Carson’s Silent Spring. Rachel Carson can be credited with leading the effort to eliminate dichlorodiphenyltrichloroethane (DDT) and chlorinated hydrocarbon pesticides from commercial use in the United States, as well as leading the public to recognize the environmental hazards, including cancer, associated with certain commercial chemicals.
A substantial number of animal cancer bioassays were conducted in the 1960s and 1970s. Examples exist of long-term cancer studies of 7 and 10 years in dogs and monkeys, respectively, being used by pharmaceutical companies during development of drugs such as birth control agents (eg, levonorgestrel and norelgestromin). However, standardization of practices did not become widespread until the 1970s and 1980s. In 1982, the United States Food and Drug Administration (FDA) Red Book, the first regulatory document with detailed carcinogenicity testing guidance, was published by the Bureau of Foods. Other key events affecting carcinogenicity hazard assessments included the discovery and development of the Ames test (a bacterial test system for the detection and classification of mutagens and carcinogens) in 1973 9 and the implementation of the Good Laboratory Practice (GLP) Act in 1978 (Title 21, Code of Federal Regulations, Part 58).
The 1980s and 1990s were characterized by growing standardization and refinement of the rat and mouse lifetime cancer test process and application of results. An early example of the latter is the realization that a positive response in the rodent cancer assay is a poor predictor of human cancer risk for compounds with robust CYP450 enzyme induction. In an FDA Advisory Committee meeting in February 1987, the FDA considered the relevance of the multi-site carcinogenesis seen across species associated with robust CYP450 enzyme induction in lifetime cancer studies of loratadine, cetirizine, and doxylamine in rats and mice. After a thorough review of the data, the FDA approved loratadine and cetirizine as nonsedating antihistamines and elected not to respond in terms of restrictive labeling or restricting sales of the marketed over-the-counter sedating antihistamine doxylamine. Subsequently, this mouse liver/rat thyroid tumorigenesis syndrome no longer created a barrier to approval. Loratadine and cetirizine quickly generated billions of dollars in revenue, and doxylamine sales continued unabated.
High-Level Evaluation of the Effects of Public Policy on Cancer Incidence
An assessment of the effectiveness of public policy relevant to environmental xenobiotics should include a review of the trends in life expectancy and cancer rates for the population at risk. In the United States, across the entire population, a continuous increase in life expectancy started in 1900 and continued through 2006, representing an extraordinary period of accomplishment in medical research and in the agricultural, chemical, and pharmaceutical industries in concert with the associated regulatory authorities. 10
As populations live longer, the incidence of cancer increases. In the United States, the incidence of cancer peaked in the early 1990s, and the incidence of age-adjusted cancer has slowly declined since then. Probable reasons for this decrease include the increased recognition of the risks associated with smoking, as well as other lifestyle decisions. In 2006, 559,888 people in the United States died of cancer, making it the second most frequent cause of death. Heart disease represented the most frequent cause of death; together, heart disease and cancer accounted for 1,191,524 deaths in the United States in 2006. 26
To explore the potential role of pharmaceutical agents in cancer causality, this review examines the top 10 cancers as determined by numbers of new diagnoses per year. These cancers account for >75% of all new cancer cases and two-thirds of all cancer deaths per year. In Table 1 , the generally recognized risk factors associated with each of these cancers are identified. Three pharmaceutical risk factors, immunosuppressants, cancer chemotherapies, and hormonal modulation therapies, are potentially significant for certain cancer types. Two of these risk factors, immunosuppression and hormonal modulation, are increasingly understood, although they have not been consistently identified as carcinogens in animal cancer studies, as detailed below.
Top 10 United States Cancers in Terms of Mortality and the Generally Recognized Risk Factors Associated With Each of These Cancers in 2010 1
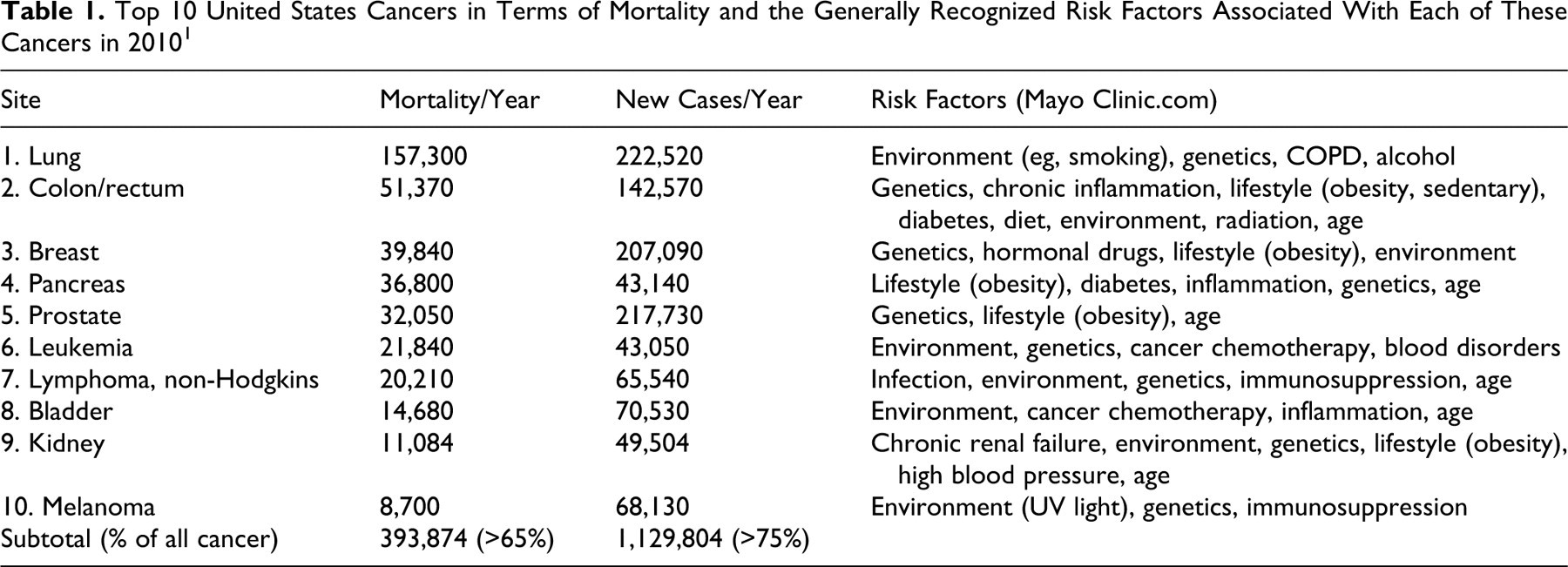
It is clear that pharmaceuticals, with the potential exception of hormonal and immunosuppressive agents, have a minor impact on cancer rates on a population basis. However, the challenge in pharmaceutical toxicology is to identify the risk for the individual, regardless of the population-based impact. To enable enhanced understanding of the effectiveness of the lifetime (2-year) rodent carcinogenesis study, this article reviews the PDR, which provides the labels for all currently marketed pharmaceuticals. As required by the FDA, the drug label provides all known information (public or proprietary) considered relevant for the attending physician and the patient regarding cancer hazard. Significant pharmaceutical cancer hazard has generally been recognized as consistently associated with 1 or more of 4 biologic activities of the inducing drug: (1) genotoxicity, (2) hormonal modulation, (3) immunosuppression, and (4) chronic toxicity. 17 This review challenges the belief that assays for these 4 biologic effects suffice as indicators of cancer hazard.
