
Abstract
The diagnosis of idiopathic multicentric Castleman disease typically requires serum interleukin-6 testing—often inaccessible in resource-limited settings. A female patient in her early 40s presented with 6-month progressive generalized pruritus, low-grade fever, fatigue, and painless multiregional lymphadenopathy. Laboratory studies showed systemic inflammation, computed tomography/ultrasound-confirmed bilateral axillary lymphadenopathy, splenomegaly, and serositis. Lymph node biopsy revealed hyaline-vascular features: atrophic germinal centers with a “lollipop-like” vasculature and “onion skin–like” lymphocyte proliferation. Interleukin-6 testing was unavailable. CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) chemotherapy (nine cycles) combined with thalidomide (100 mg/day, cycles 6–9) was administered. Significant lymph node regression normalized the levels of inflammatory markers, and partial remission was observed at the 1-year follow-up. Rigorous histopathology combined with dynamic inflammatory markers enables the diagnosis of idiopathic multicentric Castleman disease without interleukin-6 testing. CHOP–thalidomide is an effective alternative therapy in resource-constrained regions.
Keywords
Introduction
Castleman disease (CD), also known as vascular follicular lymph node hyperplasia and giant lymphadenopathy, is a rare lymphoproliferative disorder characterized by heterogeneous clinical and pathological features. 1 Its idiopathic multicentric variant (idiopathic multicentric Castleman disease (iMCD)) presents with systemic inflammation, lymphadenopathy, and life-threatening multiorgan dysfunction, often mimicking lymphoma or autoimmune conditions. 2 Although dysregulated interleukin (IL)-6/JAK/STAT3 signaling is central to the pathogenesis of iMCD, 3 its low incidence (∼5000 cases worldwide) and nonspecific presentations frequently lead to diagnostic delays—with >60% of cases initially misdiagnosed. 4 Histologically, iMCD is classified into hyaline-vascular, plasmacytic, and mixed subtypes. 5 iMCD encompasses three clinical subtypes: (a) TAFRO (thrombocytopenia, anasarca, fever/reticulin fibrosis, renal dysfunction, organomegaly) syndrome, characterized by acute onset, thrombocytopenia, anasarca, and rapid multiorgan failure; (b) idiopathic plasmacytic lymphadenopathy (IPL), characterized by chronic plasmacytic infiltration and mild inflammation; and (c) not otherwise specified (NOS)—the most common subtype, manifesting as subacute lymphadenopathy and systemic inflammation (e.g. elevated C-reactive protein (CRP) levels). 4 Our patient’s symptoms fulfilled the diagnostic criteria for iMCD-NOS, which is distinct from TAFRO’s life-threatening course. Current international diagnostic criteria include serum IL-6 elevation as a supportive biomarker but not a mandatory requirement for iMCD diagnosis. 2 Notably, serum IL-6 is not often significantly elevated in patients with iMCD owing to its buffered regulatory system, which limits its diagnostic sensitivity.6,7 In clinical practice, CRP is more widely used as a practical indicator of systemic inflammation in most centers, especially in resource-limited settings. Treatment relies on anti–IL-6 monoclonal antibodies (e.g. siltuximab), although cost and availability restrict global use. 5
Here, we present a case of hyaline-vascular iMCD diagnosed without IL-6 testing. The patient was admitted to the hospital with the initial symptoms of generalized pruritus, periodic malaise, persistent low-grade fever, and painless progressive enlargement of multiple lymph nodes. The patient ultimately met the diagnostic criteria for iMCD after imaging and pathologic tissue biopsy. We highlight a multiparameter diagnostic model integrating pathology, inflammatory markers, and imaging. Moreover, we demonstrated CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone)–thalidomide as an effective alternative therapy in resource-constrained settings. This underscores the need for adaptable strategies in iMCD management.
