
Abstract
The objective of this study was to investigate the effects of chronic inhibition of nitric oxide synthase (NOS) on cyclooxygenase-2 (COX-2) expression in the macula densa (MD) of swine, as well as the effects on expression of related proteins. Adult female Yucatan swine were given either tap water (control, n = 6) or water with N
G-nitro-
Keywords
The macula densa (MD) is composed of specialized morphologically distinct tubular epithelial cells located on the glomerular side of the distal tubule. By sensing electrolyte concentration in the distal tubule, the MD provides feedback to control preglomerular resistance and renin secretion, thereby participating in control of electrolyte and water homeostasis. Activity of the Na-K-2Cl cotransporter in the MD is thought to initiate a series of reactions that control glomerular filtration rate and renin secretion. Cyclooxygenase-2 (COX-2) and neuronal nitric oxide synthase (nNOS) are colocalized in the MD. The products of these enzymes—prostaglandins (PGs) and nitric oxide (NO), respectively—have been reported to be modulators of this series of reactions, counteracting afferent arteriolar vasoconstriction mediated by tubuloglomerular feedback (TGF), as well as TGF-induced inhibition of renin secretion.7,12,21,23,28,31 In addition, both enzymes are upregulated with interventions, such as low salt diets, administration of loop diuretics, and renovascular hypertension. That these enzymes colocalize in the MD suggests that nNOS might be involved in mediating expression of COX-2 and vice versa. 5 Interestingly, hemodynamic responses to COX-2 inhibition, as seen after nonselective NOS blockade, could not be duplicated by selective nNOS blockade, thus suggesting that other NOS isoforms could be involved. 3 Castrop et al proposed that MD-dependent renin secretion does not require intact nNOS or endothelial NOS (eNOS) activity but, rather, NO generated by global NOS activity. 4 In addition to eNOS and nNOS, the other isoform of NOS—inducible NOS (iNOS)—may participate in this process. 30 The isoform is constitutively expressed in rodent kidney; its renal expression is less certain in large animal models. 20
Initial data from in vitro and in vivo studies suggested that NO enhances production of COX-2-derived products and so acts as a modulator of the COX-2 pathway.5,9 These data led to the hypothesis that NO bioavailability is obligatory for COX-2 expression and/or activity (Fig. 1a). Studies to the contrary have been reported. There was no reduction in COX-2 immunoreactivity in the MD cells of nNOS knockout mice. 27 Indeed, studies have demonstrated that COX-2-derived PGs play an important role in buffering renal vasoconstriction secondary to a reduction in NO bioavailability.3,24
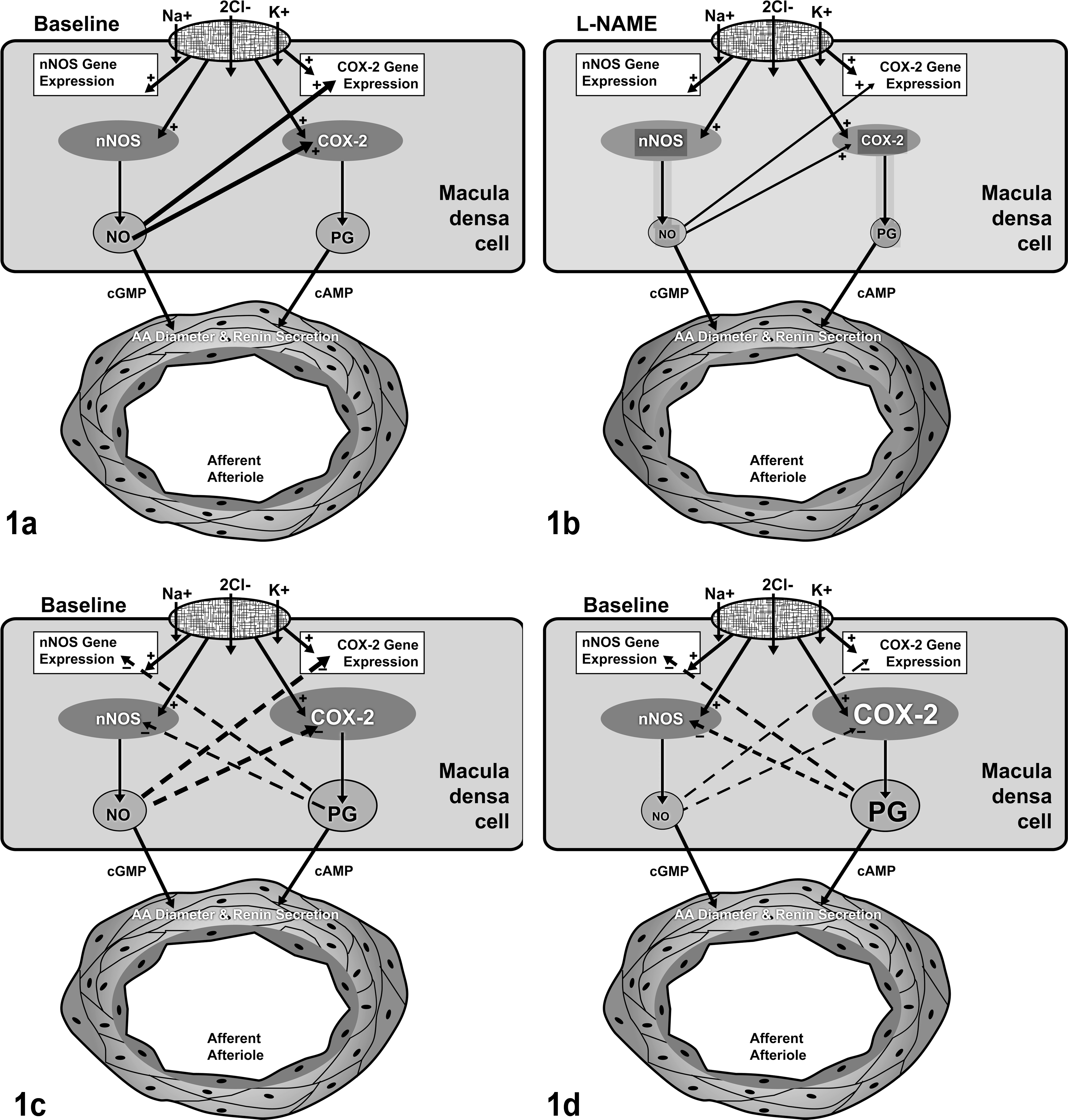