Current Regulatory Standards in the Cancer Hazard Identification Process
In April 1990, the International Conference on Harmonization (ICH) was launched at a meeting of the International Federation of Pharmaceutical Manufacturers and Associations (EFPIA) in Brussels, which included regulatory authorities from Europe, the United States, and Japan. Among other things, the ICH has greatly enhanced standardization of regulatory expectations for the lifetime cancer bioassay. The Cancer Assessment Committee (CAC), created in 1991 by the FDA, more directly affects carcinogenicity testing practices in the United States. The CAC established a carcinogenicity protocol review process, as well as a process for adjudication of outcome assessments, to ensure continuity in standards across the drug divisions in the Center for Drug Evaluation and Research (CDER) at the FDA. Since 2002, animal carcinogenicity studies have been considered pivotal studies and are subject to the FDA Special Protocol Assessment (SPA) guidance document. Upon request, the SPA guidance provides for FDA evaluation, within 45 days, of protocols and issues relating to the protocols to assess whether they are adequate to meet scientific and regulatory requirements identified by the sponsor. 2
Today the regulatory requirements, in terms of indication for and execution of the carcinogenicity assessment, have been carefully described in ICH guidance documents including: M3, Guidance on Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals; S1A, Guideline for the Need for Carcinogenicity Studies of Pharmaceuticals; S1B, Testing for Carcinogenicity of Pharmaceuticals; S1C, Dose Selection for Carcinogenicity Studies; S2, Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended for Human Use; and S6, Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals. Animal carcinogenicity studies are typically not required for registration of biopharmaceuticals under ICH S6.
In general, animal carcinogenicity studies of small organic molecules are required for drugs that would be used continuously in humans over a 6–month time period and may be required for drugs that would be used repeatedly and intermittently over 6 months. Carcinogenicity studies may also be recommended for some pharmaceuticals if there is concern about their carcinogenic potential. For example: (1) previous demonstration of carcinogenic potential that is considered relevant to humans in the product class, (2) structure-activity relationship suggesting carcinogenic risk, (3) evidence of preneoplastic lesions in repeat-dose toxicity studies, and (4) long-term tissue retention of parent compound or metabolite(s) resulting in local tissue reactions or other pathophysiologic responses.
Subchronic toxicity studies (13 weeks in duration) are necessary for dose setting for lifetime rodent carcinogenicity bioassay protocols, as the top dose is expected to be one of the following: (1) the maximum tolerated dose (MTD), defined as the dose at which mean body weight is 10% less than that of the concurrent control group and/or the dose inducing lesions that would not be anticipated to shorten the animal’s life span; (2) an upper limit of 1000 mg/kg/day (with some exceptions or permutations); (3) the dose at which the exposure achieved is more than 25 times the efficacious exposure in humans; (4) the dose at which saturation of exposure occurs; or (5) the maximum feasible dose. Three dose groups plus one or more concurrent control groups are appropriate; the low dose should approximate or slightly exaggerate the human efficacious exposure, and the middle dose should represent a logarithmic relationship between the high and low doses. For a 2-year study in rats, the sample size should be sufficient to ensure that approximately 25 animals per sex per dose level are alive at 24 months, with 50 as the minimum total starting number of animals per sex per dose level.
Two species must typically be studied, with at least 1 studied in a long-term (2-year) bioassay. The default option of the rat as the first species leaves an option for the second study to be a lifetime mouse study, as done in the past, or a short- to medium-term study, as is done currently. The latter option could be done in a transgenic mouse model, an initiation-promotion model, or a neonatal rodent model. Two transgenic mouse models, the rasH2 and p53+/-, are being used with increasing frequency. (These assays are discussed in more detail in the following section.) The rasH2 and p53+/- cancer bioassays typically have a sample size of 25 mice per sex per dose and duration of 6 months and include the use of positive controls. In any in vivo study, the route of administration should be the intended clinical route.
Once the results of the carcinogenicity studies are in hand, a pharmaceutical toxicologist/pathologist addresses the human risk in the decision to progress the molecule for registration and labeling on the basis of the weight of the evidence. The considerations include: (1) trans-species response; (2) whether multiple tissues are affected; (3) dose-responsiveness; (4) tumor latency; (5) malignant versus benign tumor induction; (6) tumor multiplicity, progression, and metastasis; (7) unique versus common tumor induction; and (8) statistical significance. Equally critical is the assessment of biological plausibility, including consideration of rodent-specific mechanisms and consideration of the biologic attributes of the test article (DNA reactivity, hormonal modulation, chronic tissue injury, immunosuppression, and cell cycle or growth perturbation).
Even if the considerations listed in the previous paragraph indicate that carcinogenicity is unlikely in humans, a positive response in a carcinogenicity study still weighs significantly in the business decision to progress the molecule, since the label will include the findings even when they are generally recognized as nonpredictive for human risk.
Development of the safety assessment strategy relevant to carcinogenic risk is a complex process that depends on the nature of the chemical matter; the pharmacologic action of the drug candidate; the results of in vitro assessments including genetic toxicity studies; the results of acute, subchronic, and chronic toxicity studies; and the indication of the drug candidate. Consultation and collaboration with the regulatory authority on the need, strategy, and tactics for cancer risk assessment is considered good drug sponsor practice.
Science-Based Improvements in the Carcinogenicity Hazard Identification Process
Toward the end of the 1990s, a notable improvement in the cancer hazard identification process occurred with the advent and institutionalization of an alternative to the standard lifetime mouse bioassay (mentioned briefly in the previous section). Genetically modified mice with the insertion or deletion of genes relevant in human cancer were approved as alternatives to the lifetime mouse bioassay in ICH S1B. This approval followed an extraordinary international coalition of industry, academic, and regulatory scientists in a collaboration that directed a large prospective research effort to improve the predictive utility of nonclinical carcinogenicity studies. Manuscripts from the consortium are published in total in Toxicologic Pathology, volume 26, number 4, 1998.
The predictivity of transgenic mouse models has been chronicled by workers in the National Toxicology Program (NTP). The transgenic alternatives, specifically the combined predicitivity data of p53+/- (for genotoxic molecules) and rasH2 (for nongenotoxic molecules), provide comparable predictiveness for human response to known (Group 1) and probable (Group 2A) human carcinogens to the rat and mouse lifetime bioassays, alone or in combination. The Group 2A molecules tested in the NTP survey all tested positive using a combined p53+/- and rasH2 test system. Similarly, of the 14 Group 1 carcinogens tested in this combined rasH2 and p53+/-test system, 11 yielded positive responses, 1 yielded a negative response, and 2 were untested. In contrast, when applying the conventional 2–species lifetime rodent test system to this same list of 14 Group 1 carcinogens, 5 positive tests and 1 negative test were reported, with most (8) untested. 32 These data support the belief that the transgenic alternatives provide comparable ability to detect Group 1 and 2A carcinogens as the lifetime combined rat and mouse lifetime bioassays.
