
Abstract
Disseminated superficial porokeratosis with dermal amyloid deposits is exceptionally rare, with only 12 documented cases worldwide. The pathogenesis involves keratinocyte degeneration, resulting in amyloid deposition. A 76-year-old Chinese male presented with a 10-year history of multiple annular brown pruritic macules on the extremities and trunk, along with dense pruritic papules on the bilateral anterior tibial regions. Histopathological examination revealed cornoid lamella, dyskeratotic cells, and dermal amyloid deposits. Treatment with topical corticosteroids, 3% boric acid, and oral acitretin improved pruritus and pigmentation over 4 months; however, lesions recurred after discontinuation. Concomitant hyperuricemia (523 μmol/L) suggests a potential novel role of purine metabolism in amyloidogenesis, an association that has not been previously reported.
Keywords
Introduction
Porokeratosis (PK) refers to a group of keratinization disorders, with most forms exhibiting an autosomal dominant mode of inheritance resulting from abnormal clonal proliferation of epidermal keratinocytes. Clinical variants include not only PK of Mibelli but also disseminated superficial, disseminated actinic superficial, linear, punctate, and palmaris et plantaris disseminata. Various types can coexist simultaneously, presenting as mixed PK.1–3
Amyloidosis can be subdivided into localized cutaneous amyloidosis and systemic amyloidosis with cutaneous involvement.4,5 In both groups, amyloid precipitates are observed in the extracellular space of the dermis. In localized cutaneous amyloidosis, amyloid accumulates in the papillary dermis, whereas in systemic amyloidosis with cutaneous involvement, it accumulates in subpapillary layers, dermal appendages, and blood vessels.6,7
Localized cutaneous amyloidosis is characterized by amyloid protein deposition in the dermal papilla, resulting in specific skin manifestations. These deposits originate from keratin peptides of necrotic keratinocytes, distinct from systemic amyloidosis derived from immunoglobulins or serum proteins. 4 This condition manifests in two primary forms. Primary localized cutaneous amyloidosis (PLCA) features amyloid confined to the papillary dermis without systemic involvement and results from dysregulated epidermal keratinocyte metabolism. It comprises three subtypes: lichen amyloidosis (LA), presenting as intensely pruritic, grouped, skin-colored or hyperpigmented, dome-shaped papules typically on extensor surfaces; 8 macular amyloidosis (MA), characterized by “wavy” pigmented macules predominantly in the scapular region or lower extremities, often coalescing; 9 and the less frequent nodular amyloidosis (NA), featuring solitary or multiple firm nodules or plaques of variable size associated with dermal thickening and occurring anywhere on the body. 10 Secondary cutaneous amyloidosis denotes clinically inapparent amyloid deposits within the skin that are associated with pre-existing dermatoses or neoplasms, such as basal cell carcinoma, PK, or mycosis fungoides. 11 Alterations in keratinocytes are central to the pathogenesis of cutaneous amyloidosis, although the mechanisms remain incompletely understood. Pathogenetically, keratinocyte damage, apoptosis, and necrosis, as observed in conditions such as disseminated superficial porokeratosis (DSP), may initiate amyloidogenesis. Necrotic keratinocytes are phagocytosed by dermal macrophages and fibroblasts, transforming into amyloidogenic substances. Immunohistochemically, dermal amyloid deposits consistently associate with epidermal keratin, as evidenced by positive AE3 staining, indicating origin from high–molecular-weight epithelial keratins. 12 Herein, we report an unusual DSP variant with histologically confirmed dermal amyloid deposits.
Case report
A 76-year-old Chinese male presented with a 10-year history of multiple annular brown pruritic macule lesions involving the extremities and trunk along with dense, markedly pruritic papules on the bilateral anterior tibial regions. The brown macules appeared on the chest and back and progressively spread to the arms and thighs. The light-brown papules on the anterior tibia of both lower legs gradually extended to the buttocks and back. These papules were discrete, symmetrically distributed, and accompanied by marked pruritus.