Schematic illustration of cyclooxygenase-2 (COX-2) regulation: 1a and 1b, the obligatory nitric oxide (NO) hypothesis; 1c and 1d, the reciprocal regulation hypothesis. Abbreviations: nNOS, neural isoform of NO synthase; PG, prostaglandin; AA, afferent arteriole; L-NAME, N
G-nitro-
Constitutive expression of low levels of COX-2 in the MD of normal kidney has been documented in several laboratory animal species. 14 Initial studies in humans failed to detect COX-2 in the MD; 15 more recent studies have documented its expression in MD of normal human kidneys. 1 Markedly increased COX-2 protein was detected in the MD of elderly humans, children with Bartter-like syndrome, and hypertensive patients.13,16,19 Decreased endogenous NO production is associated with aging and is known to contribute to hypertension.10,11 Thus, an alternative to the obligatory NO hypothesis noted above has emerged—namely, that COX-2 expression and NOS expression are reciprocally regulated in MD (Fig. 1c). This hypothesis suggests that in populations characterized by reduced NO bioavailability, COX-2 is upregulated in response to, and to compensate for, a relative lack of NO (Fig. 1d).
We hypothesized that, with chronic inhibition of NOS, COX-2 in the MD would be upregulated to compensate for diminished production of endogenous NO. We used a large animal model—the Yucatan miniature porcine model—to test this hypothesis. We use this model in our laboratory because the anatomy and physiology of its cardiovascular system exhibit many similarities to those of the human. In several of our studies, we have noted that certain aspects of renal function (eg, renal blood flow) faithfully model human physiology. 17
Methods and Materials
Animals and Treatment Protocol
Eleven female Yucatan miniature swine (30.4 ± 1.3 kg) were included. Animals were obtained from Sinclair Research Farm (Columbia, MO) and singly housed in pens. All experimental protocols were approved by the University of Missouri Animal Care and Use Committee. The swine were given either tap water to drink (control, n = 6) or water with N
G-nitro-
Assessment of Treatment Efficacy
Assessment of effectiveness of chronic L-NAME treatment was done by measuring plasma NO metabolite (NOx) concentration and mean arterial pressure, as done previously. 17 The former was determined via VCl3-induced reduction of NOx to NO and its subsequent reaction with O3, producing chemiluminescence (Model NOA 280i, Sievers). Mean arterial pressure was determined via an indwelling arterial catheter.
Sample Collection
Kidney samples were obtained following euthanasia. A piece of the kidney was snap frozen in liquid nitrogen and stored in a RNAse-free bag at –80°C until analysis. Another piece of the kidney was fixed in 10% neutral buffered formalin for at least 24 hours and embedded in paraffin for immunohistochemistry (IHC). In addition, plasma and serum samples were stored at –80°C until determination of plasma NOx concentration, plasma renin activity (PRA), and serum electrolyte concentrations.