Of course, the rat and mouse lifetime responses predict for rat and mouse carcinogenicity, thus predicting with precise accuracy for possible (Group 2B) human carcinogens, since all positive rodent responses are automatically classified as Group 2B by IARC and NTP, even in the absence of plausibility. 30,32 The rasH2 and the P53+/- lifetime models do not predict as well for the rat and mouse lifetime bioassay outcomes; thus these 2 model systems reduce the high false-positive rate described below, while not compromising the ability to identify known (Group 1) and probable (Group 2A) human carcinogens. That is, these models substantially improve the precision in the identification of human cancer risk. However, these same NTP workers critique the transgenic alternative as not identifying all of the human Group 2B (possible) carcinogens. Unfortunately, this statement lacks rigor because numerous drugs that had rodent responses known to be nonpredictive are listed as IARC Group 2B agents, including phenobarbital and benzoyl peroxide. Thus, the consideration of IARC Group 2B agents in the assessment of concordance of the rodent cancer study response with human risk appears to be a catch-22 conundrum for those invested in rodent carcinogenicity testing. 32
Review of Drug Labels in the Physicians' Desk Reference
Careful review of the drug label for results of animal testing and correlative effects in humans forms one measure for assessing the effectiveness of the safety assessments of the past for cancer risk. To inform the patient and the patient’s physician of the known risks, the label contains all of the information relevant to the cancer hazard of the drug. Information in the label relevant to cancer risk may be specific to the drug or may represent information in the public literature or from FDA knowledge of a class liability. In creation of the drug label, the FDA represents an impartial arbiter for all published and unpublished (proprietary) data relevant to cancer hazard associated with a marketed drug. Standard FDA practice is to include all carcinogenicity test results in the label, regardless of the plausibility of the data for human risk, the likelihood of repeatability, and the nature of the association of the test article with the findings. Over the past decade, there has been an increasing tendency to include additional, more interpretive comments in the label to help the physician or patient position the relevance of the animal findings.
For the purposes of this review, all drugs with any mention of cancer hazard concern in the “Warnings” or “Precautions” section of the label (before the animal test results) were considered of enhanced concern. The animal testing results can be found at the end of the precautions or in a separate section called “Carcinogenesis.” Thus, through the label, the level of concern, based on data as assessed by the FDA and the sponsor, can be ascertained and the animal cancer testing results of drugs associated with greater FDA concern can be explored to assess the effectiveness of the current cancer hazard identification paradigm. Occurrence of rodent cancers linked with known nonpredictive mechanisms will be reported in the label under “Carcinogenesis” without any mention of cancer risk in “Warnings,” or before “Carcinogenesis” in “Precautions.” Of course, the absence of human evidence for cancer risk does not necessarily indicate the existence of sufficient evidence to confirm absence of risk. Identifying carcinogenic drugs is difficult because of the typically long latent period of cancer, the inability to distinguish drug-induced cancers from those induced by other agents and conditions, and the general lack of epidemiologic surveillance for drug-induced cancer. 38 However, a major clinical epidemiologic study from 1969 to 1973 reviewed 215 drugs, of which 31 had a positive association with cancer risk and 41 had a negative correlation, or potentially a protective effect. These results may have discouraged significant further epidemiologic research funding on pharmaceutical carcinogenesis. 36
Data Collection From the Label Review
For this review, drug labels were derived by searching the commercially available Thompson Micromedex electronic version of the PDR. 4 This database requires that the company marketing the drug pay a fee for their drug to be listed. According to Thompson staff, an estimated 75% to 80% of all approved drugs are included, in contrast to the printed version of the PDR, which contains approximately 95% of approved drugs. Bias introduced by this incomplete listing, if any, is indeterminate. The list of compounds was assembled by conducting a full-text search of the label in the electronic PDR for the following terms: carcinogenesis, cancer, tumor, neoplasia, carcinogenicity, carcinoma, malignant, malignancy, and metastasis. The terms were searched within the entire database using the conjunction “or.” At the time that the search was conducted (July 2009), the resulting list included 791 drugs. Several drugs listed in the PDR are actually combination products or different salt forms of the same active pharmaceutical ingredient (API). Since combination products are typically not tested in combination, the constituent APIs were considered separately for this review, and the combination product was not included. A single API used in different salt forms in different products was considered only once. Vaccines, dietary supplements, herbal remedies, homeopathic formulations, vitamins, whey proteins, and compounds not evaluated by the FDA were excluded from this review. The resultant number of API labels in this review totaled 533.
Data Recording From the Label Review
The label of each compound was carefully examined for rodent carcinogenicity and genotoxicity testing results and enhanced human carcinogenicity concern. Any presentation in the label of rodent cancer responses was treated as a positive response, regardless of rationalization. In circumstances in which the label stated that a rodent cancer test was inadequate, the drug test was considered as “none” for that specific rodent model. If the tumor response and/or the genotoxicity test results in the PDR were listed as “equivocal” or if there were conflicting results (eg, dapsone), the drug was considered “positive” for this review. Results of genotoxicity assessments were categorized as: Ames test results, in vivo clastogenicity test results, in vitro clastogenicity test results, and in vitro mutagenicity test results (separate from the Ames test results). If genotoxicity test results were not included in the label, genotoxicity testing was considered “not reported.”
In instances where a racemic mixture was tested, but the results for the active enantiomer were not specifically reported, the carcinogenicity results for the racemic mixture test were included. If a metabolite of the prodrug was tested, but not the prodrug itself, then the prodrug was listed as “none” (not tested). Animal carcinogenicity results were considered positive or negative based on results of testing, but not based on general class-related statements of cancer risk for a drug. That is, if a generic statement that the class of drugs is carcinogenic to animals was included, but no test results were presented for the specific API, then the drug was listed as “none” (not tested).
Rodent and human cancer tissue target(s), mechanism of pharmacologic action, and drug indication(s) were also tabulated. The “Warnings” and “Precautions” sections were searched for any mention of cancer hazard, and the information was recorded.
Results From Review of 533 Drug Labels
(A supplemental file including results of every drug label reviewed from the PDR is available at http://ganc.sagepub.com/supplemental.)
Of 533 distinct APIs, 287 (54%) had been tested for carcinogenic hazard in rodents. Of the 287 drugs tested for carcinogenicity hazard, 161 (56%) tested positive. All drugs that the FDA considers of enhanced concern for cancer hazard in patients taking the drug were identified by search of the label for any mention (no matter the nature) of cancer in “Warnings” or “Precautions,” antecedent in the label and unassociated with the report of the animal testing results under “Carcinogenicity” (Table 2 ). The basis for enhanced concern presented in the label varied substantially from label to label, but primarily included: (1) statement that the significance of the animal cancer findings is unknown that is, the reason for concern is based exclusively on the animal response without data on the human response (eg, albuterol, dexlansoprazole, esomeprazole, and teriparatide; (2) references from the published literature relevant to cancer risk (eg, arsenic, paliperidone, risperidone, molindone, and olanzapine); (3) a listing of human cancers known to be induced by the specific drug (eg, chlorambucil, infliximab, and rituximab); (4) epidemiologic data indicating potential risk (eg, all therapies for Parkinson disease, since patients with Parkinson have an increased risk for melanoma independent of the therapeutic modality); (5) results from a clinical study conducted outside of the United States (eg, the World Health Organization [WHO] study of fenofibrate); or (6) a plausible concern based on the mechanism of action of the drug (eg, epoetin, filgrastim, and sargramostim).
