
Abstract
The pathogenesis and virulence of Bovine enterovirus-1 (BEV-1) in cattle is largely unknown. Reports concerning its virulence suggest that there might be an association between BEV-1 infections and a range of diseases in cattle that vary from respiratory to enteric to reproductive disease and infertility. In the current study, the pathogenesis associated with acute infection of BEV-1 in calves experimentally inoculated with the Oklahoma isolate of BEV-1 was described. Although interpretation of the study was limited by lack of an effective control group, results suggest that an association between inoculation of BEV-1, virus localization, and the potential development of lesions in the brain and heart probably exists. In the experiment, BEV-1 virus localized to the terminal ileum, ileocecal and cecocolonic junctions, spiral colon, and ileocecal lymph nodes; BEV-1 virus was detected in the cytoplasm of enterocytes, lamina propria macrophages, endothelium, neurons of the submucosal and myenteric plexi, and lymphocytes of the submucosal lymphoid tissue. Although no clinical signs were noted following acute infection, BEV-1 was localized in the cerebellar white matter of a calf with encephalitis and in the heart of another calf with coronary arteritis. The current study suggests that the BEV-1 isolate is infectious to young calves and that BEV-1 potentially can have a similar pathogenesis to that observed in natural or experimental enterovirus infections in other species.
Keywords
Bovine enterovirus (BEV) belongs to the family Picornaviridae (picornaviruses), which consists of small (28-30 nm), nonenveloped viruses with an icosahedral capsid that encloses a single copy of positive-sense RNA genome. Bovine enterovirus is in the genus Enterovirus, along with poliovirus, human enteroviruses, coxsackieviruses, swine vesicular disease virus, echovirus 11, and others. Originally classified into several serotypes, only 2 serotypes, BEV-1 and BEV-2, are now recognized.10,15,19,21 Because of the unavailability of type-specific antisera or a commercially available diagnostic test, a genotypic classification that supports previously recognized serological distinctions has been proposed. 7
The pathogenesis and virulence of BEV-1 in cattle are largely unknown. Early reports concerning the virulence of BEV suggested that there might be an association between BEV infections and a range of diseases in cattle, as viruses were isolated from animals with clinical signs that varied from respiratory to enteric to reproductive disease and infertility.3,4,14,22 However, there are no published reports that describe, in animals experimentally infected with virulent BEV-1, the lesions associated with infection and disease or its pathogenesis in cattle. Obtaining knowledge about the susceptibility of cattle to challenge, the pathology associated with infection, and the prevalence of BEV-1 infection in herds is essential to the understanding of infection and disease in cattle.
Recently, in the first report of BEV in more than 20 years, BEV-1 was isolated from a 2-year-old pregnant Aberdeen Angus in Oklahoma with fatal enteric disease. 1 The current study reports the results of an experimental infection in calves using the BEV-1 isolated from the Oklahoma heifer.
Methods
Experimental Design
Twelve calves were inoculated intranasally with 107 tissue culture infective doses (TCID50) of BEV-1 in the form of Madin Darby bovine kidney (MDBK)–infected cell culture supernatant fluid (1 ml), and 6 calves each were inoculated with uninfected MDBK cell culture supernatant fluid to serve as controls.16,17 To identify potential route of entry and early changes, 2 controls and 4 infected calves were necropsied at 4 hours (day 0) post inoculation (PI) and on days 5 and 10 PI. The response of calves to BEV-1 infection was followed by clinical observation, complete blood cell counts, serology, fecal cultures, necropsy, and histopathology. Infection of calves was confirmed by polymerase chain reaction (PCR), virus isolation in MDBK cell cultures, and in situ hybridization (ISH) using a riboprobe corresponding to BEV-VP1 protein.
Calves
Eighteen (17 males and 1 female) 8- to 12-week-old dairy calves were acquired from facilities operated by the College of Agriculture and Environmental Sciences, University of Georgia, and were maintained during experiments at facilities operated by the Animal Resources Department at the College of Veterinary Medicine. Calves were tested for the presence of BEV-1 titers before being admitted to the experimental housing facilities. Before inoculation with BEV-1 isolate, calves, except for 1, had low (≤1:4) to negative serum neutralizing (SN) titers to BEV-1; calf No. 16, from group 3, had a titer of 1:8. 8 Calves were cared for by University Research Animal Resources staff under a protocol approved by the Institutional Animal Care and Use Committee in accordance with specifications detailed in the Institute for Laboratory Animal Research (ILAR) Guide for the Care and Use of Laboratory Animals. The University Research Animal Resources departments at the University of Georgia are fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International (AAALAC-I).