IHC
COX-2 protein expression in MD was determined by IHC with Dako’s Envision system. Fixed kidney samples were sectioned serially at a thickness of 5 μm, collected onto positively charged slides, deparaffinized, and steamed in Target Retrieval (Dako) for 30 minutes and cooled for 20 minutes at room temperature. Sections were incubated in 3% (vol/vol) hydrogen peroxide for 5 minutes to inhibit endogenous peroxidase, then treated with protein block for 10 minutes. Sections were incubated with a polyclonal COX-2 antibody (1:800; Cayman) for 45 minutes. Negative controls were incubated with nonimmune serum instead of antibody. After washing, sections were incubated with labeled polymer–horseradish peroxidase (Dakocytomation Envision+) for 30 minutes. To immunostain iNOS and renin, the Dako LSAB+ system was used. The same procedure as above was followed with a few modifications. The sections were incubated overnight with a polyclonal iNOS antibody (1:600, Transduction Labs) or a polyclonal renin antibody (1:60,000, Innovative), then with biotinylated anti-sheep immunoglobulin G and labeled polymer–horseradish peroxidase. All proteins were visualized with diaminobenzidine, counterstained with hematoxylin, dehydrated, and coverslipped. The slides were stained manually; a Tris buffer or water wash was performed after each step. Sections were photographed with an Olympus BX40 microscope and Spot Insight color camera (Diagnostic Instruments).
Laser Capture Microdissection
Laser capture microdissection (Pixcell IIe, Arcturus) was used to obtain MD from control and L-NAME porcine kidneys. The frozen kidney was cryosectioned to 8-μm thickness, collected on slides, and stored at –80°C until further analysis. The sections were stained with hematoxylin and dehydrated just before laser capture microdissection. The MD—a plaque of distinct tubular epithelial cells located near the glomerulus—was identified on the basis of morphology and collected on high-specificity caps. Total RNA was extracted (RNA Isolation Kit, PicoPure) and stored at –80°C until further analysis.
mRNA quantification
Quantitative real-time reverse transcription polymerase chain reaction was performed to determine COX-2 mRNA expression in MD. cDNA was synthesized from mRNA by reverse transcription, and polymerase chain reaction was performed as described by Tharp et al. 26 Briefly, COX-2 cDNA was amplified with primers 5′ GGC TGC GGG AAC ATA ATA GA 3′ (forward) and 5′ AAA AGC AGC TCT GGG TCA AA 3′ (reverse) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA, with primers 5′ TCA AGA AGG TGG TGA AGC AG 3′ (forward) and 5′ TGT CGT ACG AGG AAA TGA GC 3′ (reverse). The reaction mixture consisted of RNAase-free water, Sybergreen master mix, 100 nM of primers, and 1.0 μl of cDNA. The conditions for amplification of cDNA were 95°C for 10 minutes, followed by 50 cycles of 95°C for 5 seconds, 56°C for 20 seconds, and 72°C for 30 seconds. Relative amounts of COX-2 DNA, as normalized by GAPDH DNA, were calculated from threshold cycle numbers using the 2-ΔΔct method.
Measurement of Serum Electrolytes and PRA
Serum Na+ and K+ concentrations were measured by an ion-specific electrode (Model AU400, Olympus). A 125I-radioimmunoassay of generated angiotensin I (Ang I) was used as an index of PRA (DiaSorin). Antibody-coated tubes (rabbit anti–Ang I), renin activity controls, Ang I standards, and 125I Ang I tracer were used according to the manufacturer’s instructions. Preincubation at pH 6.0 for 3 hours with phenylmethylsulfonyl fluoride was done to inhibit angiotensin-converting enzyme and other angiotensinases. Calibrator curves were prepared at both 4°C and 37°C, with the former (background) subtracted from the latter. PRA was calculated with a log-logit program. Interassay and intra-assay coefficients of variation were 8.5% and 6.2%, respectively, whereas sensitivity of the assay was 0.25 ng/ml.
Statistics
Results are expressed as mean ± standard error of the mean. Data were analyzed by the unpaired t test using Sigma Stat 2.0 software.
Results
Assessment of Treatment Efficacy
Swine in the present study were a subset of those included in a larger study examining multiple effects of chronic NOS inhibition. 17 In that study, swine of the L-NAME-treated group consumed 8.0 ± 0.4 mg/kg of L-NAME per day (n = 24) via drinking water. L-NAME-treated swine were hypertensive (control, 87 ± 5 mmHg, n = 9; L-NAME, 118 ± 7 mmHg, n = 8; P = .002); in addition, plasma NOx concentration was reduced by L-NAME treatment (control, 12.5 ± 1.9 μM, n = 21; L-NAME, 8.1 ± 1.2 μM, n = 19; P = .032).
IHC localization of COX-2 in MD
There was intense COX-2 immunoreactivity in the MD of 3 L-NAME swine, focally intense immunoreactivity in a fourth, but little immunoreactivity in the fifth. In controls, there was focally moderate immunoreactivity in 1 animal and none in the other 5. Representative sections from each group are shown in Figure 2 .