The patient denied a history of systemic disease, and his family history was also negative. Physical examination revealed that his general condition was good. No obvious abnormalities were found during the systemic examination. Clinical examination revealed hyperkeratotic annular plaques of size 1–15 mm. These lesions showed slightly elevated borders with a central area of normal or mildly atrophic skin, which appeared on his trunk and limbs (Figure 1(a) and (b)). Several hemispherical reddish-brown papules measuring 1–2 mm with a rough papillomatous surface were observed on the buttocks and back (Figure 1(c)). There were dense, hemispherical, light-brown verrucous papules approximately 2 mm in size on the bilateral anterior tibial regions. These rashes were nonconfluent and symmetrically distributed (Figure 1(d)). Laboratory findings showed elevated serum uric acid (523 μmol/L), whereas all other routine parameters were within normal limits. Histopathologic examination of skin lesions in the right forearm showed typical findings of superficial PK, including dyskeratotic cells beneath the cornoid lamella, with absence of granular layer, and perivascular inflammatory cells infiltrate in the superficial dermis. In the papillary dermis, prominent deposits of amorphous eosinophilic globular material were observed (Figure 2(a) and (b)). Crystal violet staining revealed metachromatic reddish-purple globular deposits within the papillary dermis, consistent with amyloid deposition (Figure 2(c) and (d)). Topical desonide cream, halometasone cream, and 3% boric acid solution, combined with oral acitretin capsules were initiated, which resulted in a modest reduction in size and number of lesions along with partial control of pruritus. Nevertheless, upon cessation of the medication, signs of recurrence were detected.