Housing and Treatment Groups
All calves were housed indoors under controlled temperature and humidity. Calves were grouped according to their scheduled day of euthanasia; an all-in all-out practice and disinfection was carried out between treatment groups. Between groups there was a 4- to 5-week period with no animals in the animal rooms. A total of 6 calves per treatment group were housed at any one time; 6 calves were kept in 2 separate rooms; each room had 3 pens (1 calf/pen) with solid partitions in between, and each pen had a separate water source, but all 3 pens shared ventilation. No direct contact existed between calves. A water bath containing dilute solution of sodium hypochlorite (bleach) was placed at the entrance door of each animal room. Personnel wearing approved personal protective equipment (PPE) fed the calves individually. Blinded to the contents of the inoculum, calves in each room were randomly inoculated intranasally; 2 calves received 1 ml of infected MDBK cells (107 TCID50/ml), and 1 calf received 1 ml of uninfected cell culture supernatant. Calves from treatment group 1 were euthanatized with an intravenous overdose of sodium pentobarbital at 4 hours (day 0) PI, group 2 calves were euthanatized at day 5 PI, and group 3 calves were calves euthanatized at day 10 PI.
BEV-1 Isolate
The Oklahoma isolate of BEV-1 was grown in MDBK cells that were free of pestiviral contamination, as previously reported. 1 Briefly, the cells were grown in 75-cm 2 flasks in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) until they formed a confluent cell monolayer. The media was removed, and each flask was infected with 5 ml of BEV-1 in DMEM without FBS at a multiplicity of infection of about 0.1 TCID50/cell. After incubating for 2 hours, each flask received 25 ml of DMEM supplemented with 2% FBS. The reduction in the amount of FBS limited the amount of nonviral protein present in the final inoculum. When viral specific cytopathic effects (CPE) were visualized in ≥75% of the cell monolayer, the flasks were frozen at –70°C and then thawed, and the contents were centrifuged at 2000 × g for 15 minutes to get rid of cell debris. The supernatant was titrated following the Reed and Muench method and used as the inoculum. 17 The inoculum was diluted to contain 107 TCID50/ml. Negative control cell culture supernatant was prepared in the same way using MDBK cell cultures mock-infected with 5 ml of DMEM without FBS.
Clinical Observations
All calves were examined prior to inoculation and then daily after inoculation for clinical signs of disease, including lethargy, anorexia, fever, oculonasal discharge, diarrhea, respiratory distress, and decreased skin turgor (dehydration).
Collection of Blood and Fecal Samples and Analysis
Baseline blood samples were collected at the time of initial physical exam and then prior to euthanasia and necropsy. Blood samples were collected in EDTA-coated tubes for complete blood count and differential. Blood was also collected in plain tubes and allowed to clot, and the serum was extracted and stored at –20°C for subsequent use in the serum neutralization (SN) test for antibody detection. 8
Fecal samples were obtained at the time of necropsy with 2 sterile, plain swabs from the rectum. The rectal swabs were placed in sterile plain tubes, stored at 4°C, and subjected to virus isolation. 8
Serum Neutralization Test
The SN test was performed following the standard method. 8 Briefly, serial 2-fold dilutions (1:2 through 1:256) of sera were mixed with an equal volume (25 μl) of BEV-1 containing 100 TCID50 of virus in a 96-well microtiter plate and incubated for 1 hour. Mixing diluted sera with virus resulted in a 1:4 through 1:512 dilution series. Then 150 μl of a cell suspension containing 125,000 MDBK cells/ml was added. The plates were incubated for 3 days at 37°C in 5% CO2 and examined for virus-specific CPE. The titer of each serum was expressed as the reciprocal of the highest dilution that inhibits virus-specific CPE.
Viral Culture
Lung and intestinal tissues collected at necropsy, and rectal swabs were used for virus isolation. 8 Briefly, about 1 g of tissue or 2 rectal swabs were homogenized in 9 parts of DMEM containing antibiotics. After centrifugation and filtration through a 0.45-μm filter, 50 μl of inoculum was adsorbed onto 1-day-old monolayers of MDBK cells in a 24-well plate for 2 hours at 37°C. The monolayers were refed with DMEM containing 5% FBS and incubated at 37°C in 5% CO2. The plates were examined daily for evidence of viral CPE for 7 days, after which they were scored as positive (presence of virus-specific CPE) or negative (absence of CPE).
Polymerase Chain Reaction on Tissue Samples
RNA extraction
Three 2-mm squares from small and large intestine, mesenteric lymph node, tonsil, lung, and spleen were cut and ground together in 2 ml of double distilled (DD) water. Additionally, the mucosal surfaces of the intestine were swabbed and used for extraction. The MagMAX-96 Viral RNA Isolation Kit (Ambion, Austin, TX) and Qiagen RNeasy Kit (QIAGEN Inc, Hilden, Germany) were used following the manufacturer’s instructions with the following modifications: the turbo DNase treatment step was omitted and replaced by Qiagen BioSprint 96 (Thermo Corp for Qiagen, Finland) to carry out the extraction. RNA was stored at –20°C until use.