Case presentation
A female patient in her early 40s presented in June 2021 with a 6-month history of generalized pruritus, persistent low-grade fever (37.5°C–38.5°C), fatigue, and progressive painless lymphadenopathy. Symptoms began acutely in January 2021 with a 40°C fever, temporarily relieved by antipyretics. Her medical history included type 2 diabetes and moderate anemia. Physical examination revealed multiple enlarged lymph nodes in bilateral cervical, axillary, and inguinal regions, with the largest lymph node measuring 2.5 cm in the right axilla, which was firm and mobile without tenderness. The patient was admitted to the Lincang People’s Hospital, Lincang City, Yunnan Province, China, in June 2021. Written informed consent for treatment was obtained from the patient prior to initiating chemotherapy. Additionally, written informed consent for the publication of clinical details, histopathological images, and imaging data was separately obtained from the patient. We have de-identified all patient details, including age (reported as a range) and exact dates (generalized to month and year) to protect patient privacy. The reporting of this study conforms to the Case Report (CARE) guidelines. 8
Laboratory studies demonstrated significant systemic inflammation (Table 1).
Key laboratory examinations.

Serum protein electrophoresis and IgG and IgG4 testing were not performed due to the limited laboratory resources at our institution at the time of admission. These tests are recommended for the differential diagnosis of plasmacytic disorders 9 ; however, they were not clinically necessary in this case considering the presence of the classic hyaline-vascular histopathology.
Computed tomography (CT) confirmed bilateral axillary lymphadenopathy (Figure 1), splenomegaly, and serositis (pleural/pericardial effusions). Ultrasound detected enlarged lymph nodes (Figure 2). Bone marrow aspiration showed trilineage hyperplasia without malignancy.
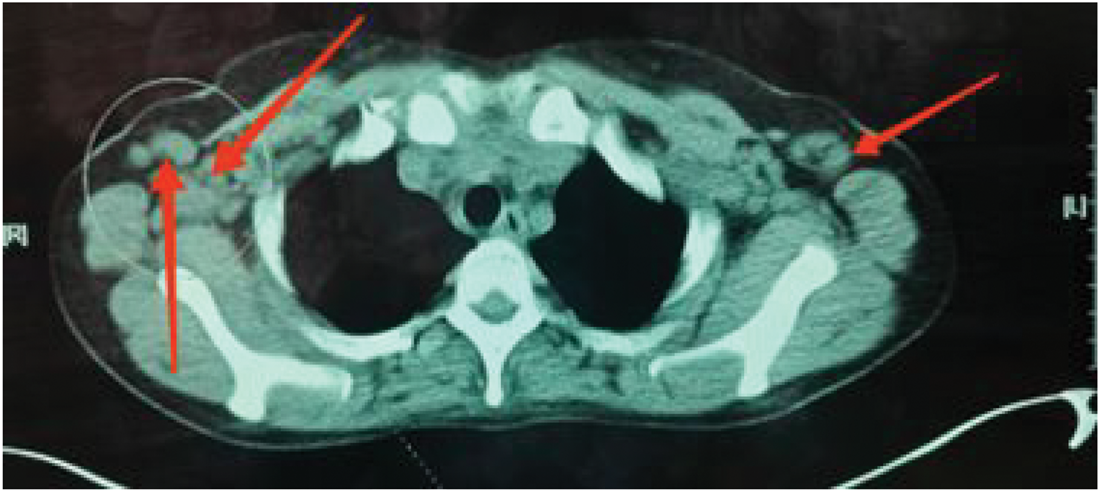
Computed tomography of the patient’s bilateral enlarged axillary lymph nodes.