(a and b) Hyperkeratotic annular plaques measuring 1–15 mm in size, characterized with slightly elevated borders and a central area of normal or mildly atrophic skin, appeared on the trunk and limbs (black arrow). (c) Several hemispherical reddish-brown papules with a rough papillomatous surface, measuring 1–2 mm in size, appeared on the buttocks and back (black arrow). (d) Dense, hemispherical, light-brown verrucous papules, approximately 2 mm in size, were present on bilateral anterior tibial regions (red arrow).
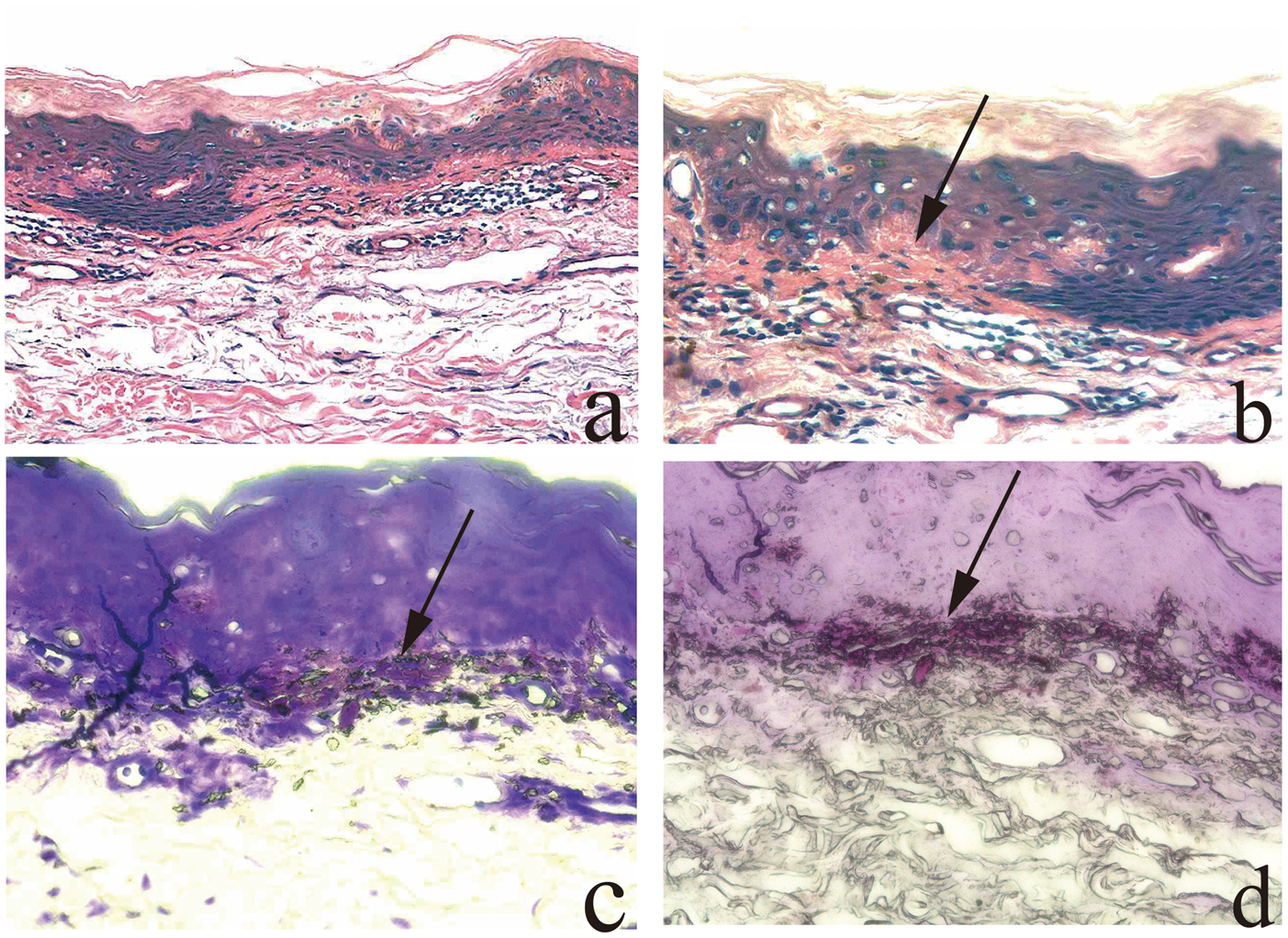
Histopathology of a right forearm lesion. (a) Beneath the cornoid lamella, some dyskeratotic cells were observed, the granular layer was absent, and perivascular inflammatory cells infiltrated the superficial dermis (hematoxylin & eosin stain, original magnification 20×). (b) In the papillary dermis, prominent deposits of amorphous eosinophilic globular material were observed (black arrow; hematoxylin and eosin stain, original magnification 40×). (c and d) Metachromatic, reddish-purple globular deposits were observed in the papillary dermis (crystal violet stain; original magnification 40×).
Discussion
DSP is a skin disease with distinctive clinical and histological characteristics. Its pathogenesis remains incompletely understood, and it is often indirectly associated with other skin diseases, increasing the complexity of clinical diagnosis and treatment.