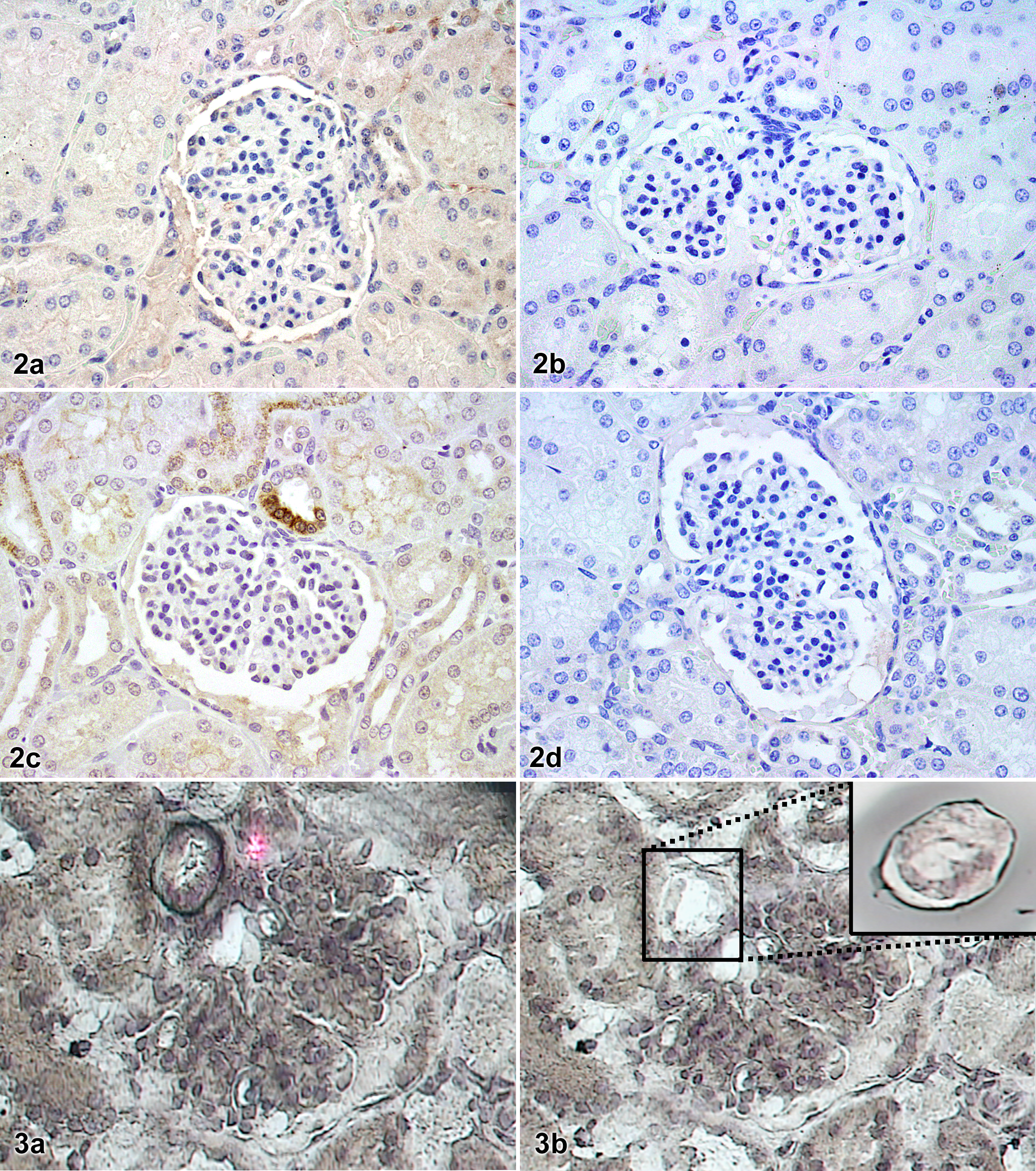
Whole kidney sections; control swine (2a, 2b) and swine treated with N
G-nitro-
COX-2 mRNA Expression
Although IHC studies are helpful for localization of protein expression, they have certain limitations for quantification of expression. To confirm IHC findings, we used laser capture microdissection to obtain MD cells, as shown in Figure 3 . Figure 4 shows expression of COX-2 mRNA (normalized to that of GAPDH) in microdissected MD from control and L-NAME swine. There was a significant increase in COX-2 mRNA expression in the MD with L-NAME treatment (P = .0082).