Drugs With Enhanced Concern for Human Cancer Risk a
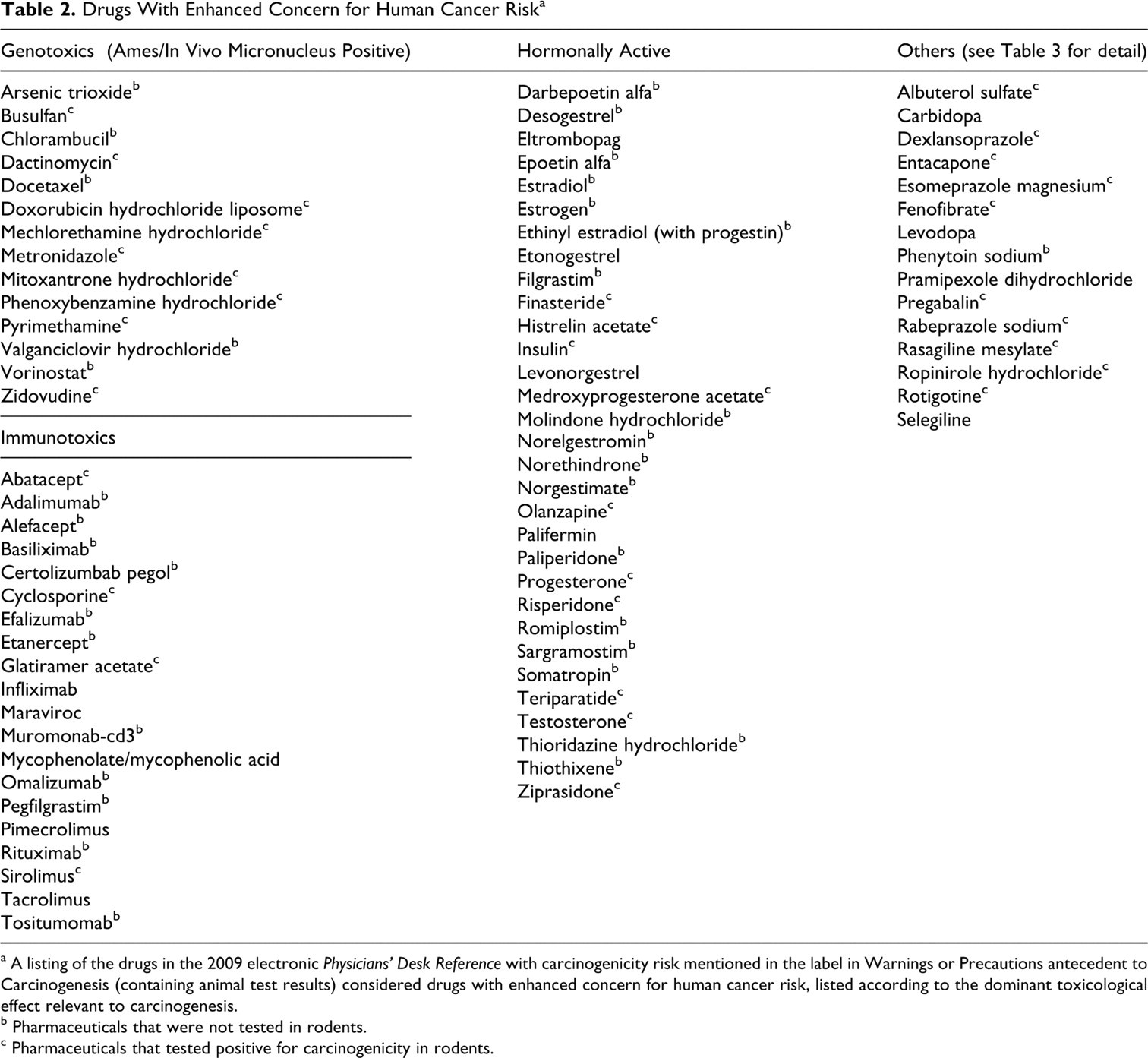
a A listing of the drugs in the 2009 electronic Physicians' Desk Reference with carcinogenicity risk mentioned in the label in Warnings or Precautions antecedent to Carcinogenesis (containing animal test results) considered drugs with enhanced concern for human cancer risk, listed according to the dominant toxicological effect relevant to carcinogenesis.
b Pharmaceuticals that were not tested in rodents.
c Pharmaceuticals that tested positive for carcinogenicity in rodents.
The drugs with enhanced levels of concern for human cancer risk totaled 78 out of the 533 (15%) drug labels surveyed. These drugs included hormonal agents, immunosuppressives, Ames- and in vivo micronucleus-positive genotoxins (predominantly cancer chemotherapeutics), and a few that did not fit into any of these 3 categories and were designated “other” (Table 3 ). Of these 78 drugs, 32 (41%) had tested positive in the rodent bioassay and 12 had tested negative. Thus, the false-negative rate for drugs of enhanced concern was 15% (Table 4 ). Thirty-four (44%) had not been tested. Out of all the drugs examined, most drugs that tested positive in rodents were listed without recognition of enhanced human risk. As mentioned previously, 161 tested positive in rodents, out of which only 32 were indicated to be of enhanced human concern, for a hypothetical false-positive rate of 80%.
Plausibility of Cancer Risk for Drugs of Enhanced Concern That Are Not Immunosuppressive, Hormonally Active, or Genotoxic: the “Other” Category a
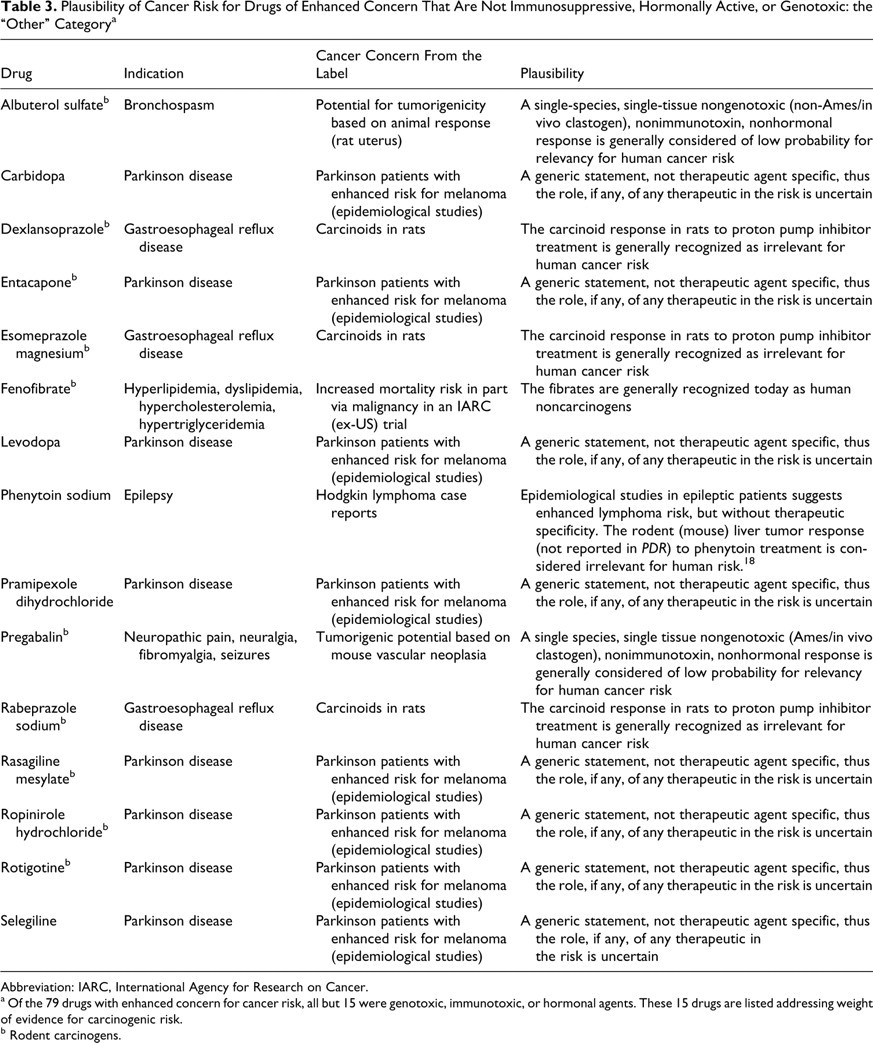
Abbreviation: IARC, International Agency for Research on Cancer.
a Of the 79 drugs with enhanced concern for cancer risk, all but 15 were genotoxic, immunotoxic, or hormonal agents. These 15 drugs are listed addressing weight of evidence for carcinogenic risk.
b Rodent carcinogens.