Previous studies have reported an association between DSP and cutaneous amyloidosis. Although the exact mechanism remains unclear, 12 documented cases support this association (Table 1).12–21 Analysis of these cases indicates a predominance of middle-aged and older males, with a chronic disease course lasting several years. Lesions most commonly involve the limbs and trunk, including the back and chest, with approximately 40% of cases exhibiting pruritus to varying degrees. And some cases are accompanied by systemic diseases, such as pulmonary tuberculosis and hypertension. Furthermore, approximately 30% of cases exhibit a familial clustering tendency (such as children or siblings being affected), suggesting a possible genetic predisposition. The pathological characteristics include a hyperkeratinized cornoid lamella layer in the epidermis, accompanied by the disappearance of the granular layer. Dyskeratotic cells and vacuolated keratinocytes can be observed. Eosinophilic, amorphous amyloid deposits are evident in the dermal papillary layer, predominantly located beneath and surrounding the angular sample layer, with partial extension around the superficial dermal blood vessels. Inflammatory cell infiltration, primarily comprising lymphocytes and histiocytes, is noted, with some cases showing an increase in eosinophils and melanocytes (pigment incontinence). Apple-green birefringent amyloid materials were observed with Congo red staining, displaying bright yellow fluorescence under a fluorescence microscope with thioflavin T staining. Additionally, Periodic acid–Schiff (PAS) staining revealed oxidized glycoprotein elements with weak positivity. Approximately 66.7% of cases underwent immunohistochemical analysis, revealing positive reactions for acidic keratins (56.5 kDa), basic keratins (65–67 kDa), high–molecular-weight keratins (58–67 kDa), and basal layer keratins. In contrast, immunostaining was negative for immunoglobulin light chain (AL type) and amyloid A (AA) protein, a systemic amyloidosis biomarker. These findings indicate that amyloid deposits in cutaneous amyloidosis primarily originate from the basal layer of the epidermis and adjacent cells rather than from systemic amyloidosis. Cryotherapy has demonstrated a 70% efficacy rate in treating these deposits, particularly in cases of isolated or localized skin lesions. Topical dimethyl sulfoxide (DMSO) may directly affect amyloid accumulation, with histological evidence supporting reduction or elimination of amyloid deposits after treatment. Etretinate has shown efficacy in certain generalized cases, although associated side effects must be considered. Conversely, topical glucocorticoids primarily alleviate pruritus and may result in recurrence after discontinuation. Additionally, topical retinoids primarily target hyperkeratinization without significantly affecting amyloid deposition.
Summary of reported cases of DSP with amyloid deposits.

DSP: disseminated superficial porokeratosis; N.d.: not described; DMSO: dimethyl sulfoxide; F: female; M: male.
In our case, the patient exhibited hyperkeratotic annular plaques of size 1–15 mm. The lesions showed slightly elevated borders with a central area showing normal or mildly atrophic skin and appeared on the trunk and limbs. Skin biopsy sections revealed dyskeratotic cells beneath the cornoid lamella with absence of the granular layer and perivascular inflammatory cell infiltration in the superficial dermis. Combined with the patient’s clinical manifestations, physical examination, and pathological findings, these results were consistent with the pathogenic characteristics of DSP. Meanwhile, hematoxylin and eosin (H&E) staining of lesional skin from the right forearm showed prominent amorphous eosinophilic globular deposits in the papillary dermis. Crystal violet staining demonstrated metachromatic reddish-purple globular deposits within the papillary dermis, suggesting localized cutaneous amyloid deposition in this area. As mentioned earlier, cutaneous amyloidosis can be classified into two distinct forms: primary and secondary, with the latter being closely associated with pre-existing dermatological conditions. In the present case, the patient had a decade-long history of DSP and presented with pruritus at the lesional sites. The chronic inflammatory milieu of the primary disease, combined with mechanical trauma secondary to repetitive scratching, likely resulted in injury to epidermal keratinocytes in the right forearm. These damaged keratinocytes desquamated into the superficial dermis, where they were phagocytosed by cutaneous macrophages or fibroblasts, leading to the formation of protein bodies deposited in this area. Histopathological evaluation further revealed that amyloid deposition was localized exclusively beneath the lesional epidermis, sparing the neighboring uninvolved or distant normal skin. This spatial specificity strongly indicates a role of the lesional epidermis in amyloid formation. Additionally, the biopsy site was atypical for primary cutaneous amyloidosis, further supporting the theory that amyloid formation in DSP occurs via prolonged scratching and rubbing. Taken together, these findings support a diagnosis of secondary localized cutaneous amyloidosis arising in the context of DSP.
