
Abstract
Objective
Accurate prognostication is crucial for managing human immunodeficiency virus (HIV)-associated cutaneous T-cell lymphoma. In this study, we aimed to develop an improved machine learning-based prognostic model for predicting the 5-year survival rates in HIV-associated cutaneous T-cell lymphoma patients.
Methods
We derived and tested machine learning models using algorithms including Extreme Gradient Boosting (XGBoost), Light Gradient Boosting Machine (LightGBM), and Random Forest. Our study involved data from a US population-based cohort of patients diagnosed with HIV-associated cutaneous T-cell lymphoma between 1 January 2000 and 31 December 2018, which were extracted from the Surveillance, Epidemiology, and End Results database. The primary outcome was the prediction of 5-year overall survival. Model discrimination was assessed using the area under the receiver operating characteristic curve (AUC), and calibration was assessed using Brier scores.
Results
A cohort of 381 HIV-associated cutaneous T-cell lymphoma patients was analyzed. Multivariate logistic regression identified age ≥60 years (odds ratio = 4.88), regional stage (odds ratio = 10.31), distant stage (odds ratio = 28.37), and chemotherapy (odds ratio = 4.71) as significant independent risk factors for 5-year mortality. Among seven machine learning models developed, the XGBoost model demonstrated the highest discrimination for 5-year overall survival (AUC = 0.867), followed by LightGBM (AUC = 0.835). Both models exhibited good calibration with low Brier scores (XGBoost = 0.130, LightGBM = 0.109). Support Vector Machine performed optimally in ten-fold cross-validation, logistic regression showed the lowest Brier score (0.106), and XGBoost provided the best balance of discrimination and robust performance.
Conclusion
Our novel machine learning approach produced prognostic models with superior discrimination for 5-year overall survival in HIV-associated cutaneous T-cell lymphoma patients using standard clinicopathological variables. These models offer potential for more accurate and personalized prognostics, potentially improving patient management and clinical decision-making.
Keywords
Introduction
Lymphoma is a malignant neoplasm originating from the lymphatic system, categorized into Hodgkin lymphoma and non-Hodgkin lymphoma. 1 Human immunodeficiency virus (HIV)-associated cutaneous T-cell lymphoma (CTCL) is a rare but severe subtype of non-Hodgkin lymphoma, commonly observed in HIV-infected individuals. 2 Patients with this condition face unique challenges because an HIV infection not only affects the immune system but may also accelerate lymphoma progression. 3 Clinical presentations of HIV-associated CTCL vary from localized skin lesions to systemic disease, often with a poor prognosis. 3 Current treatment strategies include antiretroviral therapy, chemotherapy, radiotherapy, and targeted therapies; however, treatment outcomes remain suboptimal.3,4 Given the disease complexity and prognostic uncertainty, accurate prediction of patient survival rates is crucial. Established prognostic factors for HIV-associated CTCL include age at diagnosis, disease stage (such as localized, regional, or distant spread), and treatment modalities such as radiotherapy and chemotherapy, which have consistently shown significant associations with patient survival outcome.2,5,6
The Surveillance, Epidemiology, and End Results (SEER) database, a project of the National Cancer Institute, provides comprehensive cancer-related data, including incidence, mortality, and survival rates. Covering approximately 28% of the US population, it offers valuable resources for cancer research. 7 Multiple internationally recognized tools exist for risk prediction models. Most tools categorize patients into different risk groups, derived from the D’Amico three-tier system. 8 However, these models primarily focus on intermediate indicators such as biochemical recurrence rather than survival rates. 8 Research indicates that effective prognostic models can be created using simple factors such as prostate-specific antigen, grade, and stage, while refined stratification systems can enhance a model’s discriminatory ability. Additionally, using continuous rather than categorical data allows for more accurate and personalized predictions, aiding clinical decision-making. 9 For instance, the PREDICT Prostate tool and Memorial Sloan Kettering Cancer Center nomogram demonstrate high discriminatory ability in predicting survival rates and are available as web-based decision aids for patients and clinicians. 9 However, specialized prediction models for HIV-associated CTCL remain scarce. Given the uniqueness and complexity of this disease, developing a dedicated predictive tool is crucial for improving patient management and prognosis.