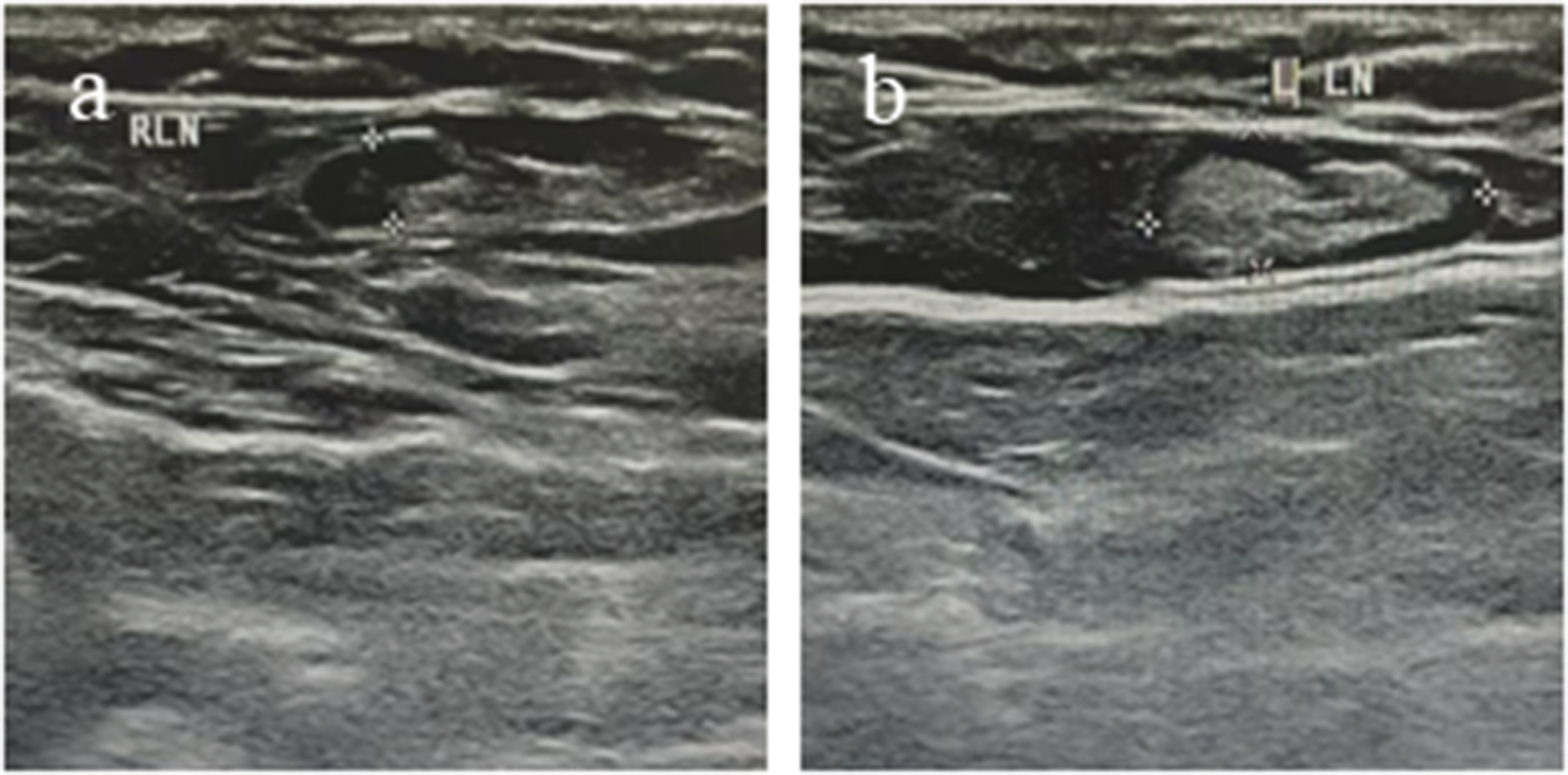
Admission ultrasound images: (a) enlarged right axillary lymph node (ultrasound) with a size of 0.6 × 1.7 cm and (b) enlarged left axillary lymph node (ultrasound) with a size of 0.7 × 2.2 cm.