Notably, compared with previously documented cases, our patient exhibited clinical manifestations resembling those of LA in PLCA, characterized by dense, hemispherical, light-brown verrucous papules (approximately 2 mm in size) on the bilateral anterior tibial regions. These lesions were nonconfluent and symmetrically distributed. The onset of these papules was close to the onset of DSP. Laboratory tests, including liver function, renal function (blood urea nitrogen (BUN), creatinine, and estimated glomerular filtration rate (eGFR)), and urinalysis, showed no significant abnormalities, indicating no involvement of visceral organs. Although histopathological biopsy of the papular lesions was not performed, the combination of the patient’s medical history, physical examination, and laboratory findings does not exclude the possibility of primary localized amyloidosis. Therefore, based on the patient’s histopathological findings and characteristic clinical manifestations, we believe that this patient with DSP may have coexisting PLCA and secondary localized cutaneous amyloidosis. As reported in the literature, gout has been associated with cutaneous AA amyloid deposition, a phenomenon arising from chronic gout–induced persistent elevation of serum amyloid A (SAA). Persistently high concentration of SAA degrades into insoluble AA protein within tissues, forming β-pleated sheet amyloid fibrils that deposit in the skin. 22 Extrapolating from this mechanism, the patient’s hyperuricemia could potentially accelerate amyloid formation and deposition in the superficial dermis, influencing the likelihood of coexisting primary and secondary localized cutaneous amyloidosis in patients with DSP. This association has not been reported in previous cases of DSP with dermal amyloid deposits, suggesting a potential role of purine metabolism dysfunction in amyloidogenesis. Currently, there is no definitive treatment for PK. Treatment outcomes vary considerably, and no method can prevent recurrence. Common treatment approaches include drug therapy and physical therapy. Drug therapies include topical glucocorticoids, calcineurin inhibitors, retinoids, 5-fluorouracil, and vitamin D3 derivatives, among others. Oral medications include retinoids and antihistamines. Physical therapies comprise dermabrasion, liquid nitrogen cryotherapy, phototherapy, laser treatment, and curettage, among others. In the present case, the patient was treated with topical desonide cream, halometasone cream, and 3% boric acid solution, combined with oral acitretin capsules. After 4 months of treatment, pruritus improved and the color of the skin lesions lightened. Nevertheless, upon cessation of medication, signs of recurrence were observed.
In summary, DSP with dermal amyloid deposits represents a rare and complex dermatological entity with poorly understood pathogenesis, posing challenges for clinical diagnosis and management. This report describes a case of DSP complicated by dermal amyloid deposits, exhibiting dual clinical and pathological features of both disorders. Histopathological analysis supported a diagnosis of secondary localized cutaneous amyloidosis in the context of DSP; however, the coexistence of PLCA could not be fully excluded. Notably, the patient’s hyperuricemia suggests a potential role of purine metabolism dysfunction in amyloidogenesis, offering a novel perspective on the DSP–amyloid association. A literature review summarizing the clinical characteristics, pathological features, and treatment outcomes of this entity highlights the need for further clinical and basic research to elucidate its mechanisms and develop effective therapies.
Footnotes
Acknowledgments
We would like to thank Xianxu Yang from the Department of Pathology at the Fifth People’s Hospital of Hainan Province for professional assistance in the preparation, examination, diagnosis, and imaging of histopathological slides.
Author contributions
All authors made a significant contribution to the work, including the conception, study design, execution, acquisition of data, analysis, and interpretation; participated in drafting, revising, or critically reviewing the article; approved the final version for publication; agreed on the journal to which the article was submitted; and accept accountability for all aspects of the work.
Data availability statement
The datasets generated and analyzed during the current study are available in the article’s supplementary material. Additional data can be requested from the corresponding author upon reasonable request, subject to applicable confidentiality agreements or ethical restrictions.
Disclosure
The authors report no conflicts of interest in this work.
Ethics approval and consent to participate
This study was approved by the Medical Ethics Committee of the Fifth People’s Hospital of Hainan Province (approval number: 2025(010)). This study was conducted in accordance with the Declaration of Helsinki, and the reporting of this study conforms to CARE guidelines. 23 Written informed consent was obtained and published from the patients prior to study commencement. We have deidentified all patient details have been deidentified. In addition, written informed consent was obtained for the publication of this case report.
Funding
This work was supported by the Construction Project of Hainan Province Clinical Medical Center.