However, enhanced personalized tools often rely on traditional statistical modeling methods, which typically use pre-specified variables and assume particular forms of interactions. In contrast, machine learning (ML), as a data-driven application of artificial intelligence, learns and improves from data automatically without explicit programming. Consequently, these algorithms can mine datasets to identify not only pre-established risk factors but also complex, nonlinear relationships and subtle patterns often overlooked by traditional methods. Although ML applications in healthcare (e.g. developing novel prognostic models) are rapidly growing, the use of ML in the specific and rare context of HIV-associated CTCL remains limited. Notably, to the best of our knowledge, there are no prior reports of ML-based prognostic models for HIV-associated CTCL utilizing the comprehensive SEER database, particularly involving a systematic comparison of multiple algorithms. Therefore, the core innovation of this study lies not only in addressing this gap by being among the first studies to apply such an approach in this specific patient cohort using SEER data but also in systematically developing and comparing multiple advanced ML models (including ensemble methods such as Extreme Gradient Boosting (XGBoost) and Light Gradient Boosting Machine (LightGBM), alongside other classical algorithms) to predict 5-year survival. We hypothesize that these approaches can generate superior predictive models by more effectively integrating known prognostic factors (e.g. age, stage, and treatment) and uncovering their complex interplay, thus achieving enhanced discrimination and calibration over traditional models. Our aim is to develop this improved model and rigorously compare its performance against established benchmarks. Ultimately, this research may provide crucial evidence for more accurate and personalized prognostic assessment, thereby improving patient management and individualized treatment decisions for this challenging condition.
Materials and methods
Data source
Clinical information of patients diagnosed with HIV-associated CTCL from 2004 to 2017 was extracted from the SEER database using SEER*Stat software (version 8.4.3). The SEER database, an authoritative source for cancer statistics in the US, provides cancer-related data including clinicopathological characteristics, treatment, cancer-related incidence, and survival. For this study, the inclusion criteria were as follows: (a) site recode ICD-O-3/WHO 2008 classified as lymphoma; (b) Collaborative Stage (CS) site-specific factor 1 (varying by schema from 2004 to 2017) coded as 001 or 010; (c) lymphoma confirmed by microscopic examination; (d) complete tumor staging and diagnostic data. Patients were randomly divided into validation and training sets at a ratio of 7:3. The outcome variable was 5-year survival rate.
Construction and validation of ML models
Variables identified as statistically significant through univariate and multivariate logistic regression (LR) analyses were incorporated into the ML models. Seven ML models were constructed in this study, including K-Nearest Neighbors (KNN), Random Forest (RF), Support Vector Machine (SVM), LightGBM, LR, decision tree (DT), and XGBoost. The efficacy of the ML models was validated using receiver operating characteristic (ROC) curves, precision–recall (PR) curves, decision curve analysis (DCA), ten-fold cross-validation, and calibration curves. The clinical decision-making capability of the models was assessed through DCA curves. Calibration curves were employed to evaluate prediction performance. Additionally, metrics such as accuracy, sensitivity, specificity, precision, positive predictive value, negative predictive value, recall, and F1 score were utilized to assess the ML models. The significance of ML models and ML validation tool in this study is shown in Table 1.
Significance of ML models and ML validation tool in this study.

HIV: human immunodeficiency virus; ML: machine learning; AUC: area under the ROC curve.
Statistical analysis
Categorical variables were expressed as percentages and compared using Fisher’s exact test or chi-square test. Risk factors were identified using multivariate LR analysis. A p-value <0.05 (two-sided) was considered to indicate statistical significance. All analyses were performed using R software (version 4.2.3, http://www.r-project.org).
Results
Baseline population characteristics
This study analyzed the clinical characteristics of 381 patients with HIV-associated CTCL, including 265 patients in the training set and 116 in the validation set. Baseline demographic characteristics and clinical features of the training and validation cohorts are detailed in Table 2. The patient population was predominantly aged <60 years (55%–60%), male (58%–66%), and married (54%–56%), with a majority being Caucasian (70%–73%). Most patients (86%–89%) were diagnosed between 2011 and 2017, indicating an increased diagnostic rate in recent years. Over half of the patients were from middle- to high-income households (annual income >US$74,999). Regarding disease characteristics, skin not otherwise specified was the most common primary site (53%–55%), and approximately 80% of patients were diagnosed at a localized stage. In terms of treatment, most patients did not undergo surgery (78%–80%), radiotherapy utilization was low (9%–13%), and chemotherapy usage was approximately 21%–23%. Notably, no statistically significant differences (p > 0.05) were observed between the training and validation sets for all variables, suggesting balanced data partitioning and good representation of the overall population by the validation set.