Primer design
Primers were designed using Primer BLAST designing tool (http://www.ncbi.nlm.nih.gov/tools/primer-blast/; last accessed July 23, 2010) to amplify a 484-base pair (bp) fragment chosen from the polyprotein gene of the Oklahoma BEV isolate: BEV-F 5′-ACC TTT GTA CGC CTG TTT TCC-3′ and BEV-R 5′-GAT TAG CAG CAT TCA CGG C-3.′ The primers were manufactured by Integrated DNA Technologies (IDT, Coralville, IA).
RNA amplification
Qiagen One Step Reverse Transcriptase Polymerase Chain Reaction (RT-PCR) Kit (QIAGEN Inc, Hilden, Germany) was used according to the manufacturer’s instructions using the following procedure: 50°C 30 minutes → 94°C 10 seconds, 54°C 10 seconds, 72°C 35 seconds 30 cycles in the BioRad Mycycler (BioRad, Hercules, CA). Amplified products were then visualized by agarose gel electrophoresis. The BEV-1 Oklahoma isolate grown in MDBK cells was used as positive control for all RT-PCR tests; DD water was used as negative control.
In Situ Hybridization in Tissues
Preparation of probe for ISH
Bovine enterovirus-1 genomic RNA sequence (GenBank D00214), total length 7479 bp, was used to design PCR primers with insert length 526 bp on the target VP1. BEVF1: 5′-GAT GAG AGC ATG ATT GAG ACC-3′; BEVR: 5′-CGA GTG TCC TAA AGT ACA TGA-3′. The primers were manufactured by Integrated DNA Technologies, Inc. (IDT, Coralville, IA). The BEV-1 RNA was extracted from infected MDBK cell culture supernatant as described in the previous section. The RT-PCR used the above primers. First-strand cDNA synthesis was done using Superscript II (Invitrogen, Carlsbad, CA). The PCR was performed using the following procedure: 94°C 5 minutes, 94°C 30 seconds, 50°C 30 seconds, 72°C 30 seconds 40 cycles, 72°C 7 minutes 4°C. The PCR product was purified from an agarose gel and then ligated into the TA vector (pGEM-Teasy vector, Promega, Madison, WI). Ligation products were introduced into Escherichia coli by heat shock. Positive colonies were identified by PCR and then sequenced at the Molecular Genetics Instrumentation Facility (University of Georgia, Athens, GA) in both forward and reverse directions to obtain the complete sequence of the 5′ and 3′ gene fragments and verify a match with the Genebank sequence for BEV-VP1. A confirmed positive was cultured and plasmid DNA prepared using a Promega mini-prep kit. The resulting constructs were cut with restriction enzyme (SpeI), followed by in vitro transcription with T7 RNA polymerase generate an negative-sense RNA of approximately 526 nucleotides in length. These negative-sense riboprobes were used for ISH of formalin-fixed, paraffin-embedded tissue samples to detect BEV-1. A sense probe, cut by SacII followed by in vitro transcription with SP6 polymerase, was used as negative control for BEV-1 detection.
In situ hybridization in tissues
A standard method for riboprobe ISH was used as follows: slides were first heated at 70°C for 10 minutes and dewaxed in Hemo-DeTM (Fisher Scientific, Pittsburgh, PA). 2 Slides were then air-dried thoroughly, and tissue sections were rehydrated in 5 mM magnesium chloride (MgCl2) in phosphate-buffered saline (PBS) solution and digested with proteinase K (100 μg/ml) in proteinase K buffer (100 mM Tris pH 7.5 + 2 mM calcium chloride, CaCl2) for 15 minutes at 37°C. The enzymatic reaction was stopped with 0.1 M glycine in 0.2 M Tris, pH 7.5. Prehybridization solution (5× standard sodium citrate containing 48% [vol/vol] formamide, 5% [wt/vol] blocking reagent [Roche, Madison, WI], 0.1% N-lauroylsarcosine, and 0.02% sodium dodecyl sulfate) was added to sections for 1 hour at 42°C. A total of 70 μl hybridization solution (2 μl probe in 70 μl prehybridization solution) per slide was applied directly onto the section, which was covered with a siliconized coverslip, and the edges were sealed with nail varnish. Hybridization occurred overnight at 42°C in a humidified chamber. The next day, after stringent washes at 50°C, a 1 in 300 dilution of sheep anti-digoxigenin -AP Fab′2 (Roche) was applied for 2 hours at 37°C. Following washing, the signal was developed with the substrate and chromogen 4-nitro-blue-tetrazolium chloride (NBT, Roche) and 5-bromo-4-chloro-3-indolyl-phosphate (BCIP, Roche), respectively. Development was allowed to progress for 1 to 6 hours. Slides were counterstained lightly with hematoxylin (Gill’s, Vector Laboratories, Burlingame, CA) and coverslipped with Permount (Fisher).