Rodent Test False Negatives a
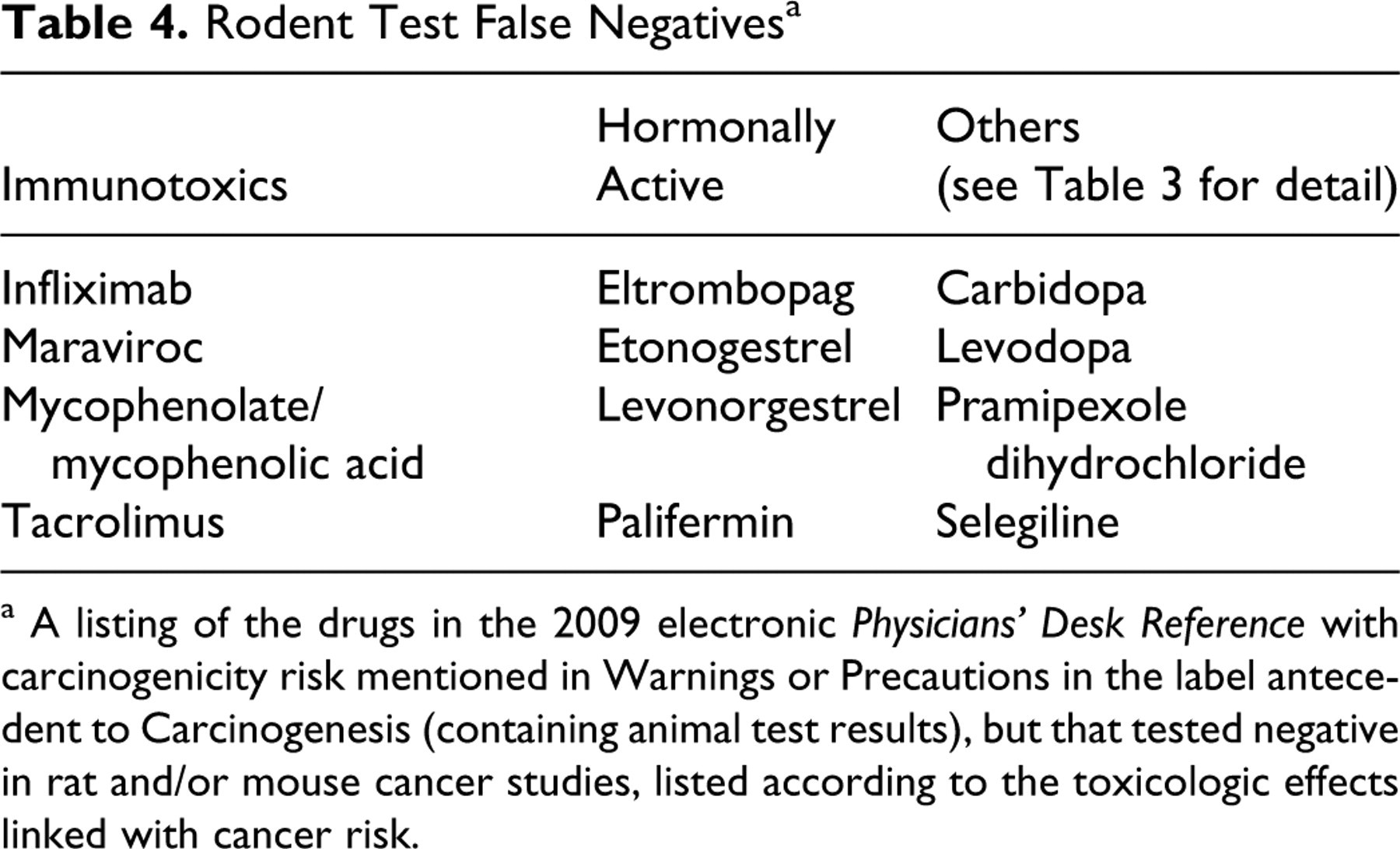
a A listing of the drugs in the 2009 electronic Physicians' Desk Reference with carcinogenicity risk mentioned in Warnings or Precautions in the label antecedent to Carcinogenesis (containing animal test results), but that tested negative in rat and/or mouse cancer studies, listed according to the toxicologic effects linked with cancer risk.
Several drugs were positive for carcinogenic response in toxicology studies, including alefacept (immunomodulatory molecule causing B-cell lymphoma in cynomolgus monkeys positive for lymphocryptovirus [LCV], which can lead to B-cell lymphomas), calcitonin, micafungin, pimecrolimus, and progesterone. Several molecules were tested in multiple rodent cancer studies, sometimes using different test systems, including benzoyl peroxide, levofloxacin, penicillamine, pimecrolimus, and tretinoin. When tested repeatedly, most molecules, including dapsone, metoprolol, sirolimus, and pimecrolimus, had conflicting test results; this was considered evidence of poor test reproducibility.
Several drugs were tested in the transgenic mouse model, including benzoyl peroxide, dapsone, eszopiclone, frovatriptan, maraviroc, methylphenidate, palifermin, olmesartan, pantoprazole, rabeprazole, ribavirin, sunitinib, and tretinoin. A few molecules, such as norelgestromin, were tested in 7-year dog and/or 10-year monkey carcinogenicity studies. The results from these models are not included in the determinations that follow.
The sensitivity of these results was determined by taking the number of APIs that had positive rodent test results and were of enhanced human carcinogenicity concern (32) divided by the total number of APIs that had positive rodent test results and were of enhanced human carcinogenicity concern (32) plus the number of APIs that had false-negative rodent test results (12), for a sensitivity value of 32/44 = 73%. Positive predictivity was determined by taking the number of APIs that had positive rodent test results and were of enhanced human carcinogenicity concern (32) divided by the number of APIs that had positive rodent test results (161), for a positive predictivity value of 32/161 = 20%. Negative predictivity was determined by taking the number of APIs that had negative rodent test results and were not of enhanced human carcinogenicity concern (114) divided by the number of APIs that had negative rodent test results (126), for a negative predictivity value of 114/126 = 90%. The hypothetical false-positive rate was determined by taking the number of APIs that had positive rodent test results and were not of enhanced human carcinogenicity concern (129) and dividing that number by the number of APIs that had positive rodent test results (161), for a hypothetical false-positive rate of 129/161 = 80% (Fig. 1 ).