Baseline characteristics of patients with HIV-associated cutaneous T-cell lymphoma in the training and validation cohorts.

HIV: human immunodeficiency virus.
“1” refers to the training cohort, and “2” refers to the validation cohort.
Univariate and multivariate LR analyses
Univariate and multivariate LR analyses identified risk factors for 5-year survival in patients with HIV-associated CTCL. The detailed results of these analyses are presented in Table 3. Age emerged as a significant prognostic factor, with patients aged ≥60 years exhibiting a 4.88-fold increased mortality risk (odds ratio (OR) = 4.88, 95% confidence interval (CI): 2.04–11.68, p < 0.001). Disease stage significantly impacted prognosis; compared with localized stage patients, regional stage patients showed a 10.31-fold increased mortality risk (OR = 10.31, 95% CI: 4.24–25.11, p < 0.001), while distant stage patients demonstrated a 28.37-fold increased risk (OR = 28.37, 95% CI: 1.58–507.67, p = 0.023). Radiotherapy was significantly associated with an increased mortality risk in univariate analysis (OR = 5.61, 95% CI: 2.29–13.72, p < 0.001) but showed no statistically significant association in multivariate analysis (p = 0.267). Chemotherapy was significantly associated with a high mortality risk in multivariate analysis (OR = 4.71, 95% CI: 1.97–11.28, p < 0.001). Female sex potentially exerted a protective effect, as indicated by univariate analysis (OR = 0.50, p = 0.049); however, this result was not significant in multivariate analysis (p = 0.318). Marital status, race, diagnosis year, household income, primary site, diagnosis-to-treatment time, and surgery did not demonstrate significant effects in univariate analysis. These findings provided crucial variable selection basis for constructing ML models to predict 5-year survival rates in HIV-associated CTCL patients.
Univariate and multivariate logistic regression analyses of factors associated with 5-year survival in patients with HIV-associated cutaneous T-cell lymphoma.

HIV: human immunodeficiency virus; OR: odds ratio.
“0” represents patients who were alive at 5 years, and “1” represents patients who died within 5 years.
Construction and validation of ML models
Seven ML models were constructed to predict the 5-year survival rates in patients with HIV-associated CTCL. XGBoost demonstrated the highest area under the ROC curve (AUC) value (0.867), followed by LightGBM (AUC = 0.835), LR (AUC = 0.8), RF (AUC = 0.757), SVM (AUC = 0.751), KNN (AUC = 0.737), and DT (AUC = 0.691) (Figure 1). The PR-AUC values for LightGBM, XGBoost, LR, RF, SVM, DT, and KNN were 0.527, 0.498, 0.48, 0.471, 0.415, 0.405, and 0.352, respectively (Figure 2). DCA indicated minimal differences in the net benefit across all ML models (Figure 3). Results from ten-fold cross-validation (Figure 4) revealed that SVM performed optimally in predicting the 5-year survival rate of patients with HIV-associated CTCL. Lower Brier scores indicated higher consistency between predictions and reality, indicating superior performance. As illustrated in Figure 5, LR exhibited the lowest Brier score (0.106), followed by LightGBM (0.109), KNN (0.114), SVM (0.122), RF (0.129), DT (0.129), and XGBoost (0.13).

ROC curves for seven machine learning models predicting the 5-year survival rate in HIV-associated cutaneous T-cell lymphoma patients. XGBoost demonstrated the highest AUC (AUC = 0.867), followed by LightGBM (AUC = 0.835), logistic regression (AUC = 0.8), Random Forest (AUC = 0.757), Support Vector Machine (AUC = 0.751), K-Nearest Neighbors (AUC = 0.737), and decision tree (AUC = 0.691). ROC: receiver operating characteristic; HIV: human immunodeficiency virus; AUC: area under the ROC curve; XGBoost: Extreme Gradient Boosting; LightGBM: Light Gradient Boosting Machine.