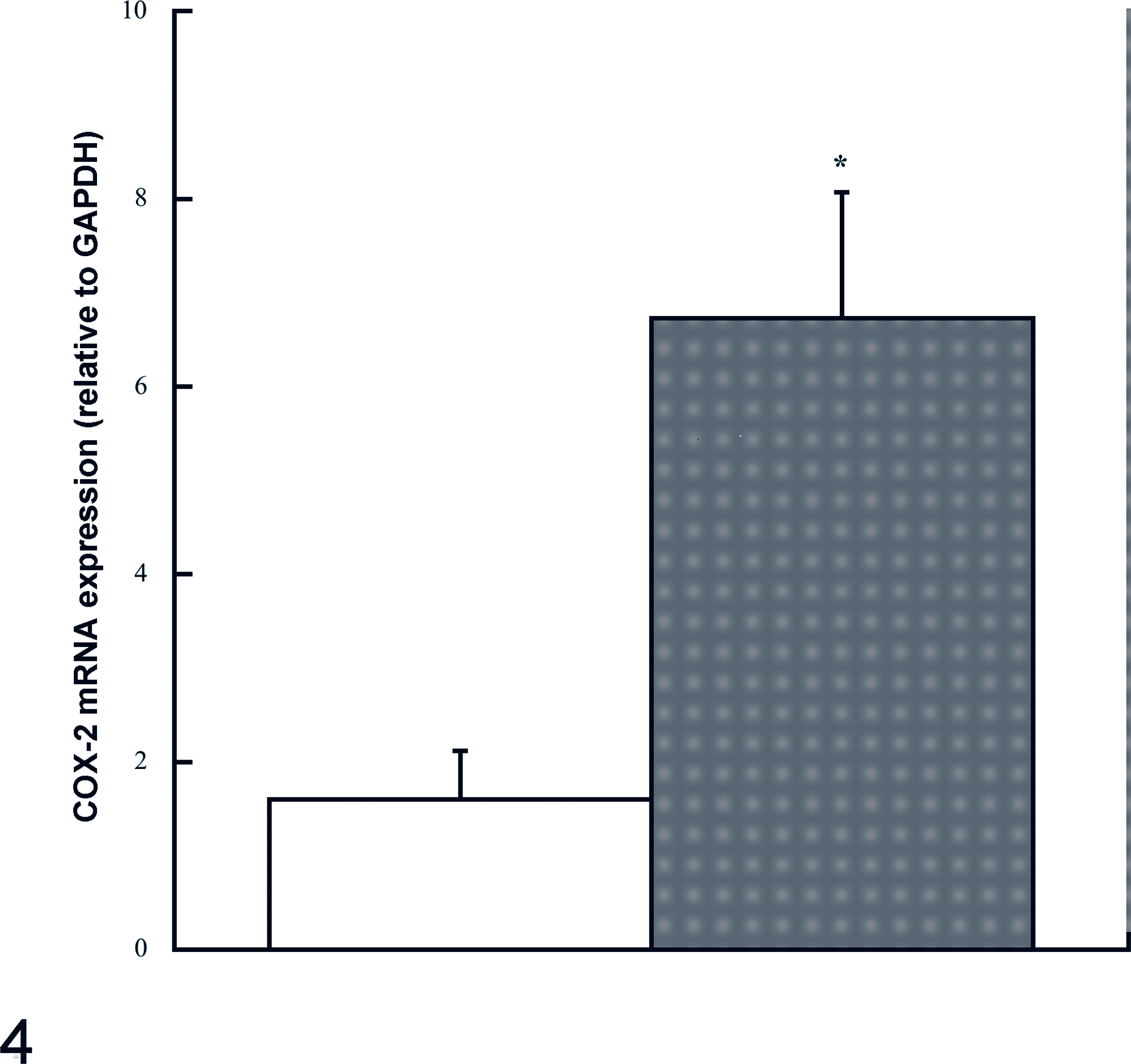
Reverse transcription polymerase chain reaction quantification of cyclooxygenase-2 (COX-2) mRNA expression in the macula densa of swine treated with N
G-nitro-
IHC Detection of iNOS
To determine whether altered iNOS expression was associated with increased expression of COX-2 in the MD, IHC for iNOS protein was performed. iNOS positive reactions were not observed in the MD of either control or L-NAME-treated pigs (Fig. 5). However, glomerular macrophages showed positive iNOS immunoreactivity, which did not appear different between groups.
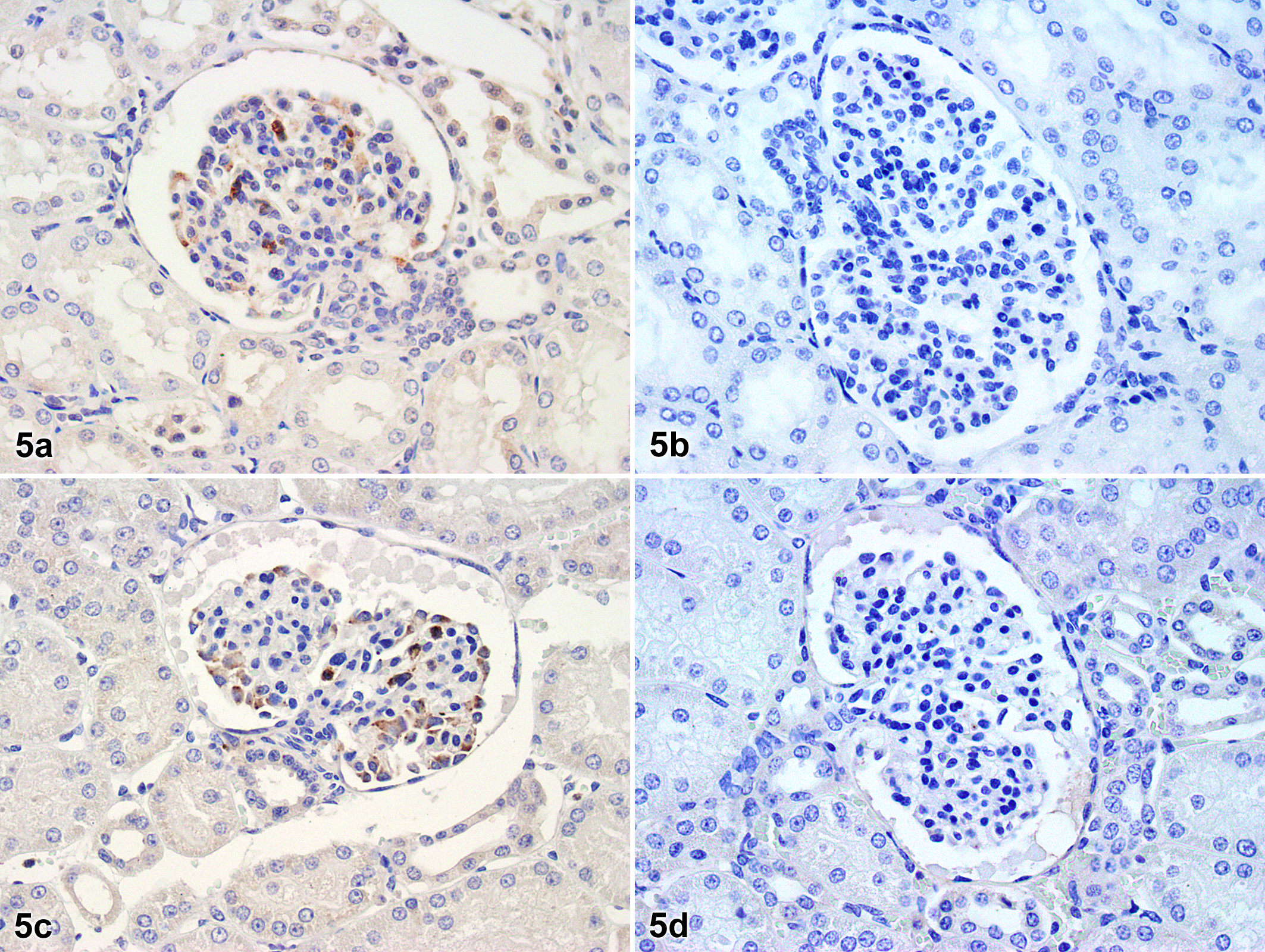
Whole kidney sections; control swine (5a, 5b) and swine treated with N
G-nitro-
Renin Expression in Juxtaglomerular Apparatus
IHC was performed to determine whether a change in the renin expression in the juxtaglomerular apparatus (JGA) was associated with chronic NOS inhibition. Figure 6 shows JGA renin in control and L-NAME-treated