Right axillary lymph node biopsy revealed classic hyaline-vascular features: atrophic germinal centers penetrated by hyalinized vessels (lollipop sign), concentric lymphocyte proliferation (onion skin–like pattern) (Figure 3(a) to (c)), and immunohistochemical evidence of CD21+ follicular dendritic cell networks with BCL2− germinal centers (Figure 3(d) to (f)). Serum IL-6 testing was unavailable. The diagnosis was confirmed using the 2021 Chinese iMCD criteria, 3 which align with the international consensus (Fajgenbaum et al.) 10 as follows: (a) histopathological evidence of CD (hyaline-vascular features in this case); (b) multicentric lymphadenopathy; (c) systemic inflammatory manifestations (CRP >30 mg/L; erythrocyte sedimentation rate (ESR), >100 mm/h); and (d) exclusion of infection, malignancy, and autoimmune diseases. After a multidisciplinary review, the patient met the 2021 Chinese iMCD diagnostic criteria, confirming hyaline-vascular iMCD.

Lymphatic tissue biopsy findings. (a, b) Perifollicular lymphocyte proliferation arranged in a multilayered ring, forming an “onion skin–like” pattern. (c) Some of the proliferating blood vessels grew into the ingrowth center, forming a “lollipop”-like appearance. (d) CD20 testing showed an increase in the number of lymphatic follicles. (e) CD21 testing showed concentric proliferation of follicular dendritic cells and (f) BCL2 testing showed a widening of the sleeve area, and BCL2 was not expressed in the center of the growth.
Following initial supportive care for fever and hyperglycemia, treatment commenced with CHOP chemotherapy: cyclophosphamide (1 g on day 1), vincristine (2 mg on day 1), doxorubicin (70 mg on day 1), and dexamethasone (10 mg on days 1–5) every 3 weeks over nine cycles. Thalidomide (100 mg nightly) was added during cycles 6–9 for immunomodulation. The third chemotherapy cycle induced hypertension, which was controlled with nifedipine. During cycle 7, herpes zoster infection on the chest was resolved with acyclovir (Figure 4). Generalized pruritus was persistent for 6 months, with paroxysmal worsening at night (visual analog scale (VAS) score, 8/10). Possible mechanisms include IL-6–mediated activation of cutaneous sensory nerves and mast cell degranulation. After two cycles of CHOP, the pruritus improved (VAS, 3/10); by cycle 6 (with thalidomide), it resolved completely. The patient returned to the hospital for regular check-ups, but her condition did not improve substantially. Remission was defined and evaluated according to the international consensus criteria for iMCD, 11 which require the following: (a) ≥50% reduction in the size of measurable lymphadenopathy compared with baseline; (b) normalization of systemic inflammatory markers (CRP, <5 mg/L; ESR, <20 mm/h); (c) resolution of systemic symptoms (e.g. fever and pruritus); and (d) absence of new lesions or organ dysfunction. In this case, follow-up assessments at 3, 6, and 12 months included the following: (a) imaging: ultrasonography showed that the size of the bilateral axillary lymph nodes reduced from 2.5 cm (baseline) to 0.7–1.3 cm (12 months) (Figure 4); (b) laboratory tests: the CRP level decreased from 32.44 to 3.2 mg/L, ESR from 123 to 18 mm/h, and ferritin level from 1357 to 142 ng/mL; and (c) clinical symptoms: complete resolution of fever and pruritus. Based on these findings, the patient achieved partial remission at the 1-year follow-up.
Initial ultrasound review after the completion of chemotherapy. (a) Enlarged right axillary lymph node with a size of 0.7 × 0.4 cm and (b) enlarged left axillary lymph node with a size of 1.3 × 0.6 cm.
Discussion
CD is a rare disorder of the lymphatic system that is often clinically unremarkable and easily confused with other lymphatic disorders. Herein, we reported the case of a female patient in her early 40s who was admitted to the hospital with generalized pruritus, persistent low-grade fever and malaise, and painless progressive enlargement of multiple lymph nodes, which led to the diagnosis of hyaline-vascular CD. This case of hyaline-vascular iMCD illustrates critical diagnostic and therapeutic challenges in resource-constrained settings.
