
Abstract
Situs inversus (SI) is a congenital condition characterized by left–right transposition of thoracic and visceral organs and associated vasculature. The usual asymmetrical positioning of organs is established early in development in a transient structure called the embryonic node. The 2-cilia hypothesis proposes that 2 kinds of primary cilia in the embryonic node determine left–right asymmetry: motile cilia that generate a leftward fluid flow, and immotile mechanosensory cilia that respond to the flow. Here, we describe 3 mouse SI models that provide support for the 2-cilia hypothesis. In addition to having SI, Dpcd/Poll–/– mice (for: deleted in a mouse model of primary ciliary dyskinesia) and Nme7–/– mice (for: nonmetastatic cells 7) had lesions consistent with deficient ciliary motility: Hydrocephalus, sinusitis, and male infertility developed in Dpcd/Poll–/– mice, whereas hydrocephalus and excessive nasal exudates were seen in Nme7–/– mice. In contrast, the absence of respiratory tract lesions, hydrocephalus, and male infertility in Pkd1l1–/– mice (for: polycystic kidney disease 1 like 1) suggested that dysfunction of motile cilia was not involved in the development of SI in this line. Moreover, the gene Pkd1l1 has considerable sequence similarity with Pkd1 (for: polycystic kidney disease 1), which encodes a protein (polycystin-1) that is essential for the mechanosensory function of immotile primary cilia in the kidney. The markedly reduced viability of Pkd1l1–/– mice is somewhat surprising given the absence of any detected abnormalities (other than SI) in surviving Pkd1l1–/– mice subjected to a comprehensive battery of phenotype-screening exams. However, the heart and great vessels of Pkd1l1–/– mice were not examined, and it is possible that the decreased viability of Pkd1l1–/– mice is due to undiagnosed cardiovascular defects associated with heterotaxy.
Although vertebrates may appear symmetrical externally, there is marked left–right (LR) asymmetry in the positioning of almost all their internal organs. 11,39 The term situs solitus is used to describe the usual asymmetric positioning of the cardiac atria and the viscera; however, in approximately 1 in 10,000 people, there is a mirror-image reversal of organ positioning, called situs inversus (SI). 45,61 The terms situs ambiguus and heterotaxy are used to describe other variations in organ placement owing to abnormal LR patterning during embryonic development. Based on the position of the cardiac atria, SI has been further classified into SI with dextrocardia (SI totalis) and SI with levocardia (a form of heterotaxy), the latter of which is frequently associated with congenital heart defects. These distinctions are important because although SI totalis is generally asymptomatic, complex congenital cardiac defects and transposition of the great vessels of the heart are often associated with situs ambiguus or heterotaxy. 8,9,32
The association among SI, bronchiectasis, and sinusitis in humans was first reported in 1933, 30 but more than 40 years passed before Afzelius linked this classic triad of lesions of Kartagener syndrome with the defective structure and function of cilia. 2 In addition to the classic Kartagener triad, patients with ciliary defects may develop nasal polyps, impaired olfaction, recurrent otitis media, hearing loss, reduced fertility (females), and/or infertility (males). Abnormalities in the structure and/or function of both motile and sensory cilia have been linked to a range of diseases now classified as ciliopathies. 40 Dysfunctional or absent motile cilia and the related spermatic flagellum lead to chronic sinopulmonary disease, hydrocephalus, male infertility, congenital heart disease, and laterality defects, whereas defects in sensory (immotile) cilia can cause polycystic kidney disease, nephronophthisis, polydactyly, retinal degeneration, obesity, sensorineural deafness, and anosmia. 17,41 The complexity of ciliary structures and functions is reflected by the myriad clinical presentations associated with ciliary abnormalities. 62
Cilia are complex organelles that project from the cell surface and contain a microtubule cytoskeleton (axonemes) surrounded by a ciliary membrane. The ciliary membranes contain receptors and ion channels that regulate motility or respond to external mechanical or chemical stimuli, which in turn affect cell differentiation, migration, and growth. 56 Depending on their function, cilia can be divided into motile and primary (immotile) cilia. The more familiar motile cilia, found on multiciliated cells of mammalian epithelia, have a 9 + 2 microtubule arrangement of their axonemes, with 2 central microtubules surrounded by 9 doublet microtubules. These are found along surfaces of the respiratory tract, oviduct, epididymis, and brain ventricles (ependyma); their main function is to move extracellular fluids along surfaces. The term primary ciliary dyskinesis (PCD; previously known as immotile cilia syndrome) is used to describe diseases associated with dysfunctional motile cilia or flagella, whether accompanied by SI or not. In mice, PCD causes reduced mucociliary clearance leading to chronic respiratory tract infections, impaired flow of cerebrospinal fluid in the brain (leading to hydrocephalus), infertility syndromes, and randomization of body situs. 24,54 In contrast, individual immotile or primary cilia have a 9 + 0 axoneme, which lacks the central 2 microtubules. Immotile primary cilia are present on almost every cell type in the body and, until the 1990s, were thought to be vestigial organelles. They are now known to have important physiological roles in mechanosensation and chemical sensation, signal transduction, and control of cell growth in many tissues (see review 60 ).