swine. Few renin-positive cells were observed in afferent arterioles of either control or L-NAME-treated pigs. There was no apparent difference between groups in intensity of renin staining. PRA, however, was greater in L-NAME-treated swine (control, 0.34 ± 0.08 ng/ml, n = 10; L-NAME, 1.26 ± 0.03 ng/ml, n = 7; P = .00000003).
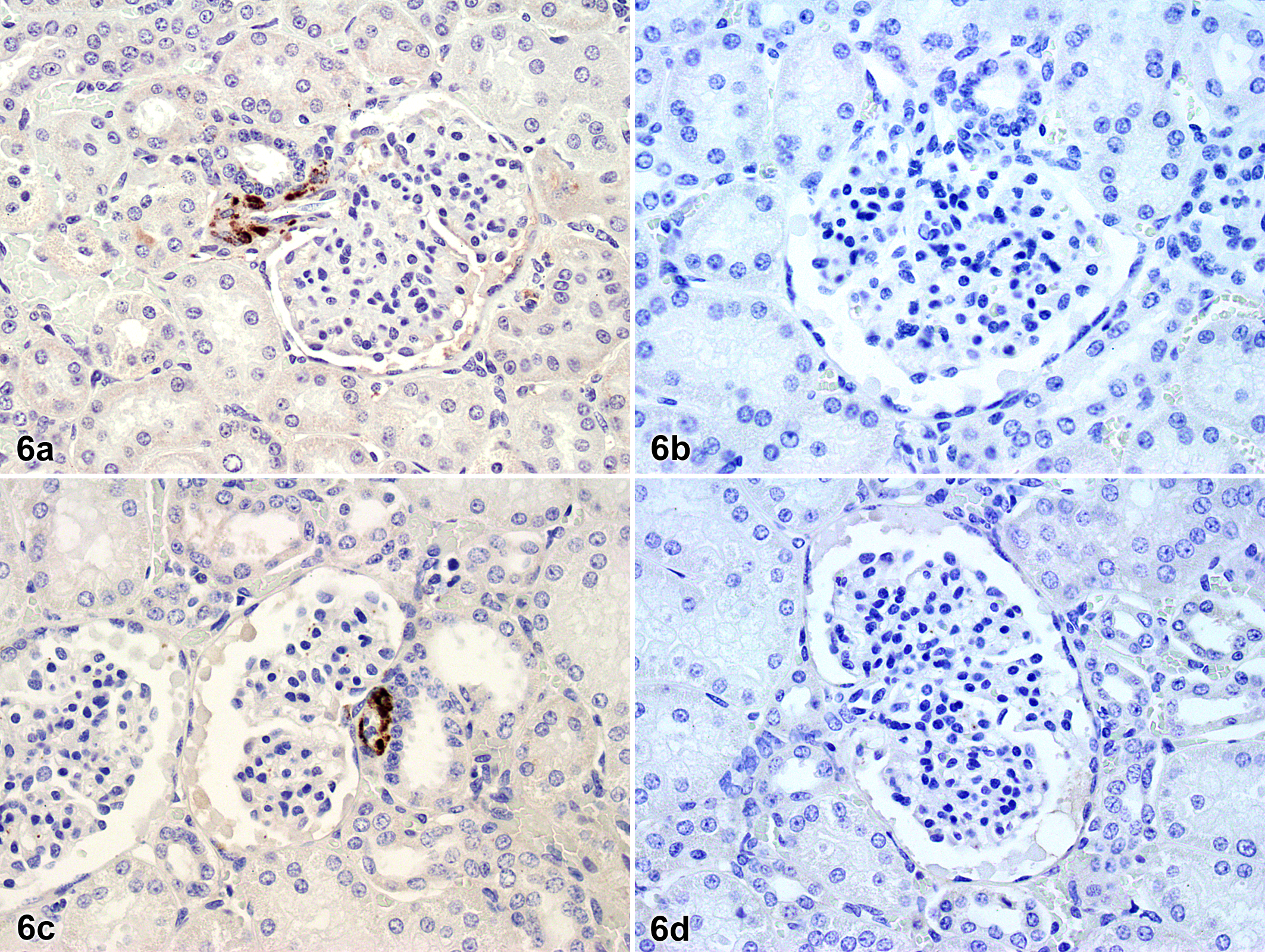
Whole kidney sections; control swine (6a, 6b) and swine treated with N
G-nitro-
Serum Electrolyte Concentrations
Serum Na+ and K+ concentrations were within the normal range for all swine. In addition, there were no significant differences between control and L-NAME-treated groups (Table 1).
Effect of Chronic L-NAME Treatment on Serum Electrolyte Concentrations a
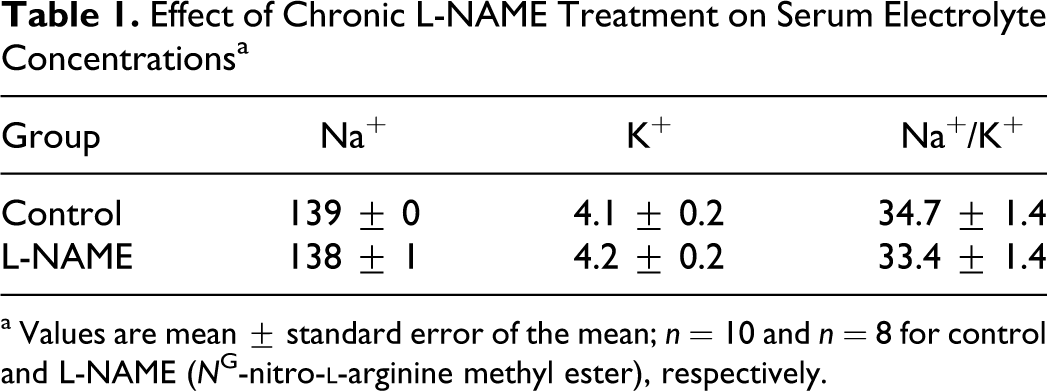
a Values are mean ± standard error of the mean; n = 10 and n = 8 for control and L-NAME (N
G-nitro-
Discussion
COX-2 and nNOS colocalize in the MD, and significant interactions have been documented. NO has been shown to increase the production of PGs by acting at the posttranscriptional and translational levels to increase COX-2 protein. 18 In cultured microdissected rabbit thick ascending limb cells, for example, a NO donor increased COX-2 expression, whereas a NO synthesis inhibitor decreased it. 9 Synchronized expression of COX-2 and nNOS in MD-dependent events led to the hypothesis that NO bioavailability is obligatory for COX-2 expression and/or activity (Fig. 1a). We found that increased COX-2 expression in the MD—at both the message level and the protein level—was associated with chronic inhibition of NOS, contrary to a reduction in COX-2 expression, which would be predicted by the obligatory NO hypothesis (Fig. 1b). This study is, to our knowledge, the first to determine changes in renal COX-2 expression with chronic NOS inhibition. Importantly, we found an upregulation of COX-2 in MD cells of the kidney, obtained by microdissection.