PR curves for seven machine learning models. PR-AUC values were as follows: LightGBM (0.527), XGBoost (0.498), logistic regression (0.48), Random Forest (0.471), Support Vector Machine (0.415), decision tree (0.405), and K-Nearest Neighbors (0.352). PR: precision–recall; AUC: area under the receiver operating characteristic curve; XGBoost: Extreme Gradient Boosting; LightGBM: Light Gradient Boosting Machine.

DCA comparing the net benefit across all machine learning models for predicting the 5-year survival rate in HIV-associated cutaneous T-cell lymphoma patients. DCA: decision curve analysis; HIV: human immunodeficiency virus.
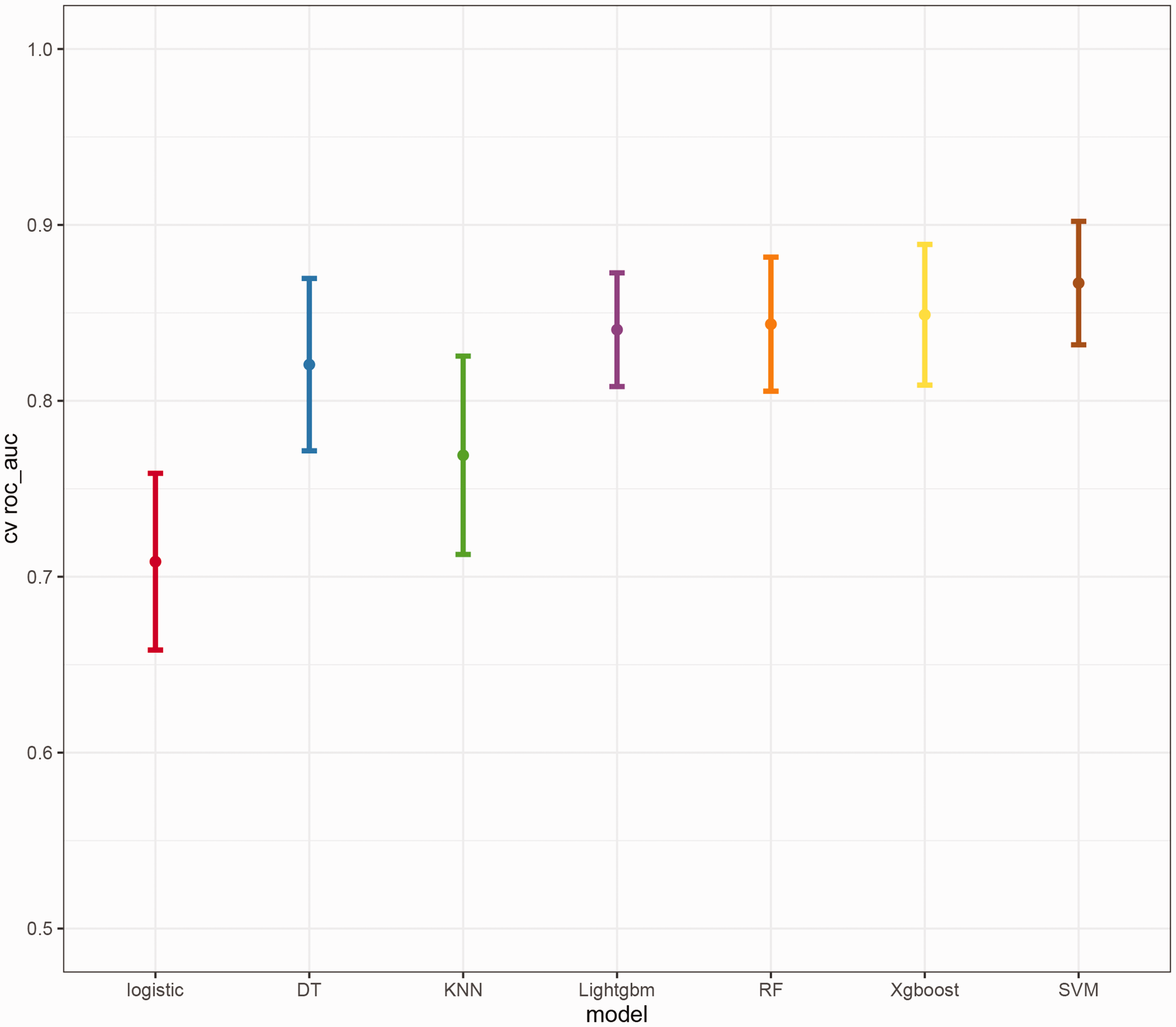
Ten-fold cross-validation results for seven machine learning models, with SVM demonstrating optimal performance in predicting 5-year survival rates. SVM: Support Vector Machine.