Different mutations affecting ciliary biogenesis, motility, or sensory functions have been linked to abnormal situs development in mice, demonstrating that ciliary function at the embryonic node (a transient midline structure formed during gastrulation) is essential for normal LR determination. 23 In the mouse embryo, each cell on the ventral embryonic node has a single projecting apical cilium. 28 These nodal monocilia are located at the center of the ventral node of embryos. 23,51 Like all other primary cilia, the nodal monocilia have 9 + 0 microtubule arrangement and lack the central pair of microtubules present in conventional motile cilia; 18,50 however, unlike all other primary cilia, these nodal monocilia are motile. Unlike other motile cilia, which are oriented perpendicular to the cell surface and move back and forth, nodal monocilia are tilted toward the posterior of the embryo 51 and have a unique rotational movement that generates a directional extracellular flow (nodal flow). 10,53 During rotation, the tips of the monocilia actually brush along the nodal surface during the rightward stroke and thus form a D-shaped, rather than circular, trajectory. 53 This rotational movement produces an overall leftward nodal flow because monocilia are close to the nodal surface during rightward movements, where higher shear forces impede fluid flow, whereas during leftward movements, the monocilia are at the top of their rotational movement and farther from the cell surface, where shear resistance to fluid flow is low. 12,13 Absence or dysfunction of embryonic nodal monocilia leads to randomization of LR body asymmetry. 50 Numerous studies have shown this nodal flow to be the primary event leading to the LR patterning. 18,42,50,52,68,70,71 Nodal flow triggers a rise in intracellular calcium in cells at the left side of the node, which subsequently leads to asymmetric gene expression and morphogenesis. 5 Interestingly, this leftward nodal flow is only required during the 1- to 6-somite stage, which spans only 6 to 7 hours of development, to establish LR asymmetry in mouse embryos. 65
Mouse models have proven useful in elucidating many of the fundamental mechanisms involved in the structure and function of motile and immotile cilia, and they have helped in identifying more than 20 genes involved in the specification of LR asymmetry. 37 The structure and function of motile cilia are often defective in SI (reviewed in Lee and Anderson 37 ). For example, mice with inactivating mutations in the kinesin Kif3A and Kif3B lack nodal monocilia 50,71 and show randomized LR situs. 50 Other mutant mice showing SI have nodal cilia that are not motile. 43 Mutant mice lacking a dynein chain protein (Mdnah5) have immotile cilia and develop chronic respiratory infections, hydrocephalus, and SI. 24 Other relevant knockout mouse models include those lacking the helix factor hepatocyte nuclear factor/forkhead homolog (Hfh-4), 14 the dynein heavy chain protein (Mdhc7), 49 the axonemal filament protein Tektin-t, 72 the sperm-associated antigen 6 (Spag6), 83 Pcdp1 (nm1054), 38 and adenylate kinase 7 (Ak7). 19 Although most human cases appear sporadically, there is clear evidence of autosomal recessive inheritance of laterality defects in humans 4,15,85 Genetic models in mice are valuable because human homologs exist for many of the LR-determining genes identified in mice, and mutations in the homologous genes are linked to similar phenotypes in humans. 63
In generating and analyzing more than 4,600 knockout mouse lines in a high-throughput mutagenesis and phenotyping process designed to characterize protein functions and identify novel drug targets, 80,81 we have discovered many pathological phenotypes in mice that may prove useful in elucidating fundamental biological processes and the pathogenesis of several genetic diseases. Here, were report the development of SI in approximately 50% of mice having functional deletions of the genes Dpcd/Poll, Pkd1l1, and Nme7. Although the occurrence of PCD and SI in Dpcd/Poll–/– was recently repoted, 34,82 the association between the genes Pkd1l1and Nme7 and the development of SI in mice has not been reported. Together, these 3 mouse SI models provide support for the 2-cilia hypothesis, which proposes the involvement of two kinds of primary cilia in the determination of LR asymmetry: motile cilia that generate a leftward fluid flow and immotile mechanosensory cilia that respond to the flow.
Materials and Methods
We generated targeted and gene trap mutations in strain 129S5SvEvBrd-derived embryonic stem (ES) cells, as described. 1,80 The Dpcd/Poll and Nme7 knockout lines were generated by gene trapping using ES cell clones from the OmniBank library. 1,80 Clones OST280355 and OST31116 were selected for microinjection based on sequence similarity to the Poll and Nme7 genes, respectively. The genomic insertion sites for both mutations were determined by inverse genomic polymerase chain reaction (PCR; Fig. 1A). 21 Gene disruption was confirmed by a direct analysis of gene expression using reverse transcription PCR (RT-PCR). RNA was extracted from spleen and kidney of wild-type (+/+) and homozygous (–/–) mutant mice, with a bead homogenizer and RNAzol (Ambion, Austin, TX), according to manufacturer’s instructions. Reverse transcription was performed with SuperScript II (Invitrogen, Carlsbad, CA) and random hexamer primers, according to the manufacturer’s instructions. PCR amplification was performed with oligonucleotide primers for Poll (5′-CTACCCCTTCCTCCCCCAATCCGTTTTCA-3′ and 5′-TCTAGCTTCTCCCACCTCCCTTTTG-3′), Dpcd (5′-AGGAAATGGCAGAAGAATATGACGA-3′ and 5′-GGGCCACAGACACACTATACACA-3′), and Nme7 (5′-CCTGTGCTGAGATCTACAAACTATG-3′ and 5′-TGGAAAGTATGAAATGAACTAGACGCTGAG-3′). In the case of OST280355, the insertion occurred within the first intron of the Poll gene near the transcriptional start site for Dpcd. RT-PCR analysis of both Dpcd and Poll genes showed that transcription of both genes was disrupted by the insertion event (Fig. 1A).