COX-2 has been reported to be constitutively expressed in the kidney of several laboratory species studied to date. 8 Initial studies in humans failed to detect COX-2 in the MD of normal adult kidney. 15 However, COX-2 expression in human MD has been demonstrated in elderly individuals and patients with a clinical history of hypertension.13,19 Diminished NO production is associated with aging and has been demonstrated to be a mechanism underlying certain forms of hypertension.29,32 Therefore, increased COX-2 expression in elderly or hypertensive patients may be secondary to decreased endogenous NO formation, suggesting an alternative to the obligatory NO hypothesis noted above; that is, COX-2 and NOS expression are reciprocally regulated in MD (Fig. 1c).
An apparent upregulation of COX-2 activity and/or expression in whole kidney tissue, possibly to compensate for diminished NO synthesis, has been documented in several studies using rats, mice, and dogs. Baylis et al reported that, in conscious rats, COX inhibition produced acute renal vasoconstriction only after acute or chronic treatment with L-NAME, thus suggesting that vasodilatory PGs compensate for renal vasoconstriction caused by NOS inhibition. 2 Similar findings were reported by Roig et al in chronically instrumented dogs. 24 Under resting conditions, COX inhibitors did not affect basal hemodynamic parameters, but elimination of the vasodilator influence of NO resulted in an apparent compensatory vasodilatory response by COX products, which buffered the vasoconstrictor tone resulting from inhibition of NOS activity. In another study, L-NAME-induced vasoconstriction appeared to be primarily compensated for by COX-2-derived PGs. 3 Renal afferent arterioles make a large contribution to renal vascular resistance, which is modulated by vasodilatory substances from MD. Thus, it may be that increased potential for vasodilatory PG formation, via COX-2 upregulation in MD, was in response to, and to compensate for, a relative lack of NO in our swine (Fig. 1d). This compensation was incomplete, however, given that we reported that renal blood flow at rest was reduced in these L-NAME-treated swine. 17 Findings consistent with our data come from rodents with deletion of nNOS in which low-salt and high-salt diets caused changes in renal COX-2 expression similar to those in wild-type mice. NO generated by nNOS may not be a necessary mediator of COX-2 upregulation in the MD. 27 Our results from swine therefore substantiate and extend previous findings from murine models.
L-NAME is not an effective inhibitor of iNOS; indeed, in chronic L-NAME hypertension, iNOS is upregulated to compensate for the loss of other NOS isoforms. 22 In cells that express both constitutive NOS (ie, eNOS and nNOS) and iNOS isoforms, NO released under basal conditions by the former 2 isoforms keeps iNOS in a quiescent state through the inhibition of nuclear factor κB, which in turn regulates iNOS transcription mechanisms. 6 To examine a possible role of iNOS for increased expression of COX-2 in the MD of L-NAME-treated swine, we performed IHC to localize iNOS expression. We were unable to detect iNOS protein in the MD of either L-NAME or control pigs. Glomerular macrophages were the only observed iNOS-positive cells, and there was no difference in expression between groups. These findings suggest that iNOS-derived NO did not contribute to increased COX-2 expression in our L-NAME-treated swine and therefore do not support the obligatory NO hypothesis. Furthermore, the findings suggest that, unlike small animal models (eg, mouse), swine do not constitutively express iNOS in the kidney. 20
Whether NO and PGs act in parallel to induce renin secretion is still controversial, but several studies have demonstrated modulatory roles for MD-derived NOS and COX-2 products in renin secretion. MD-derived PGs are potent stimulators of renin synthesis and secretion. In the present study, there was no difference in renin expression in the JGA between L-NAME and control swine, but there was a marked increase in PRA with chronic NOS inhibition. This may indicate that increased PG formation induced greater renin release from the JGA, resulting in elevated PRA. We must acknowledge that renin expression and secretion are under the control of mechanisms such as increased sympathetic nerve activity in addition to MD-derived NO and PGs.
In summary, the present findings demonstrate that chronic inhibition of NO synthesis results in upregulation of COX-2 in the kidney. The key new finding is that this increase in COX-2 expression occurs in the MD. MD-derived COX-2 products could in turn counteract renal vasoconstriction in NOS inhibition-induced hypertension and maintain renin synthesis/secretion. Selective COX-2 inhibitors are now widely used as anti-inflammatory agents, but the renal effects of these drugs are not well studied. It is important to consider the adverse effects of these drugs on kidney function, especially when there is diminished formation of endogenous NO, as in hypertension and aging.10,11,29,32 Upregulation of COX-2 in response to decreased NO may be a compensatory mechanism aimed at protecting renal vasculature from hypoperfusion and may thus explain COX-2 inhibitor-related nephrotoxicity in some patients. NO-donating nonsteroidal anti-inflammatory drugs are a new class of drugs produced by adding a NO moiety to them. 25 Inhibition of COX with concomitant release of NO may have promising effects in these patients, and further studies are required to study the renal effects.
Footnotes
Acknowledgments
We gratefully acknowledge the technical assistance of Jennifer Casati and Miles Tanner, as well as the animal care provided by Dave Harah.
The authors declared that they had no conflicts of interest with respect to their authorship or the publication of this article.
This work was supported by National Institutes of Health grant Nos. RR-18276 and HL-52490.