Necropsy and Collection of Tissues for Histopathology and Laboratory Analysis
At necropsy, organs and tissues were examined in situ, dissected free, and fixed in neutral-buffered, 10% formalin solution for 1 week. Observations noted at necropsy were recorded in the individual animal necropsy record. Animal identification was saved. Tissue samples collected for microscopic examination included brain, heart, kidneys, large intestine (cecum, cecocolonic junction, and spiral colon), liver, lungs, lymph nodes (mesenteric, retropharyngeal), small intestine (duodenum, jejunum, ileum [ileocecal sphincter]), spleen, thymus, and tonsils (palatine and pharyngeal). Fresh, unfixed samples of intestines (jejunum and spiral colon at the cecocolonic junction), kidney, liver, lung, lymph nodes (mesenteric), spleen, and tonsils were also collected. Formalin-fixed tissues were processed, embedded in paraffin, sectioned at approximately 5 μm, stained with hematoxylin and eosin (HE), and evaluated for microscopic lesions.
If microscopic lesions were present, the lesion distribution was recorded as focal, multifocal, or diffuse, with distribution scores of 1, 2, or 3, respectively. If no significant tissue alterations were present (ie, histologically normal), a score of 0 was recorded. The severity of the lesions was recorded as minimal, mild, moderate, or severe, with severity scores of 1 through 4, respectively.
Statistical Analysis
All analyses were performed using SAS 9.2 (Cary, NC). A Wilcoxon rank sum test was used to compare severity, distribution, and the sum of the severity and distribution of microscopic findings, serology, PCR, and ISH (Cecum Illeocecel Valve [CIV], Spiral Colon [SC]) data between samples from control and treated calves. Virus isolation and ISH binary findings were compared between control and treated animals using a Fisher’s exact test. SN titers were represented as the reciprocal of the lowest dilution that tested positive (eg, 1:4 = 4) or as a 0 if the 1:1 dilution was negative. Pre- and postinoculation SN values were compared using a paired t-test.
A 2-factor analysis of variance (ANOVA) was used to test for differences in complete blood count (CBC) measurements between treatment and control and between days. The full model included factors for virus and day and a virus by day interaction term. A 3-factor ANOVA was used to test for differences in CBC measurements including room as an additional factor. The full 3-factor model included factors for virus, day, room, and all 2-way and one 3-way interaction term. Multiple comparisons were adjusted for using Tukey’s test. For all significant interactions, treatment (virus) effects were examined for each factor (either room or day) separately to evaluate the impact of the interaction on main effect conclusions.
All hypothesis tests were 2-sided, and the significance level was α = .05. The Wilcoxon rank sum test was implemented using PROC NPAR1WAY in SAS, the Fisher’s exact test was implemented in PROC FREQ, the paired t-test was implemented in PROC UNIVARIATE, and the ANOVA was implemented using PROC GLM in SAS.
Results
Clinical Observations
All experimental animals survived to their scheduled euthanasia times. Through the duration of the study, a few clinical signs of illness were observed in 3 of the experimental animals belonging to group 3 and consisted of moderate diarrhea in calf Nos. 17 (mock inoculated), 16 (infected), and 15 (infected) and anorexia and respiratory distress in calf No. 15.
Infection of Calves With BEV-1 Oklahoma Isolate
Table 1 summarizes the laboratory data generated on blood, feces, and tissues. As expected, calves belonging to group 1 (4 hours PI; day 0) had no detectable SN titers against BEV-1 before or after inoculation. In group 2 (day 5 PI), SN titers ranged from 0 to 16; most BEV-1 infected calves developed a detectable titer that ranged from 4 (calf No. 8) to 8 (calf No. 11) to 16 (calf No. 9); infected calf No. 10 had no detectable titer. Although mock-inoculated (negative control) calf No. 12 did not develop SN titers, the mock-inoculated calf No. 7 had a titer of 4. In group 3, there was no evidence of seroconversion; there was either no change or a mild increase or decrease in the SN titers (≤4-fold) of mock-inoculated or infected calves, respectively.