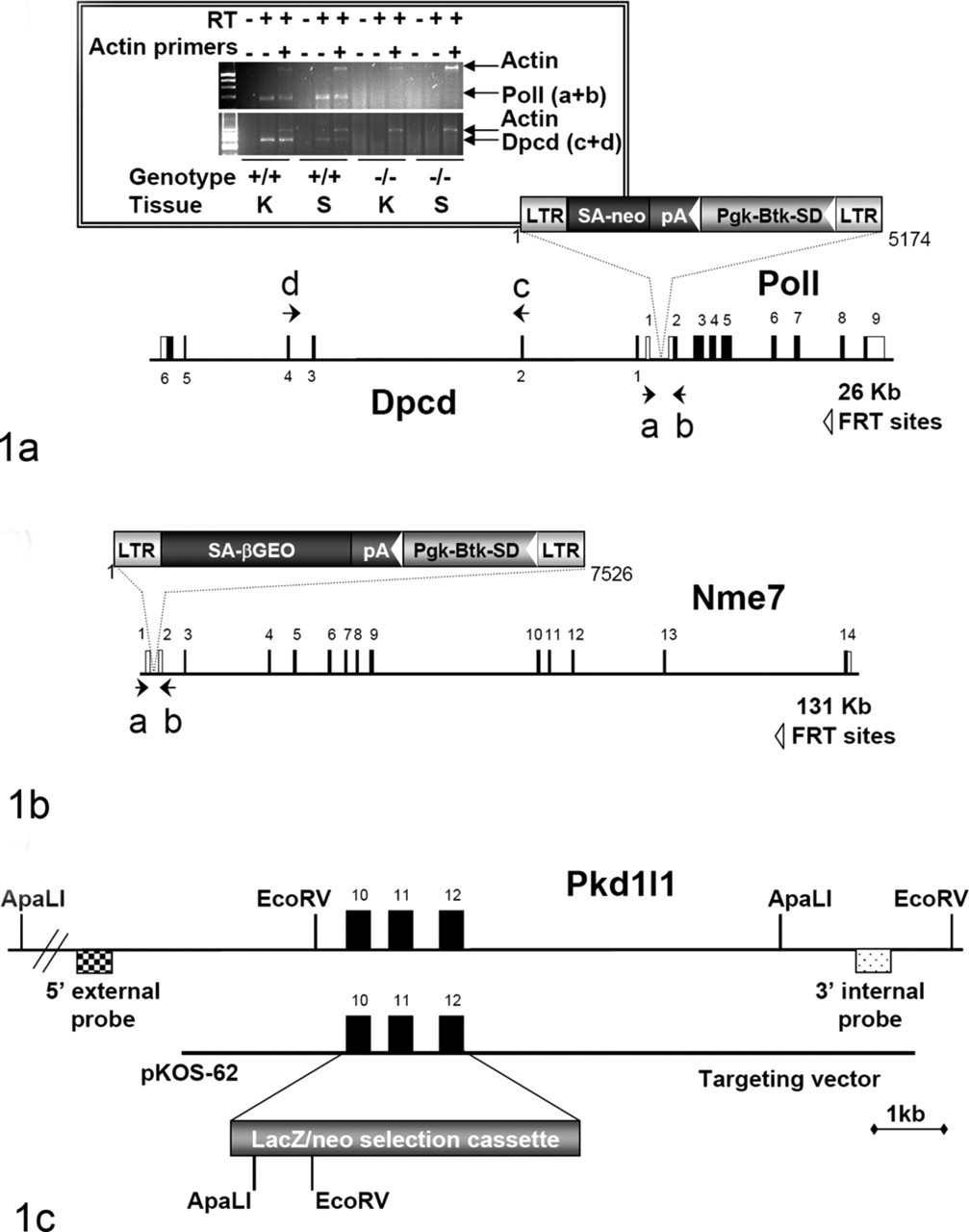
The Pkd1l1 knockout line was generated by homologous recombination, where exons 10 through 12 of the Pkd1l1 gene were deleted and replaced with a selection cassette containing both the LacZ reporter gene and the neomycin gene (Fig. 1C). A lambda KOS phage library 74 was screened by duplex PCR using oligonucleotide primers (5′-GTGCATAGGCACACACATGTGCATG-3′ and 5′-CTTATACCCAACAGTCACACCACAC-3′; 5′-GTGCAGGCTCAACTTTGGCACTG-3′ and 5′-CAGCCTGCCTTCTTTCCAGTCAAC-3′) to identify a single genomic clone (pKOS-62) containing exons 10, 11, and 12 of the Pkd1l1 gene. Gene-specific arms (5′- TCTTCTCTTGTTTCAGGCACAGTCGTGGTGCATTC-3′ and 5′- ACTGTGGGGTTTTAAGTATCGATCCTTCAGAAGGC-3′) were then appended by PCR to a yeast selection cassette containing the URA3 marker, which was then cotransformed into yeast with pKOS-62. Clones were isolated that had undergone homologous recombination to replace exons 10 to 12 with the yeast selectable marker. The yeast cassette was subsequently replaced with an ES cell selection cassette containing the reporter gene LacZ. The targeting vector was electroporated into 129S5/SvEvBrd ES cells, and G418/FIAU-resistant ES cell clones were isolated. Correctly targeted clones were identified and confirmed by Southern analysis (data not shown).
Targeted or gene-trapped ES cell clones were microinjected into C57BL/6-Tyr c-Brd (albino) blastocysts to generate chimeric animals, which were bred to C57BL/6-Tyr c-Brd (albino) females, and the resulting heterozygous offspring were interbred to produce homozygous gene-deficient mice. Knockout F2 mice used in phenotyping studies were produced by intercrossing the F1 heterozygous knockout (+/–) offspring of chimeric founder parents; wild-type, heterozygous, and homozygous F2 littermates were produced in an approximately a 1:2:1 mendelian ratio. Using the albino variant of C57BL/6 permits simple visual recognition of chimeric offspring 84 because they have dark eyes and patches of dark hair that derive from stem cells from the agouti 129S5/SvEvBrd. Animal genotypes were determined by quantitative PCR as previously described. 21 In brief, DNA isolated from tail biopsy samples was assayed by quantitative PCR for the neo gene, which is present in both the gene-targeting and gene-trapping vectors used to generate the gene knockout mutations described in this study.
Mouse Husbandry
Mice were housed in a barrier facility at 24°C on a fixed 12-hour-light/12-hour-dark cycle and were fed rodent chow No. 5001 (Purina, St. Louis, MO) ad libitum. Procedures involving animals were conducted in conformity with the Institutional Animal Care and Use Committee guidelines that are in compliance with the state and federal laws and the standards outlined in the Guide for the Care and Use of Laboratory Animals. 47 Quarterly sentinel surveillance of the source colonies was conducted (Charles River) and showed no evidence of rodent viruses, mycoplasma, or Helicobacter sp.
Phenotypic Screen
Wild-type and homozygous null mice were subjected to a comprehensive battery of phenotype screening exams, as previously described. 6,80 Screening assays included the following: behavioral tests (such as circadian rhythm, open field, inverted screen, prepulse inhibition of the acoustic startle response, tail suspension, marble burying, and context trace conditioning), fundoscopy and retinal angiography exams, blood pressure and heart rate measurements, serum chemistries, insulin levels, glucose tolerance testing, hematology, peripheral blood FACS (fluoresence-activated cell sorting) analysis, urinalysis, quantitative magnetic resonance, dual-energy x-ray absorptiometry scans, computerized axial tomography (CAT) scans, microcomputed tomography scans, fertility testing, skin fibroblast proliferation assays, and pathology.