In terms of diagnosis, in addition to the classification based on the cumulative site of the disease, CD is divided into hyaline-vascular, plasma cell, and mixed types based on the pathologic morphology. Recent expert consensus has refined iMCD subtyping into three categories: iMCD-TAFRO, iMCD-IPL, and iMCD-NOS. 4 Our patient’s symptoms aligned with the diagnostic criteria for iMCD-NOS, characterized by subacute lymphadenopathy, systemic inflammation (CRP >30 mg/L), and hypervascular histopathology—previously termed as “hyaline-vascular” (Figure 3(d) to (f)). CD itself is a rare disease, and its initial symptoms, signs, and degree of manifestation may vary from case to case due to individual differences. 3 Imaging screening considerably relies on positron emission tomography (PET)-CT, enhanced CT, and high-resolution CT. 4 Ultrasonography plays a role in locating enlarged lymph nodes and monitoring changes in size during follow-up (e.g. tracking lymph node regression after chemotherapy) (Figures 2 and 4). However, there are challenges in qualitative diagnosis, and the probability of misdiagnosis and omission of CD is even greater if the sonographer is less experienced and less knowledgeable about this type of disease. Owing to the limitations of the imaging conditions in our hospital, the imaging examination could only provide limited help in the screening of this case. The pathologic findings in this case showed partial atrophy of the lymphoid follicles and hyperplasia of the sinus histiocytes, which are consistent with the features of hyaline-vascular CD, a relatively rare and easily misdiagnosed type of lymphoma or other lymphatic system diseases. 9 Lymph node pathology has been reported in the literature as the gold standard method for diagnosing CD; hence, initial screening for the disease relies on a multidisciplinary rationalization of the examination, and confirmation of the diagnosis depends on the interpretation of pathology as well as immunohistochemistry results. 2 In this case, the timeliness and accuracy of the pathologic examination were crucial in confirming the diagnosis.
In terms of treatment, patients with unicentric CD (UCD) have a high cure rate via complete surgical resection of the lesion. 12 IL-6 has been reported to be the most critical cytokine in the pathogenesis of iMCD. Although the pathogenesis of CD has not yet been clarified, some studies in recent years have shown that it is closely related to the high inflammatory state observed in iMCD. Therefore, the frequently recommended treatment option for patients with multicentric CD (MCD) is based on chemotherapy with siltuximab, an anti–IL-6 monoclonal antibody. However, due to the prolonged intravenous administration of siltuximab and the high price of long-term application, although siltuximab has been recommended as a first-line treatment option for patients with nonheavy MCD as early as 2021, factors such as patients’ financial constraints and the influence of individual differences on the therapeutic effect have revealed that the general clinical application of siltuximab in the primary healthcare system remains far from being fully achieved. Adverse effects of CHOP–thalidomide were mild: grade 1 hypertension (controlled with nifedipine) and grade 2 herpes zoster (resolved with acyclovir). Compared with alternatives such as tocilizumab (which requires monthly intravenous infusion and costs ∼$1500 per cycle in China), CHOP–thalidomide is more cost-effective ($200 per cycle) and suitable for resource-limited settings. Rituximab was not considered due to its limited efficacy in hyaline-vascular subtypes. 5 The thalidomide–cyclophosphamide–prednisone (TCP) regimen is currently one of the first-line treatment regimens recommended by the National Comprehensive Cancer Network for iMCD. It shares similarities with the CHOP regimen reported in this case in terms of drug composition and treatment mechanism. When IL-6–targeted therapy remains ineffective, chemotherapy regimens derived from non-Hodgkin lymphoma can serve as important alternative treatment strategies. 13 A landmark phase 2 study by Zhang et al. 14 demonstrated that TCP achieved an overall response rate of 72% in iMCD patients, with 41% achieving complete remission. This supports our use of CHOP–thalidomide, as cyclophosphamide and thalidomide share synergistic anti-inflammatory and antiangiogenic effects. 