Experimental Infection of Calves With a Bovine enterovirus-1 Isolate: Results From Serology, Virus Isolation, Polymerase Chain Reaction, and In Situ Hybridization
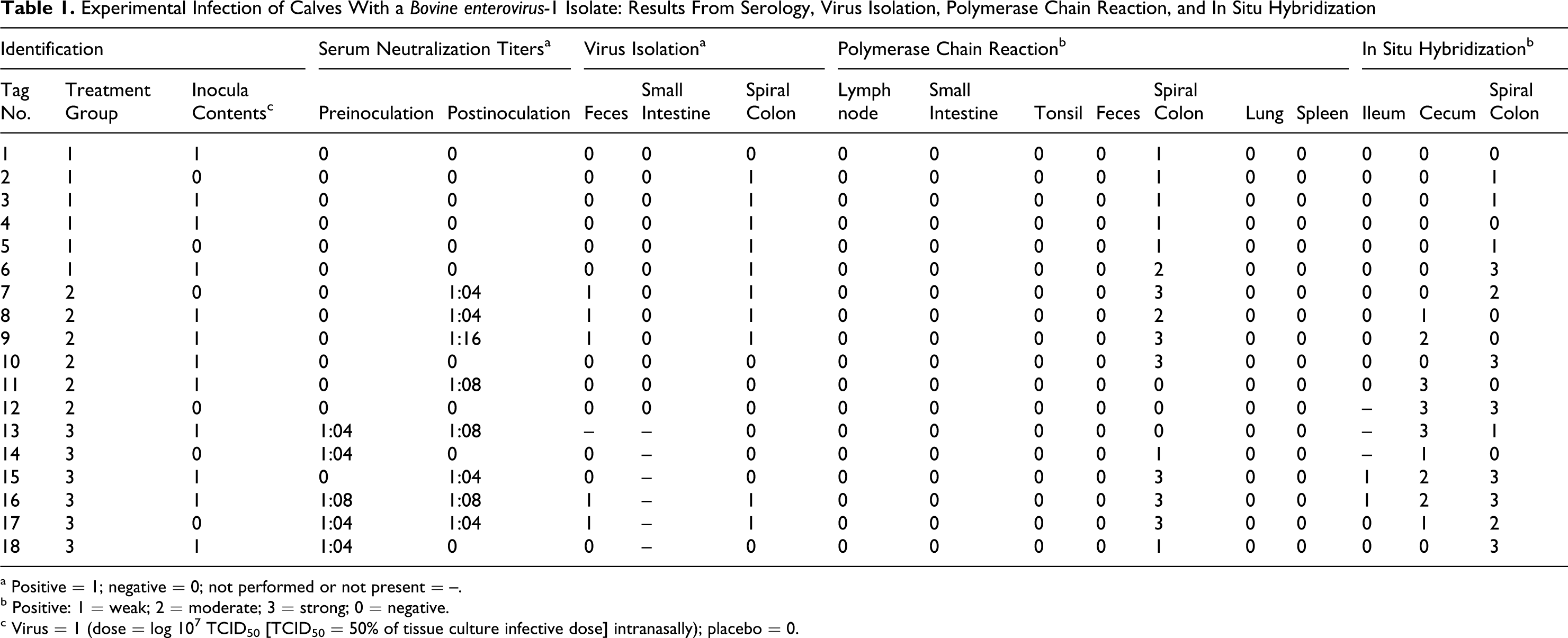
a Positive = 1; negative = 0; not performed or not present = –.
b Positive: 1 = weak; 2 = moderate; 3 = strong; 0 = negative.
c Virus = 1 (dose = log 107 TCID50 [TCID50 = 50% of tissue culture infective dose] intranasally); placebo = 0.
In group 1, BEV-1 was isolated from the spiral colon of most calves but not from the feces or from the small intestine (Table 1). In group 2, virus was isolated from both the spiral colon and feces of 2 infected calves (Nos. 8 and 9) and from 1 mock-inoculated calf (No. 7). No virus was isolated from the small intestine in this group. In group 3, virus was isolated from both the spiral colon and feces of 1 infected calf (No. 16) and from 1 mock-inoculated calf (No. 17). No virus isolation was performed from the small intestine in this group.
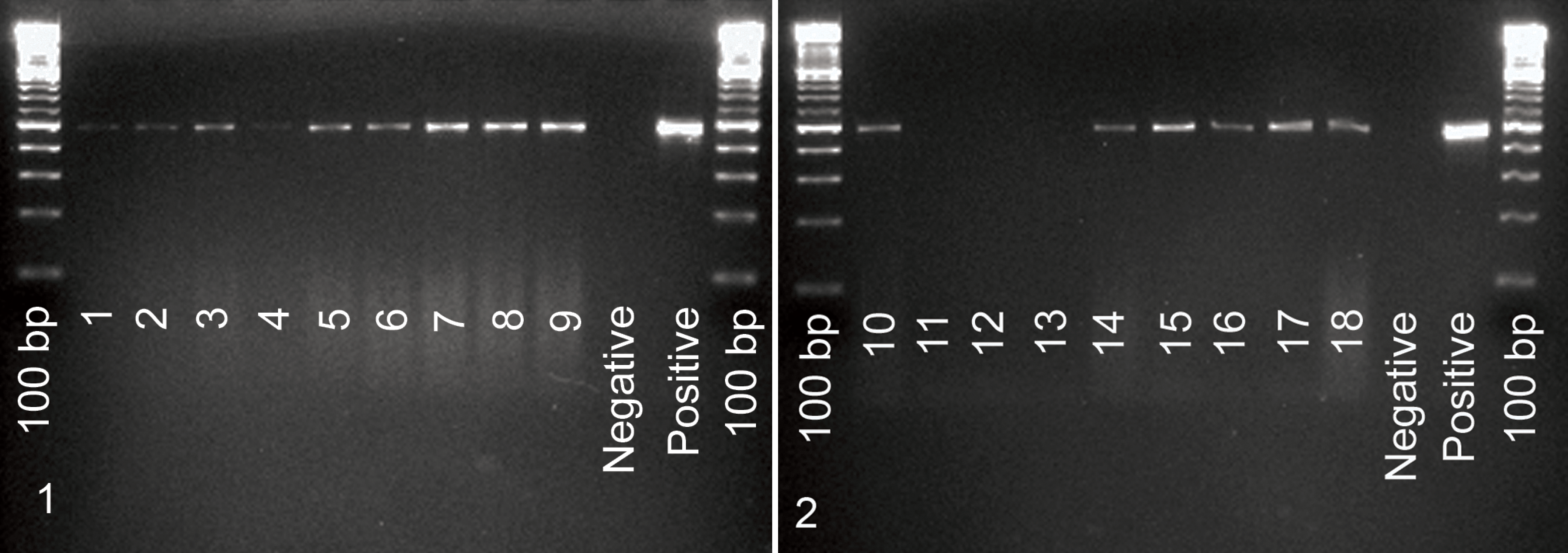