Histopathology
Immediately after euthanasia, knockout mice and age-matched normal control mice were fixed by cardiac perfusion with 10% neutral-buffered formalin. Standard tissues examined were as follows: heart, skeletal muscle, aorta, lung, kidney, renal lymph node, trachea, thyroid gland, parathyroid gland, mediastinal lymph node, adrenal gland, pituitary gland, thymus, salivary glands, cervical lymph node, esophagus, stomach, pancreas, duodenum, jejunum, ileum, cecum, colon, rectum, mesenteric lymph node, liver, gallbladder, spleen, brain, spinal cord, eyes, Harderian gland, urinary bladder, uterus, ovaries, fallopian tube, skin, mammary gland, bone, bone marrow, adipose tissue, blood, teeth, prostate gland, testes, epididymis, seminal vesicle, vas deferens, urethral glands, inguinal lymph node, inner ear, middle ear, and nasal turbinates. Tissues were collected and immersed 10% neutral-buffered formalin for an additional 48 hours, except for the eyes, which were removed and fixed by immersion in Davidson’s fixative (Poly Scientific, New York, NY) overnight at room temperature. All tissues were embedded in paraffin, sectioned at 4 μm, and mounted on positively charged glass slides (Superfrost Plus, Fisher Scientific, Pittsburgh, PA) and stained with hematoxylin and eosin (HE) for histopathologic examination.
Results
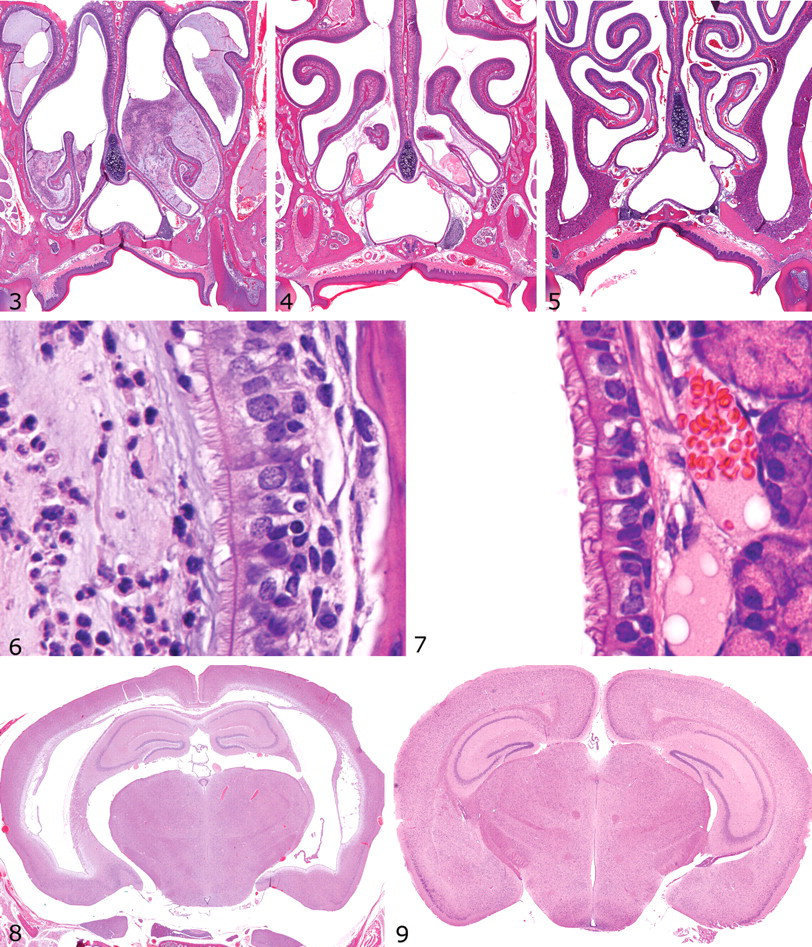
SI, as characterized by transposition of abdominal and thoracic organs, was noted at necropsy or in CAT scans (Fig. 2) in some knockout mice in all 3 lines described in this report. However, evidence for motile ciliary dysfunction differed significantly among the 3 knockout mouse lines, as shown by the diffuse severe nasal exudates in Dpcd/Poll–/– mice (Fig. 3) versus the mild multifocal exudates in Nme7–/– mice (Fig. 4) and the completely clear nasal passageways in Pkd1l1–/– mice (Fig. 5), although cilia were present in all animals (Figs. 6, 7). Surprisingly, otitis media was not present, even in mice with severe suppurative rhinitis or sinusitis. Similar nasal lesions are extremely uncommon background lesions in our C57BL/6-Tyr(c-Brd) colony, and SI has not been detected except in these 3 knockout mouse lines.