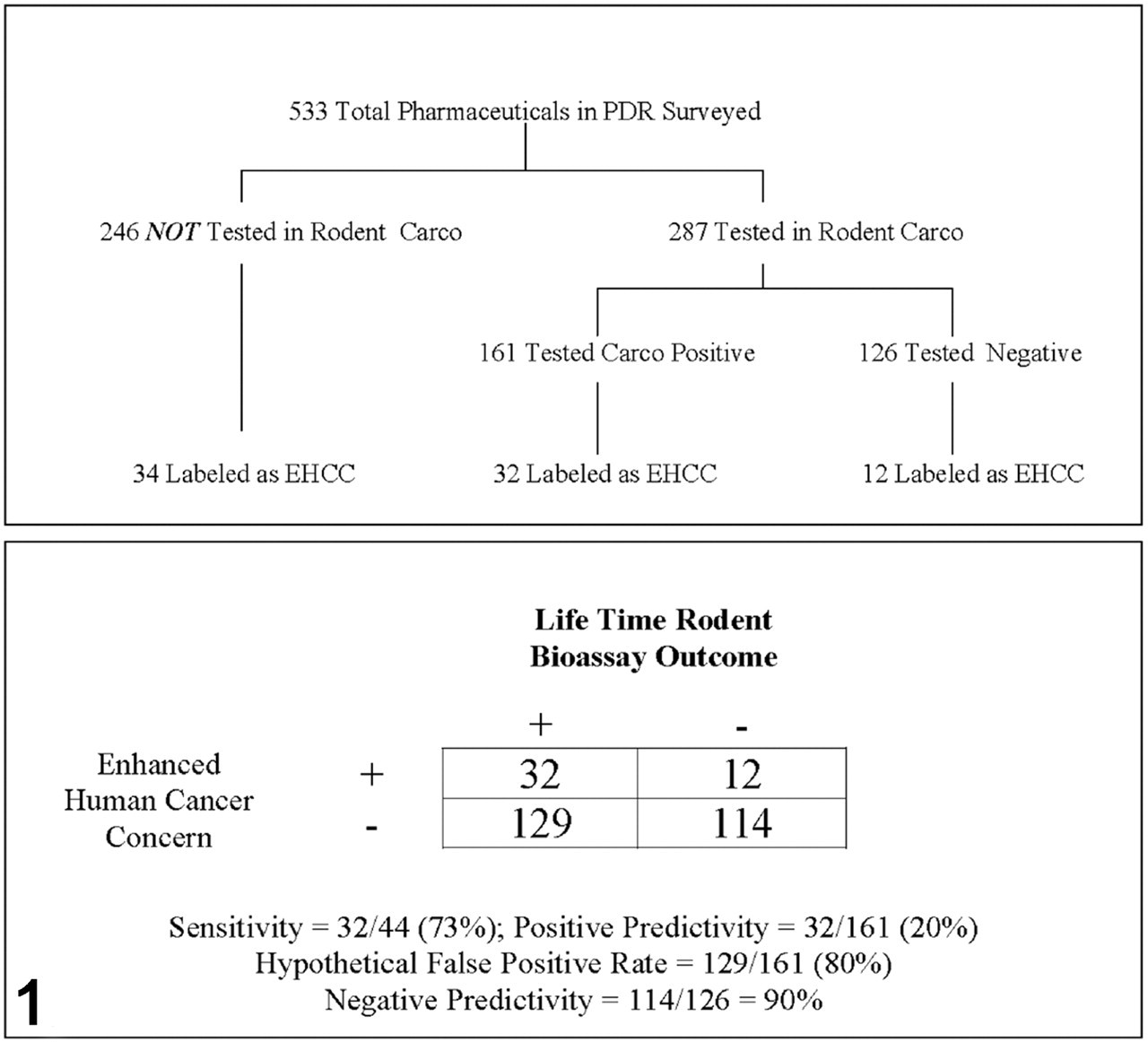
Evaluation of the effectiveness of lifetime rat and mouse lifetime bioassays considered collectively, in cancer hazard identification via review of the drug labels in the electronic Physicians' Desk Reference. Abbreviation: EHCC, enhanced human cancer concern.
Discussion of the Label Contents of Drugs With Enhanced Concern for Human Cancer Risk
Even though 78 of the 533 reviewed drugs were identified as being of enhanced concern for human cancer risk, nearly half (44%) of these had not been tested in animals. This rate of 44% untested suggests that cancer hazard often can be adequately addressed without lifetime rodent bioassays, based on the attributes of the drug’s biological effects as determined in shorter term assessments. Drugs need not be tested in lifetime or short- to medium-term cancer studies if they are one or more of the following: (1) oncologic chemotherapeutics for patients whose life expectancy is only a few years; (2) monoclonal antibodies that do not pharmacologically affect the rodent; (3) hormonal drugs with class warnings for cancer risk, attendant with generic statements of animal carcinogenic potential under “Carcinogenicity” in the label; and (4) drugs not intended for chronic use.
Review of the labels also enabled recognition of circumstances in which it appears that the FDA approved some drugs despite their having positive rodent cancer test results (that were not the typical nonpredictive tumors discussed below), presumably based on a risk/benefit decision. Examples include rasagiline and entacapone for Parkinson disease, formoterol and pentosan for COPD, raloxifene for osteoporosis, dapsone for acne vulgaris, and ziprasidone for schizophrenia. Furthermore, 3 drugs were approved that had animal cancer studies that were conducted in phase 4, including pentosan for interstitial cystitis and octreotide for acromegaly. These circumstances suggest that there may be some serious disease indications, other than cancer, in which the FDA might not consider carcinogenicity studies as critically important and may in the future consider them unnecessary, which reinforces the concept that the drug sponsor should meet with the FDA preparatory to implementation of the cancer hazard identification plan.
All of the 78 drugs with enhanced concern for cancer risk could be placed into 1 of 4 categories, based on the biological effects of the drug, as follows: (1) hormonal, (2) immunosuppressive, (3) genotoxic, and (4) other, for drugs that do not fit into any of the previous 3 categories. Most critical, in terms of more effectively predicting hazard during toxicology studies, is a determination of the attributes of the drugs in this “other” category and of the drugs that have enhanced human concern despite negative rodent carcinogenicity test results.
Attributes of the “Other” Drugs With Enhanced Levels of Concern for Humans That Were Not Immunosuppressants, Hormonal Modulators, or Genetic Toxins
In a further assessment of the 15 “other” drugs without known associated risk factors, 8 were found to be indicated for Parkinson disease. They were of enhanced concern because patients with Parkinson disease have been shown, in epidemiologic studies, to have enhanced rates of melanoma. This finding is independent of any attribute of the therapeutic modality and represents a generic statement in the label, since the causal factors are unknown. An additional 5 of these 15 “other” drugs were of enhanced concern, only because of a positive rodent tumor response. These 5 positive rodent results are probably not relevant for human risk for the reasons listed for each of these drugs in Table 3. One of the remaining 2 was phenytoin (for epilepsy), which has no rodent carcinogenicity or genetic toxicity testing in the label and probably does not pose a cancer risk for patients. Drugs indicated for epilepsy are generally associated with concerns about carcinogenicity, perhaps unfairly, because of the past practice of using thorotrast, a known human liver carcinogen, for imaging in patients with epilepsy. Some epidemiologic studies of anti-epilepsy drugs have not corrected for this exposure. 18 The last of these 15 drugs was fenofibrate. The fenofibrate label notes that a WHO study conducted outside of the United States showed evidence of increased risk for mortality with fenofibrate. The label reports a mortality rate of 5.70% versus 3.96% in the control group. The higher mortality was associated with noncardiovascular diseases, including malignancy, gall bladder disease, and pancreatitis. Although PPAR-α antagonists are recognized as potent trans-species rodent liver carcinogens, this pharmacologic activity is generally recognized to have no link with a human liver cancer response. 29 The likelihood that any of the drugs of enhanced concern in this “other” category are actually human carcinogens thus seems quite low based on contemporary understanding. Therefore, none of these 15 drugs is considered an exception to the premise that all significant pharmaceutical carcinogens are immunotoxins, genotoxins, hormonal agents, or chronic toxins.
Not included in this list of “others” are 4 therapeutics for schizophrenia: paliperidone, risperidone, molindone, and olanzapine. Each drug causes an increase in prolactin levels across species (see supplemental file). Prolactin perturbation in rodents results in specific characteristic tissue changes in the reproductive tract in subchronic and chronic rodent toxicology studies and carcinogenicity studies. 21,43 Although no actual cancer response has been detected in humans as a result of increased prolactin levels associated with treatment with these drugs, the FDA concern emanates from the fact that a significant percentage of human mammary cancers, when placed in tissue culture, are dependent on the presence of prolactin for growth. Thus, these 4 schizophrenia drugs do fit within a known biologic risk factor paradigm: hormonal modulation in toxicology studies.