Results of PCR are illustrated in Figs. 1 and 2 . The positive control used, tissue culture BEV-Oklahoma strain, had an expected PCR amplification product of 484 bp. The negative control (DD water) had no amplification product detected. Amplification products of identical molecular weight were only detected in the spiral colon; no molecular evidence of BEV-1 infection was detected in feces, lung, mesenteric lymph nodes, small intestine, spleen, or tonsils. All but 3 animals (calf Nos. 11 and 12 from group 2 and No. 13 from group 3) tested positive through PCR of the spiral colon.
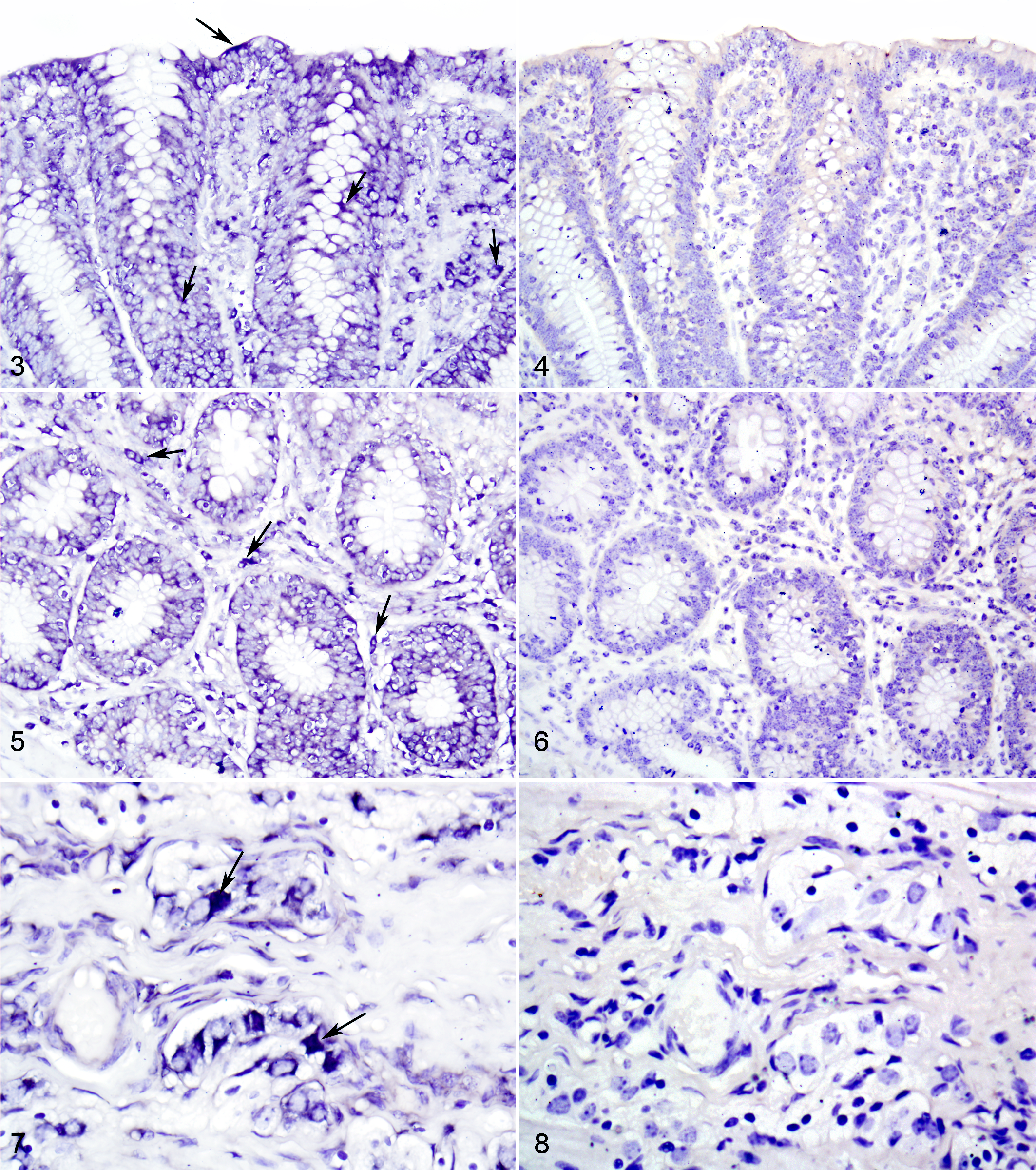
The riboprobe corresponding to BEV-VP1 protein was detected in the terminal ileum, cecum, and spiral colon (near the cecocolonic junction) of all but 2 animals (calf Nos. 1 and 41, both from group 1). Positive hybridization signal was detected in the cytoplasm of enterocytes and cells within lamina propria (probably macrophages) as well as in neurons of the submucosal and myenteric ganglia, vascular endothelium, submucosal lymphoid tissues, and ileocecal lymph nodes (Figs. 3-10).