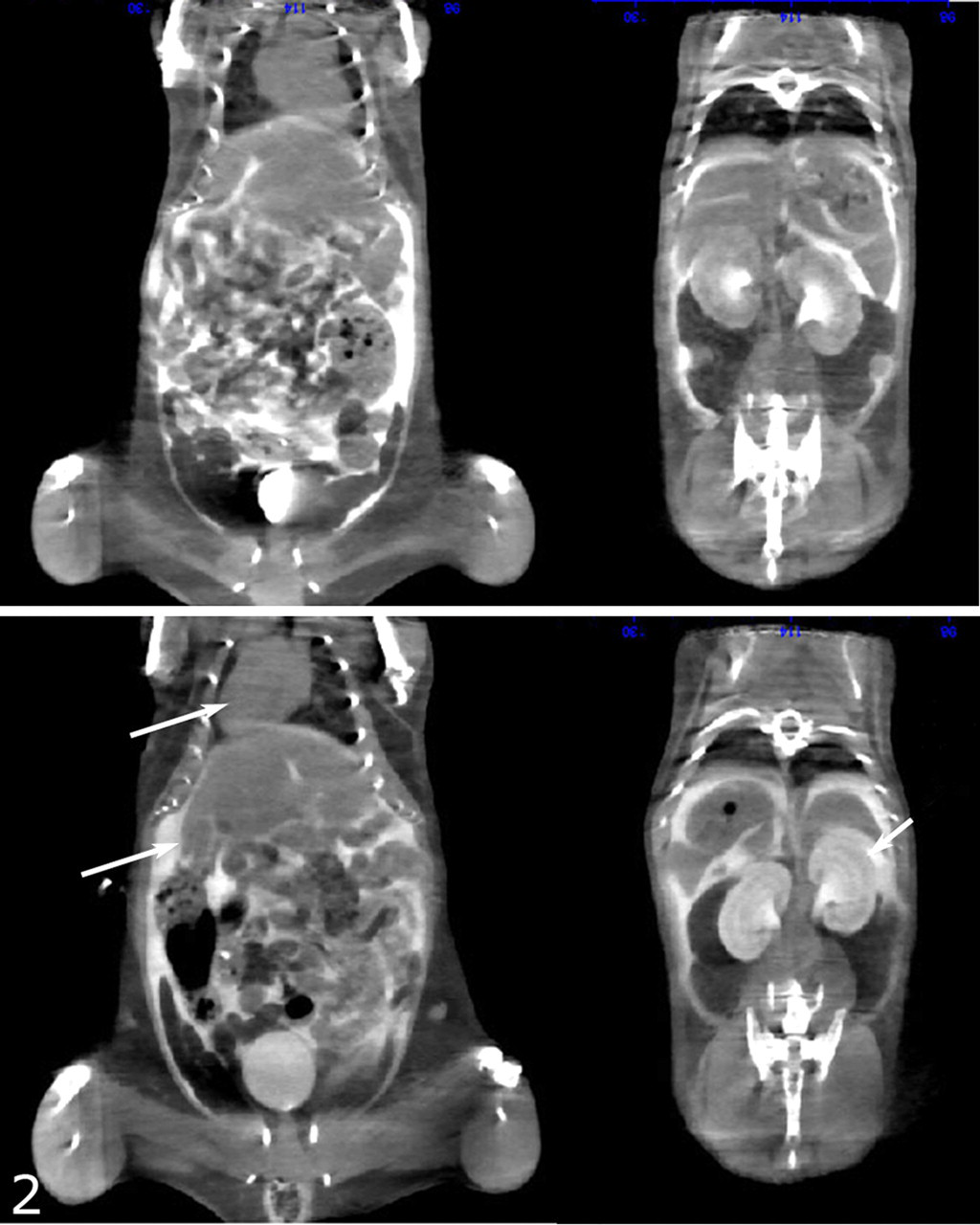
Nme7–/– mice: Top panel shows situs solitus present in approximately 50% of knockout mice. Lower panel shows transposition of the heart (top arrow, left panel) and kidneys (arrow, right panel) in a mouse with situs inversus. The reversed position of the large left lateral lobe of the liver is also evident (lower arrow, left panel). CAT scan.
Dpcd/Poll–/– Mice
Retrospective examination of the gene trap insertion site and confirmatory RT-PCR testing demonstrated that this retroviral insertion inactivated both Dpcd and DNA polymerase λ (Poll), resulting in a double knockout with a phenotype very similar to another reported previously that targeted Poll but inadvertently inactivated Dpcd. 34 However, Dpcd was later confirmed to be the gene responsible for the ciliopathies originally attributed to Poll because specific disruption of Poll without disruption of Dpcd was proven to result in no identifiable phenotype. 82 We observed that Dpcd/Poll–/– mice were smaller than wild-type littermates and were produced in slightly lower numbers than those predicted by mendelian genetics (Table 1). These data suggest marginally reduced viability of Dpcd/Poll–/– mice. SI was present in approximately 50% of Dpcd/Poll–/– mice, but several histological lesions consistent with underlying PCD were present in 100% of knockout mice. Nasal passsages and sinuses were often filled with abundant mucopurulent exudate (Fig. 3), although histologically normal respiratory cilia were present (Fig. 6). There was moderate to marked dilation of the lateral and third ventricles of the brain, but cilia on ependymal cells were histologically unremarkable (data not shown). Severely affected mice had domed skulls and were relatively inactive, but many homozygotes had only slightly domed skulls and demonstrated normal behavior in neurologic testing (data not shown). At necropsy, the cerebral cortex often partially collapsed when the calvarium was removed, and the lateral ventricles were moderately dilated with clear cerebrospinal fluid. Males were infertile, as demonstrated by the failure of 2 mature Dpcd/Poll–/– males to produce pups after each being housed with 2 fertile wild-type females and monitored for 60 days for pregnancy. Similar to previous reports, histological examinination showed apparently normal development and maturation of spermatozoa in seminiferous tubules and abundant mature spermatozoa in the epididymides; however, on wet mounts, spermatozoa were immotile and many mature spermatozoa appeared to have short tails and rounded heads (data not shown). In contrast, apparently normal fertility in female Dpcd/Poll–/– mice was demonstrated by productive breeding with wild-type mates, and both male and female Dpcd/Poll+/– mice that were used for mouse production were fertile. SI and other notable lesions were not detected in Dpcd/Poll+/– and Dpcd/Poll+/– littermates.
Genotyping results of nursing infant mice born as a result of mating heterozygous parents.