Genotoxic Drugs
Fifteen of the 78 drugs of enhanced concern for human cancer risk tested positive in the Ames and/or in vivo micronucleus assays. Most of these drugs were cancer therapeutics. A few of the genotoxic molecules were indicated for other life-threatening diseases such as AIDS. An additional genotoxic drug, metronidazole, is not intended for chronic use. The high concordance of Ames- and in vivo clastogenicity-positive responses with carcinogenicity is generally recognized, suggesting that these short-term tests might suffice without lifetime rodent testing for human cancer risk assessment. There were no false-negative results among the chemotherapeutics; however, many were not tested.
Immunosuppressive Drugs
Of the 12 false-negative responses, 4 (mycophenolate, infliximab, maraviroc, and tacrolimus) are associated with an immunosuppressive mechanism. Two other immunosuppressive drugs, cyclosporine and sirolimus, also deserve additional consideration.
The rodent carcinogenicity bioassay results for cyclosporine were reported to be negative in the peer-reviewed literature written by the sponsor. 35 As we revise historical interpretations on the basis of new knowledge and modify the label accordingly, this paper must be seen as suspect. The drug label states that there was a positive trend for lymphoma in mice. If the assay had been sufficiently robust, dependence on a trend analysis would not have been necessary to identify a response. The likelihood of repeatability of the significant trend must be considered modest at best, since the actual lymphoma incidences in all groups were within the historic norms and the control lymphoma incidence was lower than the historic mean value. The results of the 2-year carcinogenicity test of cyclosporine in mice should probably be considered equivocal when considered alone. Given the fact that cyclosporine is a known human carcinogen, pharmaceutical pathologists/toxicologists today, with the benefit of hindsight, classify the results of this assay as positive. But it is not clear that toxicologists/pathologists should base interpretations of animal studies on the human carcinogenic response. Rather, pathologists/toxicologists should challenge the utility of animal studies upon recognition of reoccurring testing mispredictions and equivocal responses to human pharmaceutical carcinogens.
A cancer test of sirolimus in rats was negative. Two 2-year cancer tests in mice were conducted, with different results. The first study resulted in liver tumors. The second study, which used slightly lower doses, was reported to be positive for lymphomas. Thus, the 2 mouse studies present discordant results and again create uncertainty as to the repeatability of either test.
Considered in total, these data indicate that lifetime mouse or rat carcinogenicity testing does not have reliable value in identifying the carcinogenic risk of immunosuppressive agents. To determine an adequate nonclinical test model for immunosuppressive drugs, it is important to understand the cause of many human cancers linked with immunosuppressive therapy. Patients taking immunosuppressive drugs often develop viral cancers, specifically, viral lymphomas and leukemias such as those associated with the Epstein-Barr virus in patients with immunosuppression. It seems that a cancer testing model with a constitutive lymphoma virus may be necessary to consistently demonstrate the carcinogenic risk of immunosuppressive drugs. One immunomodulatory drug not tested for carcinogenicity actually induced cancer in the chronic monkey toxicity study. Alefacept treatment (a psoriasis therapeutic) resulted in B-cell lymphoma in cynomolgus monkeys that tested positive for lymphocryptovirus (LCV), which causes cynomolgus monkey B-cell lymphomas (see drug label). The p53+/- mouse may provide a suitable rodent model, since it is reported to harbor a lymphoma virus and it does develop lymphomas upon chronic treatment with a prototypical immunosuppressive drug. 39 However, the robustness of the response of the p53+/- mouse to immunosuppressive therapeutics has not been confirmed. Another option for a rodent model is the AKR mouse, which has been shown to develop a lymphoma response after 3 months of cyclosporine exposure because of a constitutive lymphoma viral oncogene. 23 –25 Further exploration for a reliable model to test for immunomodulatory therapy-related carcinogenesis seems warranted. An alternative strategy might be to label a drug identified as an immunotoxin or immunosuppressive agent as a human cancer hazard without testing in the unreliable lifetime rodent bioassay. This strategy would also obviate the challenge of testing immunomodulatory antibodies that do not pharmacologically affect rodents.
Hormonal Drugs
Four drugs of the 12 false negatives are hormonally active: etonogestrel, eltrombopag, levonorgestrel, and palifermin. Even though their labels contain the generic statement that these drugs cause cancer in animals, the actual test results in rodents were negative. Thus, these results are false negatives. Several other hormonal agents were not tested in animals, and for these, the label also contains a generic statement that these agents are animal carcinogens. For the purposes of this review, these were considered as “none” (not tested) and thus did not present evidence for concordance with the enhanced human concern.
In summary, the rodent response to hormonal human carcinogens was not consistent in this data review. However, the rodent is generally recognized to consistently develop evidence of hormonal perturbation in subchronic and chronic toxicology studies of hormonal drugs. Thus, the presence of evidence of hormonal modulation in toxicology studies should probably engender a warning for cancer hazard or further testing in a transgenic mouse carcinogenicity assay. 39
Chronic Tissue Injury
Although none of the drugs reviewed was recognized to induce human cancer as a result of chronic tissue injury, chronic injury is generally recognized as a risk factor for human carcinogenesis. Examples include hepatocellular carcinoma in alcoholic cirrhosis and gall bladder adenocarcinoma associated with cholelithiasis and chronic inflammation. 34 The increased regenerative activity accompanying chronic tissue injury is widely believed to represent conditions associated with increased risk for neoplastic transformation (tumor promotion). 15 There is a strong association between chronic inflammatory conditions in a particular organ (tissue) and cancer specific to that organ (tissue). 37
Biological Attributes of Drugs Associated With a False-Negative Response in Rodents
The false-negative rate and the high rate of untested drugs that are associated with an enhanced risk for human carcinogenicity are cause for concern. The false-negative rate of the rodent carcinogenicity tests in this review of labels was 15%. This substantial incidence of false-negative responses is not generally recognized. As mentioned, 8 of the 12 false negative rodent responses to drugs of enhanced human cancer risk were linked with either hormonal or immunosuppressive action. The remaining 4 are carbidopa, levodopa, pramipexole, and selegiline, which are indicated for Parkinson disease. As mentioned previously, patients with Parkinson disease have an increased risk for melanoma regardless of the therapeutic modality. Since all of the labels of drugs to treat Parkinson disease must carry a melanoma warning, it is unclear whether rodent cancer studies add any value for identification of human risk in this therapeutic category.