14 CHOP combined with various chemotherapy regimens has been widely used in the treatment of lymphoma and has also been shown to be effective in CD. 15 Thalidomide exerts immunomodulatory effects by inhibiting the activation of nuclear factor (NF)-κB signaling pathway, which is pivotal for the transcription of proinflammatory cytokines such as IL-6 and tumor necrosis factor (TNF)-α. 16 This inhibition reduces the overproduction of IL-6 and TNF-α, thereby alleviating systemic inflammation—a key pathogenic feature of iMCD. Additionally, it suppresses angiogenesis by downregulating vascular endothelial growth factor (VEGF), which contributes to the regression of hypervascular lymph nodes observed in this case. According to relevant studies, the therapeutic effect of thalidomide combined with CHOP regimen for non-Hodgkin lymphoma in our patient was significantly better than that of CHOP regimen alone. In the implementation of CHOP combination chemotherapy, the application of glucocorticoids can be adjusted individually. 17 In this case, dexamethasone was used to replace prednisone in the conventional regimen, and a significant decrease in inflammatory indexes, such as CRP, was observed after the adjustment of the regimen. After 1 year of follow-up, there was no recurrence or metastasis, and the patient achieved partial remission according to the criteria for evaluating the efficacy of iMCD in the relevant literature, 18 which suggests the effectiveness of this treatment. In addition, studies have shown that the prognosis of patients with CD is closely related to early diagnosis and timely treatment, and timely chemotherapy can effectively improve the quality of survival and reduce the recurrence rate. 19
Study limitations included the following: (a) unavailability of equipment for testing serum IL-6, a biomarker recommended by international guidelines, 5 which hinders direct validation of the IL-6–driven inflammatory cascade; (b) single-case design, which limits extrapolation to diverse iMCD subtypes (e.g. TAFRO); and (c) 1-year follow-up, which is shorter than the 2-year period suggested for assessing long-term remission in iMCD. 11
Future studies should continue to deepen the understanding of the pathogenesis of CD and explore novel biomarkers and therapeutic approaches such as serum CXCL19, 9 with the aim of providing more effective diagnostic and therapeutic options for similar cases. By summarizing and analyzing the clinical features, diagnostic difficulties, and therapeutic effects of these cases, we can provide guidance and reference for clinicians in the management of CD and other rare diseases.
Conclusions
This case demonstrates that rigorous histopathological analysis combined with inflammatory dynamics enables accurate iMCD diagnosis without IL-6 testing. In settings where biologics are inaccessible, CHOP–thalidomide provides a cost-effective alternative with comparable remission rates. Multidisciplinary collaboration remains pivotal for managing CD’s diagnostic and therapeutic complexities. Future efforts must focus on standardizing accessible biomarker panels and expanding treatment options for global equity in CD care.
Supplemental Material
sj-pdf-1-imr-10.1177_03000605251381574 - Supplemental material for Diagnostic and therapeutic challenges in hyaline-vascular idiopathic multicentric Castleman disease: A case report
Supplemental material, sj-pdf-1-imr-10.1177_03000605251381574 for Diagnostic and therapeutic challenges in hyaline-vascular idiopathic multicentric Castleman disease: A case report by Chengbin Lu, Xiaoli Ma, Beibei Liu, Jingyan Wang, Song Luo and Qionghua Luo in Journal of International Medical Research
Footnotes
Acknowledgments
We are grateful to the patients who provided message for this study.
Author contributions
All authors have reviewed the final version to be published and agreed to be accountable for all aspects of the work.
Data availability
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Declaration of conflicting interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ethical statement and consent
This retrospective study was reviewed by the Medical Ethics Committee of Lincang People’s Hospital and was granted exemption from full review, with formal ethical approval issued. The ethical approval number is IEC/AF/61-02/2023-01.0, and the approval date is 2 April 2025. Written informed consent for treatment and publication of clinical details and images was obtained from the patient.
Funding
This study was supported by the Yunnan Province College Student Innovation Training Program Project Fund (Grant No. 202510678050X).
Informed consent statement
Informed consent has been obtained from the patient.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.