Clinical Pathology of Blood Samples
Results from the CBC data generated on blood did not provide evidence of clinical infection or disease associated with viral infection in any of the calves (infected and noninfected) from the study.
Gross and Microscopic Findings
Very few gross lesions were recorded at necropsy. Calf Nos. 8 (infected; group 2, day 5 PI) and 15 (infected; group 3, day 10 PI) had mild hyperemia of the small intestinal serosa and associated mesenteric lymphadenopathy. Mild hyperemia and focal consolidation of less than 20% of the right cranial lung lobe were detected on calf Nos. 15 (infected; day 10 PI), 17 (mock inoculated; day 10 PI), and 18 (infected; day 10 PI).
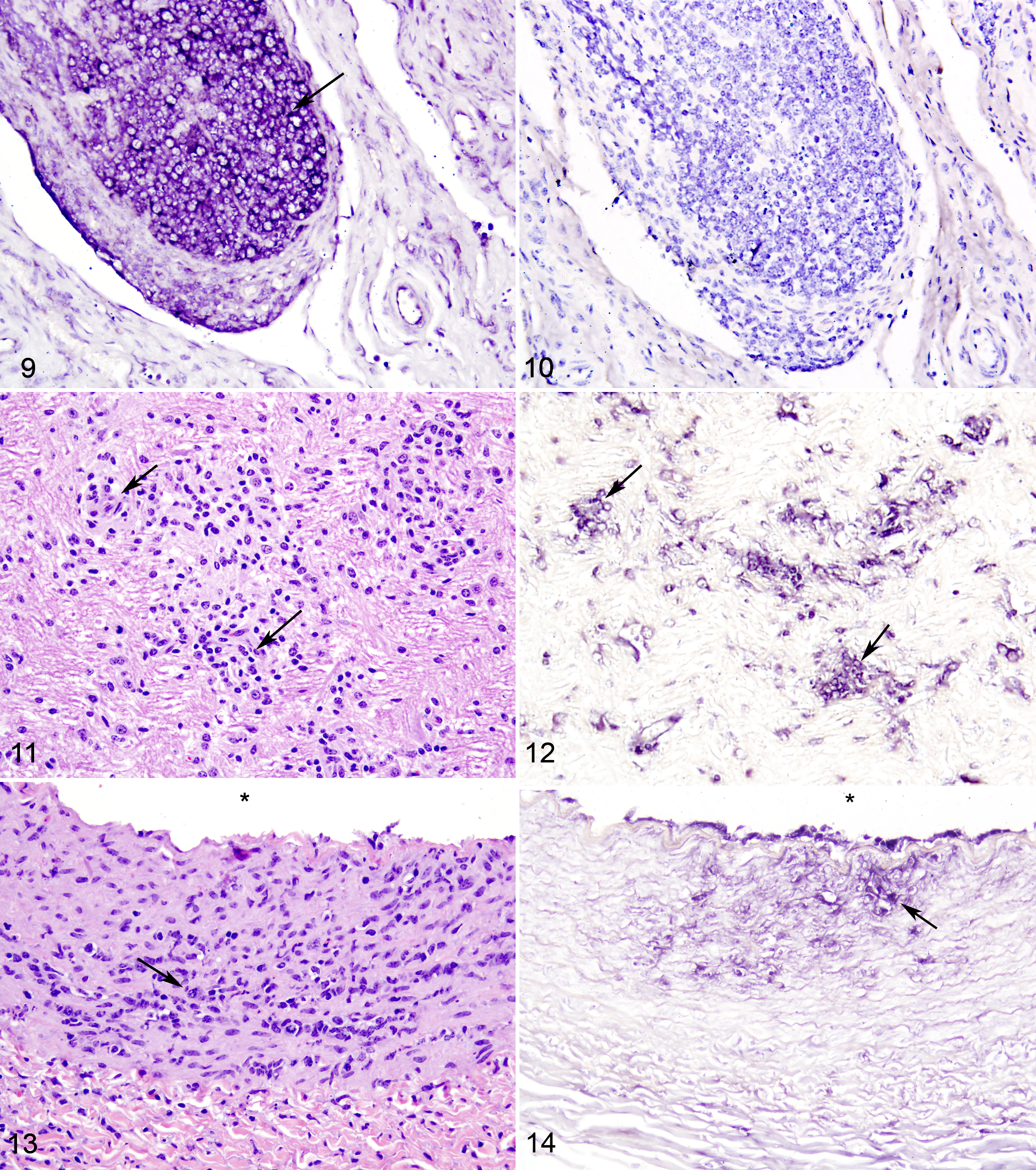
Although there were no statistically significant differences in microscopic findings between infected and mock-inoculated calves, a few biologically significant microscopic findings were documented (Figs. 3-14). Calf Nos. 8 and 9, both of which were infected and belonged to the group 2 at day 5 PI, exhibited mild, diffuse typhilitis with discrete foci of hemorrhage. Calf No. 8 had widely scattered, minimal to mild plasmacytic and lymphocytic meningoencephalitis, with a discrete focus of hemorrhage and focal gliosis in the hind brain, at the level of the cerebellar white matter, just superior to rostral medulla, and calf No. 9 had mild multifocal coronary arteritis, characterized by mild to moderate expansion of the tunica intima and media with focal collections of spindled cells, lymphocytes, plasma cells, and rare neutrophils and minimal perivascular collections of plasma cells about vessels within the tunica adventitia (vasa vasora). Detection of BEV-1 through use of negative-sense riboprobes for ISH was positive in the brain of calf No. 8 and in the coronary artery of calf No. 9 (Figs. 11-14).
Discussion
The current report describes the first pathological study in which a small group of calves were experimentally infected with the Oklahoma isolate of BEV-1. Results of the study suggest that young calves are probably susceptible to infection with this virus by intranasal inoculation and possibly through aerosol or close contact, since several noninfected, control calves had evidence of infection as determined through isolation and molecular detection of the virus from the feces and/or the spiral colon.
The finding that control calves became infected limits the interpretation of the current study. It could be argued that the calves had been exposed naturally, but the lack of serologic evidence, as groups 1 and 2 show, suggests that the calves infected each other through close contact, as indicated above and not from prior exposure. Evaluation of baseline fecal samples by PCR and virus isolation prior to inoculation would have solved this potential problem. Thus, interpretation of the study findings should be done with caution.