Brier scores for each machine learning model, indicating prediction accuracy. Lower scores represent higher consistency between predictions and actual outcomes. Logistic regression showed the lowest Brier score (0.106), followed by LightGBM (0.109), K-Nearest Neighbors (0.114), Support Vector Machine (0.122), Random Forest (0.129), decision tree (0.129), and XGBoost (0.13). XGBoost: Extreme Gradient Boosting; LightGBM: Light Gradient Boosting Machine.
Discussion
HIV-associated CTCL is a rare form of CTCL occurring in HIV-infected individuals. This condition typically manifests as skin lesions such as patches, plaques, or tumors. Owing to the compromised immune function of patients, its clinical presentation and treatment strategies may differ from non-HIV-associated CTCLs. 2 Our study developed and tested an ML-based prognostic model using a large-scale dataset to predict the 5-year survival rate in patients with HIV-associated CTCL, comparing its performance with those of multiple stratified and multivariate predictive models. To the best of our knowledge, this is the first study utilizing the SEER cohort data to compare multiple models for predicting HIV-associated CTCL-specific mortality. This research introduced an innovative mortality prediction approach, employing novel ML algorithms to automatically integrate the best features of different modeling methods, providing a new perspective on prognostic assessment in this field.
ML has shown considerable potential in research on HIV-associated health issues, with broad applications ranging from prognosticating opportunistic infections to assessing complication risks. For instance, while predicting survival for patients with HIV-associated opportunistic infections, a study utilized ML models to stratify risk in patients with cryptococcosis, achieving good predictive performance (C-index: 0.78). 10 Similarly, ML has been successfully employed to identify predictive factors for common comorbidities in individuals with HIV and construct risk models. A study on peripheral neuropathy demonstrated that ML-based approaches, such as RF, not only effectively predicted disease status (AUC > 0.80) but also identified key predictive variables missed by traditional statistical methods. 11 Regarding neurological complications, ML has been used to optimize the detection of HIV-associated neurocognitive disorders (HAND). For example, by integrating data from brief neuropsychological assessments, ML models can detect HAND with over 90% accuracy, providing a robust tool for early clinical screening. 12 Furthermore, ML models based on neuroimaging data have been used to investigate the impact of HIV infection on brain functional networks and predict cognitive status. 13 Researchers have also utilized SVM to analyze plasma extracellular vesicle phenotype data, successfully predicting cognitive dysfunction and highlighting ML’s role in integrating complex biological data for disease state assessment. 14 For HIV-associated malignancies, ML has been applied to conduct noninvasive prognostic evaluation. A study on HIV-associated lung adenocarcinoma accurately predicted the expression levels of the proliferation marker Ki-67 (test set AUC: 0.905) using radiomics features and ML models, such as SVM, demonstrating its value in oncology. 15 Collectively, these studies indicate that ML provides robust analytical tools for understanding complex pathophysiological processes in the context of HIV infection, improving disease prediction and risk stratification as well as guiding personalized management strategies. However, research employing advanced ML methods for survival prediction in HIV-associated CTCL, a specific, rare, and prognostically unfavorable tumor, remains insufficient, underscoring the necessity of the present study.
Our study developed and validated several ML models for predicting the 5-year survival rate in HIV-associated CTCL patients, with ensemble methods showing notable predictive capabilities. The favorable performance of models such as XGBoost (AUC = 0.867) and LightGBM (AUC = 0.835) may stem from their capacity to capture complex nonlinear relationships and variable interactions often missed by traditional regression methods. Gradient boosting algorithms such as XGBoost are adept at handling heterogeneous clinical data and can form robust predictors by integrating multiple base learners16,17; their strength is also noted in predicting radiation dermatitis (AUC =0.890) 16 and cardiac surgery mortality (AUC = 0.9145). 17 Our models incorporated established prognostic factors (age, stage, and chemotherapy) identified via LR, the prognostic value of which is well supported in lymphoma. The ML models’ data-driven variable weighting likely enhances predictive accuracy, and tree-based methods such as XGBoost can indicate relative feature importance, potentially identifying key prognostic drivers.