The most critical failure of the lifetime rodent bioassay is its failure to identify the carcinogenic risk of the most important human carcinogen in the western world today, cigarette smoke, a new drug but an old carcinogen. Thirteen standard lifetime rat and mouse cancer bioassays have failed to test positive for this cancer hazard. 13 A recent publication of a putative positive cancer response in a conventional mouse used an unconventionally large sample size of over 300 mice and an unconventionally long duration of nearly 3 years to achieve this end point. In addition, this report demonstrated that the cigarette smoke-exposed mice had a substantially enhanced survival rate, but the paper did not evaluate age-adjusted lung tumor rates, representing a potentially serious technical flaw. 27 It is puzzling how a modern day cancer hazard testing paradigm that fails to identify the risk of this important, widely used carcinogen could be considered satisfactory. The practices of the past—with lifetime rodent tests in 2 species—are clearly not adequate. It is possible that the rasH2 transgenic alternative model might respond positively to cigarette smoke, but this has not been confirmed as repeatable. 16
False-Positive Responses
Of the drugs that were of significant concern for human cancer risk, 41% also tested positive in rodent cancer tests. In contrast, 80% of positive rodent cancer tests involved drugs without enhanced concern for human risk. This hypothetical false-positive rate should also be considered problematic. Numerous drugs are generally recognized as safe for human use (noncarcinogens), yet they cause carcinogenic responses in lifetime rodent bioassays. Prototypical over-the-counter rodent carcinogen/human noncarcinogen drugs in this category include omeprazole, acetaminophen, sodium fluoride, benzoyl peroxide, cetirizine, loratadine, doxylamine, minoxidil, and lansoprazole. Prototypical rodent carcinogen/human noncarcinogen prescription products include clofibrate, phenobarbital, simvastatin, lovastatin, griseofulvin, sertraline, albuterol, cimetidine, gemfibrozil, and terazosin. 5,17,22,28,31,41
In several cases, the animal mode of action is recognized as not relevant for human cancer risk, including: The α2u-globulin nephropathy syndrome related to male rat kidney tumorigenesis.
7,14
The calcium phosphate urinary precipitate-linked urothelial tumorigenesis that occurs after administration of high doses of sodium salts (eg, saccharin or ascorbate) to rats.
7,14
Thyroid tumors induced by cytochrome P450 enzyme inducers that accelerate metabolism of thyroid hormones, causing feedback elevation of thyroid-stimulating hormone, which stimulates tumor development over the rodents' lifespan.
7,14
Other rodent-specific carcinogenic endocrine feedback mechanisms involving adrenal cortex/medulla, pituitary, pancreas, and gastrointestinal endocrine cells.
7,14
Soy protein (uncooked) and corn oil both induce rat exocrine pancreas cancer. The soy effect is known to be mediated by a chronically up-regulated cholecystokinin (CCK) hormonal proliferative stimulus.
11,44
Unfortunately, most of the rodent-specific responses occur through unknown mechanisms. This is the case with phenobarbital, a trans-species, multitissue, rodent-specific tumorigen and human noncarcinogen, the most studied rodent carcinogen in the history of the nonclinical pharmaceutical toxicology sciences. These facts could lead a pharmaceutical company to question the value of substantive investment in mechanistic research of tumor responses in carcinogenesis as part of the drug development process.
Calculation of a false-positive rate engenders controversy for several reasons. The most prominent reason emerges from the extremist belief that every animal cancer response identifies a human cancer risk. This point, in concert with the fact that one cannot prove a negative hypothesis, serves to prevent progress in hazard identification studies. In fact, Janet Woodcock, Director of CDER at the FDA, has identified the toxicology sciences, of any science involved in pharmaceutical research and development, as the least changing in response to the dramatic increases in biology knowledge (FDA Annual Science Forum 1998). In considering rodent false-positive responses, bear in mind that after the successful registration of loratadine, a full-page advertisement appeared in the New York Times in which a long paragraph reported the animal carcinogenicity results, and still the product quickly moved to achieve billions of dollars in sales. Patients and physicians recognize, at least to some extent, the high false-positive rate and can see that many drugs generally recognized as safe are carcinogenic in animals, which leads to general distrust of the relevance and reliability of animal cancer testing in predicting effects in humans. This high false-positive rate also has significant recognition among pharmaceutical regulatory agency staff. 40
Additional Challenges in the Application of the Rodent Lifetime Cancer Bioassay
In an extensive assessment of the repeatability of rodent cancer bioassays, results from only 57% of 121 replicates were identified as repeatable, indicating that rodent carcinogenicity assays are poorly reproducible. 20 The lack of repeatability can also be seen in this review of the PDR, for example, in the testing of dapsone, metoprolol, sirolimus, and pimecrolimus. It is unusual, in industry, to repeat rodent cancer studies; thus, the magnitude of this problem cannot be quantified for pharmaceutical cancer tests.
Other challenges include the lack of “real world conditions” in the cancer bioassay, exemplified by the sometimes highly exaggerated exposure conditions of the rodent test. In fact, several renowned toxicologic scientists believe that if the sample size and duration were increased, every bioassay would be positive because of the use of the maximum tolerated dose (MTD), with chronic toxicity as the risk factor for the positive response. Specifically, an MTD bioassay does not distinguish between human carcinogens and noncarcinogens, but rather, it fails to detect the weak tumorigens at MTD. 8 Given the standard sample size of 50, the lack of power to detect weak carcinogenic influences can be defined; an incidence of less than 10% is probably not detectable in the current rodent bioassay. The real world of medicine includes great genetic diversity, intercurrent diseases, and a highly variable environment including nutritional status; these factors are difficult to model in animal studies. In addition, the spontaneous rodent tumor rate (generally recognized as highly variable) decreases sensitivity and increases the likelihood of false-positive responses occurring by chance.
Accordingly, it has been reported that 82% of the >500 chemicals tested by the NTP have been noncommittally classified as possible human carcinogens or unclassifiable. 3 Although the NTP lifetime rodent bioassay has been described as the gold standard for predicting human carcinogenicity associated with chemicals in the environment, few attempts have been made to validate it against human carcinogenicity. 19 Since all positive rodent tests are classified by the NTP (and IARC) at minimum as possible human carcinogens and because most chemicals at MTD cause cancer in rodents, regardless of plausibility, the exercise can be considered a testing paradigm conundrum or, even worse, a nondiscriminating practice.
The number of animals required (in the thousands), the cost (in the millions of dollars), and the requisite time (3-4 years) for cancer bioassays to be completed create a significant impediment in the effort to provide needed new drugs to suffering patients. This cost and time concern is amplified because of the assay’s limited effectiveness in identifying human cancer risk.
The findings in this review support and create a framework of reference for a new multinational initiative to improve the efficiency and effectiveness of the lifetime bioassay. A very specific proposal exists and is currently undergoing a broad-based multinational challenge to develop a proof of concept. This hypothesis reflects the concept that rodent carcinogens develop premonitory tissue changes indicative of cancer risk in chronic toxicology studies. In this proposal, the absence of these specific risk factors in chronic toxicity studies would obviate the need to conduct a lifetime rat bioassay. Data supporting this hypothesis have been published, and a further challenge to this hypothesis is in progress under the auspices of a large collaboration of pharmaceutical and regulatory toxicologists and pathologists. 17,33
The Way Forward
Industry and regulatory practice in cancer hazard identification in the United States has been successfully demonstrated by the continuing increase in longevity and the decreases in age-adjusted cancer rates. Improved precision of cancer risk assessment practices will improve physician and public confidence and acceptance of the pharmaceutical and regulatory toxicology decisions involved in drug development and approval. The need for an improved cancer hazard identification strategy is now recognized internationally. By understanding and reacting to the current level of knowledge in human carcinogenesis, immediate improvements are possible. The data from this review indicate that significant human cancer risk can be effectively and efficiently assessed by demonstration of (1) immunosuppression, (2) hormonal modulation, (3) genotoxicity, (4) chronic toxicity responses (and associated exposures), and (5) rasH2 and p53+/- bioassay responses. Using these parameters, without application of lifetime rodent cancer studies, the false-negative rate identified in this review would be significantly reduced, with a substantive reduction in the false-positive rate. Furthermore, what was learned from this review may be leveraged in the future to improve the drug label for informing a higher level of concern for human cancer risk.
Footnotes
Acknowledgements
The assistance of Vilmos Csizmadia and Alexis Khalil for their help in creating this manuscript is sincerely appreciated.
The authors declared that they had no conflicts of interest with respect to their authorship or the publication of this article.
The authors declared that they received no financial support for their research and/or authorship of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.