Although infected calves did not have signs of clinical disease, calves in group 2 had evidence of seroconversion by 5 days PI (Table 1). Molecular detection of the virus through PCR and ISH methods suggests that BEV-1 potentially gains access into the body by infecting enterocytes localized on the terminal ileum, ileocecal and cecocolonic junctions, and spiral colon. Furthermore, it also suggests that cells within the lamina propria (probably macrophages) and neurons within the submucosal and myenteric plexi, endothelium, and submucosal lymphoid tissues are infected with BEV-1 and are potential points of entry of the virus for later systemic spread.
Microscopically, the acute encephalitis noted in the cerebellum of calf No. 8 and the acute coronary arteritis of calf No. 9, both from group 2 (5 days PI), suggest that BEV-1 isolate potentially spreads systemically and localizes to these organs early after infection. This finding, in light of the absence of effective negative controls, suggests that there is an association between the lesions and infection with BEV-1. If accurate, this finding would be considered very important in the pathogenesis of BEV-1 in cattle. In humans and animals, including pigs, elephants, and baboons, a similar combination of microscopic lesions has been described following enterovirus (picornavirus) infection.5,6,9,11–13,18,20
To further characterize the observed pattern of infection, microscopic lesions, and pathogenesis of this disease and to explore the host response to this infectious agent, additional studies are needed in which larger groups of animals are used in a controlled environment. Having knowledge about the susceptibility of cattle to challenge, about the pathology associated with infection, and about the prevalence of BEV-1 infection will be essential for the understanding and characterization of this pathogen in cattle worldwide.
Footnotes
Acknowledgements
The following people are recognized and acknowledged for their help and contributions: Logan Gabriel, Frank Waters, Kara Nitschke, Jeff Tucker, and the Animal Resources Department for necropsy assistance and care provided for the research animals. Rachel Steffens, Paula Bartlett, Ingrid Fernandez, and Sarah Bates are commended for their excellent assistance with the virus isolation, primer design, and PCR work.
The authors declared that they had no conflicts of interest with respect to their authorship or the publication of this article.
The study was supported by an award to Uriel Blas-Machado by the University of Georgia Veterinary Medical Experiment Station State Funding (VMES Animal Disease Project 07-001) and USDA Formula Funds-Program Award 2007-36100 (GEOV-0503).