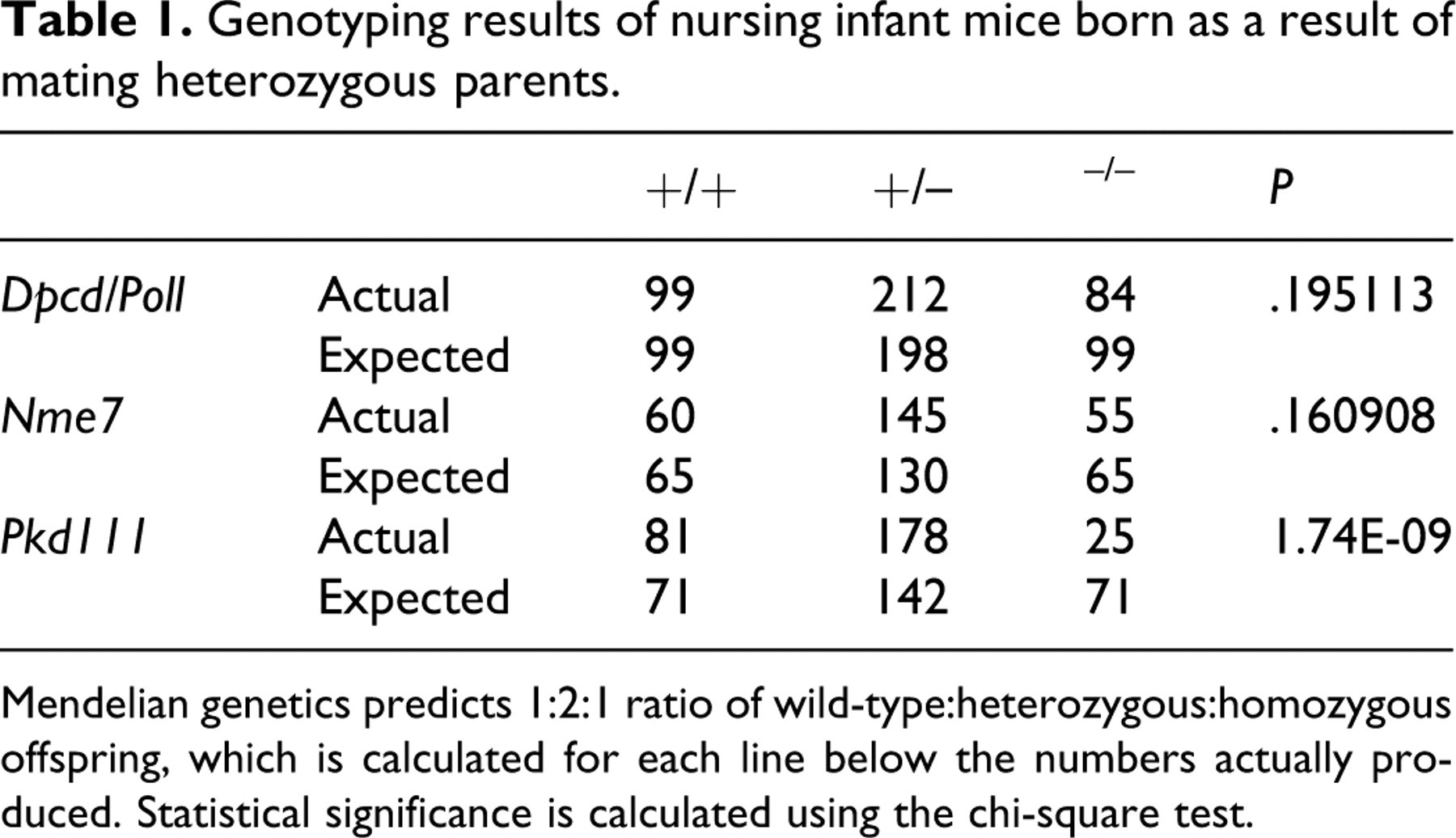
Mendelian genetics predicts 1:2:1 ratio of wild-type:heterozygous:homozygous offspring, which is calculated for each line below the numbers actually produced. Statistical significance is calculated using the chi-square test.
Nme7–/– Mice
The knockout mice did not differ in gross appearance from wild-type littermates. Although present in slightly lower numbers than those predicted by mendelian genetics, the difference was not statistically significant, suggesting that this gene is not required for embryonic development or survival to weaning. We detected SI in approximately 50% of homozygotes (Fig. 2), but mild to moderate hydrocephalus was seen in all Nme7–/– mice and was the major phenotype present that was suggestive of underlying PCD. Although suppurative sinusitis and rhinitis were not present in Nme7–/– mice, minimal to mild mucus exudates were present in a few animals (Fig. 4), suggesting a mild defect in ciliary motility. Externally, doming of skulls was undetectable or very mild in many mice, but there was moderate to marked dilation of the lateral and third ventricles of the brain (Fig. 8) in all homozygote mice. Here again, cilia on ependymal cells appeared to be histologically normal (data not shown). Despite the presence of ventricular dilatation in the brain, Nme7–/– mice demonstrated normal behavior in neurologic testing (data not shown). Similar to the Dpcd/Poll–/– mice, histologically normal cilia were present on ependymal cells and respiratory epithelia. In contrast to Dpcd/Poll–/– mice, both male and female Nme7–/– mice were fertile when bred to wild-type mates. The normal male fertility and absence of lesions in nasal passageways suggest that flagellar and ciliary functions are adequate in these organ systems. In contrast, the presence of SI and hydrocephalus suggest that both nodal flow and ependymal flow are compromised as a result of motile ciliary dyskinesis. SI and other notable lesions were not detected in Nme7+/– and Nme7+/– littermates.
Pkd1l1–/– Mice
We detected SI in approximately one third of the homozygous mutant mice available for analysis. Remarkably, no other notable phenotype was observed for the homozygous mutant mice, and no lesions suggestive of underlying PCD were present. Nasal passages and sinuses were clear in all mice (Fig. 5), and respiratory cell cilia were histologically normal (Fig. 7). Brain ventricles were normal (Fig. 9), and both male and female Pkd1l1 –/– mice demonstrated normal fertility. SI was the only significant finding in surviving Pkd1l1–/– mice that underwent a comprehensive battery of tests. However, genotyping results indicated that deletion of this gene results in significantly reduced viability during development or the early postnatal period (Table 1). The cause of decreased viability in Pkd1l1–/– mice was not determined in this study. SI, decreased viability, and any other notable lesions were not detected in Pkd1l1+/– and Pkd1l1+/– littermates.
Discussion
SI is the one phenotypic abnormality that is common to all 3 knockout mouse lines described in this report. The different lesions observed in each knockout line are reflective of the underlying structural complexity and functional diversity of cilia and so provide supporting evidence for the concept that multiple molecular mechanisms are involved in the development of LR asymmetry. The 2 alternative models proposed to explain how leftward nodal flow influences development and produces LR asymmetry in the embryo include the morphogen transport hypothesis and the 2-cilia hypothesis (see review 23 ).
The morphogen transport model proposes that LR asymmetry develops in response to chemical gradients of secreted morphogens in the node. 50 In this model, nodal flow would produce a chemical gradient of secreted morphogens, with the highest concentrations of morphogens accumulating on the left side of the ventral node. 50,53,78 Several studies support a role for morphogens in determining laterality, 46,53,73 and it was shown that fibroblast growth factor stimulates the release of membrane-sheathed particles called nodular vesicular parcels, which contain signaling molecules and possible morphogens, such as Sonic Hedgehog and retinoic acid. The nodular vesicular parcels are carried by nodal flow to the left edge, where they may release morphogens that activate the noncanonical Hedgehog signaling pathway, increase intracellular Ca++, and thus asymmetrically alter gene expression. 23,73
In the 2-cilia model, the node contains 2 types of primary cilia (motile and immotile), in which motile cilia rotate autonomously to generate the leftward nodal flow while mechanosensory immotile cilia detect and respond to the flow. 44,70 In the 2-cilia model, normal structure and function of both types of cilia are essential for the normal development of LR asymmetry. 29,57,59,64,65,78 The presence of both motile and immotile cilia at the node was demonstrated by the restricted distribution of the required motor protein LR dynein (lrd) to centrally located monocilia, whereas the peripheral nodal cilia lacked the lrd motor protein but did contain the cation channel polycystin-2 (PC-2). 44 Previous studies in knockout mice showed that both the lrd-containing motile cilia and the PC-2-containing immotile cilia are required for the establishment of LR asymmetry in mice. 44,57 A mechanosensory role for PC-2 in nodal primary cilia was suggested by the known function of PC-2 in the kidney, where it is present in the mechanosensory primary cilia of renal tubular epithelium and activates the calcium ion channel that initiates intracellular calcium signaling in response to urine flow. 58 Taken together, these findings suggested that LR asymmetry could be established solely by cilia, in which motile lrd-containing monocilia would generate a nodal flow and nonmotile PC-2 containing cilia would sense the nodal flow and initiate an asymmetric calcium signal at the left border of the node. 44 The increased Ca++ ion influx into cells on the left side of the node is linked to the asymmetric expression of Nodal and probably other genes required for the establishment of laterality. 44,73
The knockout mice described in this report illustrate the involvement of both motile and immotile cilia in the development of LR asymmetry. The gene Dpcd (for: deleted in a mouse model of primary ciliary dyskinesia) is located on mouse chromosome 19 and codes for a 23-kD protein of unknown function. 82 Originally, the development of the characteristic lesions of PCD in these mice were attributed to targeted inactivation of Poll, 34 but closer analysis of mouse chromosome 19 sequence in the region of the Poll revealed that a previously uncharacterized gene (5330431N19Rik, now named Dpcd) was transcribed from the opposite strand. 82 Dpcd was subsequently implicated as the gene involved in PCD when targeted deletion of Poll alone 7 did not result in the PCD phenotype caused by inactivation of both genes. 82 A careful examination of the targeting construct and confirmatory PCR testing demonstrated that both mouse genes were indeed knocked out by our retroviral insertion. 38,82 Our findings in our Dpcd/Poll–/– mice—which included hydrocephalus, SI, chronic sinusitis, and male infertility—exhibit the classic triad of Kartagener syndrome in mice, lesions that are clearly secondary to the absence of ciliary motility.
All Dpcd/Poll–/– mice, with or without SI, developed hydrocephalus, sinusitis/rhinitis, and male infertility, which are hallmark changes associated with PCD. Koyabashi et al reported that that the inner dynein arms of cilia from both the ependymal cell layer and the respiratory epithelium were defective in Poll-Dpcd–/– mice. 34 PCD generally results from defects in the parts of the cilia responsible for movement, such as the microtubules, dynein arms, or radial spokes. Regardless of the underlying cause, defects in structure and/or function of motile cilia can disrupt mucociliary clearance by respiratory epithelium, leading to recurrent respiratory tract infections in humans that can progress to chronic sinusitis or rhinitis and even permanent lung damage (bronchiectasis). Interestingly, mice appear to be resistant to PCD-associated pulmonary disease, and their respiratory tract lesions are restricted to the nasal passageways and sinuses. However, mice with PCD frequently develop hydrocephalus, with the dilatation of brain ventricles being attributed to disruption of the flow of cerebrospinal fluid normally generated by ciliated ependymal cells. 25 As expected with severe PCD, Dpcd/Poll–/– males were infertile, apparently owing to loss of flagellar motility in spermatozoa. However, female mice were fertile, suggesting that ciliary function in oviducts is less critical to female fertility. Similarly, reduced fertility in men with PCD is attributed to dysmotility of spermatozoa and/or reduced sperm count, whereas lack of ciliary motility in the fallopian tubes of women does not appear to have a major effect on fertility. 3
The targeted gene in our second knockout mouse is Nme7 (for: nonmetastatic cells 7). Nme7 is located on mouse chromosome 1 and codes for a nucleoside-diphosphate kinase (NDP kinase). Aliases for this gene include NDK 7, nm23-M7, NDP kinase 7, and nucleoside diphosphate kinase 7. Very little is known about the functions of this specific protein, but NDP kinases as a group are responsible for the synthesis of nucleoside triphosphates and are involved in numerous regulatory processes associated with proliferation, development, and differentiation. 36 NDP kinase 7 has not been linked to ciliary function in mammals, but this kinase has significant homology (50.7) with a protein (FAP67) that was identified in purified flagella from the green alga Chlamydomonas reinhardtii. 55 This homology is significant because the proteins involved in the function and biogenesis of cilia and flagella have been highly conserved throughout evolution. The flagellar proteome of C. reinhardtii includes many proteins with homologs that are associated with diseases such as cystic kidney disease, male sterility, and hydrocephalus in humans and model vertebrates. 55 Also, FAP67 is upregulated during flagellar regeneration 66 and was identified in a basal body proteome as BUG5. 31 Based on the development of SI and hydrocephalus in Nme7–/– mice, the protein is very likely involved in the biogenesis or function of motile cilia; however, given the absence of rhinitis/sinusitis and the normal male fertility observed in Nme7–/– mice, it appears that that the function and/or biogenesis of motile cilia/flagella in the respiratory epithelium and spermatozoa are less dependent on functional NDP kinase 7 than ependymal or embryonic node cilia are.
In contrast, the complete absence of respiratory tract lesions, hydrocephalus, and male infertility in Pkd1l1–/– mice suggests that this gene product does not primarily affect the function of motile cilia. It is possible that Pkd1l1 could be temporally limited (suggested by Unigene EST expression occuring primarily during the gastrula phase) or spatially limited in expression during development (as discussed below with iv/iv mice), but the gene name Pkd1l1 (for: polycystic kidney disease 1 like 1) indicates a sequence similarity to the gene Pkd (for: polycystic kidney disease), which provides an essential component of sensory cilia. In fact, polycystin-1L1 has a long stretch of homology with polycystin-1 (PC-1), 79 and PC-1 is clearly involved in mechanosensory functions of immotile primary cilia in the kidney. 16,59 The polycystins are mechanical stress sensor polycystins in renal tubular epithelial cells 76 that are included among many critical receptors and channels located within ciliary membranes. 56 In humans, mutations in PKD1 and PKD2 (which encode PC-1 and PC-2) result in autosomal dominant polycystic kidney disease but no other signs of ciliary dysfunction, suggesting that PKD1 and PKD2 are not required for the biogenesis or function of motile cilia. PC-1 and PC-2 are localized to primary cilia in the kidney, 76,77 where they form a multifactorial complex that functions as a flow-sensitive mechanosensor. 16,48 The PC-2 cation channel functions in a range of cells where it has been localized to a variety specific subcellular regions, including the endoplasmic reticulum and primary cilium. 33 PC-1 appears to be responsible for the proper subcellular localization of PC-2 to mechanosensory cilia in the kidney tubular epithelium; other partner proteins affecting the subcellular distribution of the PC-2 channel in cells include GSK3, 67 PIGEA-14, 22 and phosphofurin acidic cluster sorting protein. 35 These differences in subcellular distribution appear to allow PC-2 to perform multiple functions in the cell.
In renal tubular epithelium, both PC-1 and PC-2 are necessary for ciliary mechanotransduction, where bending of the cilium activates the PC-2 calcium channel. 75 However, PC-1 apparently does not have the same role in mechanosensory cilia at the embryonic node. Although PC-2 is localized to nodal cilia in mouse embryos, PC-1 is not. This finding is supported by studies showing that PC-2 is required to establish LR asymmetry in mice but that mice lacking PC-1 do not display laterality defects. 57 Taken together, these results indicate that, unlike in the kidney, PC-1 does not have a role in the embryonic node or in establishing LR asymmetry. 29 Instead, our findings suggest that polycystin 1 like 1 (Pkd1l1), clearly required in the mouse for establishment of LR asymmetry, may take the place of PC-1 in the embryonic node by combining with PC-2 in forming functional nodal cilia mechanoreceptors.
The markedly reduced viability of Pkd1l1–/– mice is somewhat surprising given the absence of any detected abnormalities (other than SI) in knockout mice subjected to a comprehensive battery of phenotype screening exams. 6,80 Although the heart and great vessels of Pkd1l1–/– mice were not examined in detail in this study, it seems plausible that the decreased viability of Pkd1l1–/– mice could be due to cardiovascular abnormalities. Dot blot analysis and RT-PCR show that human PKD1L1 is expressed primarily in testis and in fetal and adult heart. 79 The normal development of the heart is dependent on the rapid migration, expansion, and differentiation of cells in response to a variety of intra- and extracellular signaling pathways. Many congenital heart abnormalities might arise from defects in these early stages of heart development, and it is possible that Pkd1l1 has an important role in the patterning processes that shape this organ.
There is striking LR asymmetry of the heart and great vessels, and complex congenital heart defects composing the heterotaxy syndrome result when normal cardiac asymmetries fail to develop. It is well established that mutant mice with ciliopathies have a high incidence of heterotaxy in addition to pure SI. For example, mutant iv/iv mice have mutations in lrd and are similar to our Pkd1l1 –/– mice in showing SI along with normal fertility and the absence of sinusitis/rhinitis and hydrocephalus. The SI-restricted phenotype of iv/iv mice may be partially explained by lrd expression being present in the embryonic node 69 but not in respiratory epithelium. 20 Nevertheless, the iv/iv mouse also has a high frequency of complex heart defects owing to abnormal formation of the cardiac loop. Between 35% to 50% of iv/iv mouse embryos show heterotaxy associated with atrioventricular canal defects, abnormal pulmonary and systemic venous return, and abnormalities of the great vessels. 27 Abnormal origin of the coronary arteries was found in 84% of iv/iv mice. 26 In contrast, mice with mutations in the dynein heavy chain DNAH5 develop more classic features of PCD, including chronic respiratory infections and randomization of cardiac and visceral situs. 24 In addition, some Dnah5–/– mice show heterotaxy and cardiac defects. Similarly, over 6% of human PCD patients show heterotaxy with heart defects. 32 The possible causative role of heterotaxy and cardiovascular defects for the markedly reduced embryonic or neonatal viability of Pkd1l1–/– mice will need to be examined in future studies.
In this report, we describe 3 knockout mouse lines that demonstrate roles for both motile and immotile cilia in the development of SI, thus providing support for the validity of the 2-cilia model of determining LR asymmetry. These mouse models may prove useful for investigations into basic ciliary function and the pathogenesis of situs abnormalities (including heterotaxy), hydrocephalus, and PCD.
Footnotes
Acknowledgements
We thank Jeff Schrick, June Wingert, Ryan Vance, Billy Hampton, Michelle Matzig, Mary Thiel, and Kathy Henze for their invaluable support in completing these studies. We would also like to acknowledge the assistance of many technicians and scientists and the administrative support of Lexicon Pharmaceuticals, Inc., without which this work would not be possible.
The authors declared that they had no conflicts of interests with respect to their authorship or the publication of this article.
Financial support was provided solely by Lexicon Pharmaceuticals Inc.