To the best of our knowledge, this study is an early effort to systematically compare multiple advanced ML algorithms for 5-year survival prediction in HIV-associated CTCL patients using SEER data. Yang et al. 2 previously developed nomograms for this cohort using SEER data, identifying key prognostic factors and reporting good model accuracy. Our work has expanded on this by applying and comparing a suite of ML algorithms. Our XGBoost model’s high AUC (0.867) suggests strong discriminatory power. Compared with other SEER-based ML studies on lymphoma, such as Wang et al.’s DeepSurv model for primary gastrointestinal lymphoma (C-index up to 0.760), our ensemble boosting methods also demonstrated good performance using standard clinicopathological variables. 18 The versatility of XGBoost is further indicated by its successful application in other oncological predictions, including thyroid cancer risk stratification (AUC up to 0.886) 19 and non-small cell lung cancer STAS prediction (AUC up to 0.927). 20 Our systematic comparison aimed to identify suitable models for HIV-associated CTCL. These findings suggest that ML approaches merit further investigation for refining prognostication and potentially guiding personalized management in this rare malignancy.
The inherent rarity of HIV-associated CTCL considerably constrains dataset sizes, a common challenge in developing robust prognostic models for such conditions. 21 Despite these limitations, ML offers a promising avenue for prognostic prediction in rare diseases by leveraging available data to identify predictive patterns. 21 Studies tackling other rare conditions, such as primary central nervous system lymphoma 22 and diverse rare diseases using electronic health records data, 23 have demonstrated the application of advanced ML strategies to specifically address data insufficiency and still achieve meaningful predictive outcomes. This highlights a broader trend of successfully applying ML to extract valuable prognostic information even when data are scarce, a context highly relevant to our investigation of HIV-associated CTCL. Within this framework, our systematic comparison identified XGBoost as a particularly effective algorithm for this rare lymphoma subtype (AUC = 0.867). The utility of XGBoost in medical prediction, especially with potentially limited datasets, is supported by its reported success in other clinical scenarios, such as achieving higher AUCs in predicting small-for-gestational-age births 24 and diabetic nephropathy 25 compared with multiple algorithms. The ability of our XGBoost model to achieve this using only standard clinicopathological variables further underscores its practical value in rare disease research, where extensive biomarker data may be unavailable.21,26
In summary, this study utilized clinical data from 381 HIV-associated CTCL patients in the SEER database to construct and compare seven ML models for predicting 5-year survival rates, with XGBoost demonstrating strong predictive performance using standard clinicopathological variables. A key strength lies in the comprehensive multi-algorithm evaluation. This study is, however, primarily limited by its retrospective SEER-based design and the lack of external validation. Future research could expand on this work by validating these models in independent, prospective cohorts and exploring the integration of additional biomarkers. Crucially, investigating the broader applicability of these ML methodologies to other rare diseases, where data scarcity poses significant challenges, represents an important avenue for advancement. Comparing our model’s performance in detecting lymphomas not associated with HIV could also yield valuable insights. Nevertheless, this research provides a potentially useful tool for prognostic assessment in HIV-associated CTCL patients, with implications for developing more precise, individualized treatment strategies and ultimately improving patient outcomes in this disease and potentially other rare oncological conditions.
Footnotes
Acknowledgments
Not applicable.
Author contributions
Conception and design: Zhang Yongmin and Huang Weimin; Administrative support: Chen Lingzhen; Provision of study materials or patients: Tian Manwen and Ai Jing; Collection and assembly of data: Jia Lanlan, Chen Junteng, and Gan Jinying; Data analysis and interpretation: Huang Weimin; Manuscript writing: Zhang Yongmin and Huang Weimin; and Final approval of manuscript: all authors.
Data availability statement
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
Declaration of generative AI and AI-assisted technologies in the writing process
The language was polished by GPT 4o to improve the readability, and the content has been reviewed by all authors. All authors take full responsibility for the content of the publication.
Declaration of conflicting interests
The authors have no conflicts of interest to declare.
Funding
This project was supported by the Science and Technology Program of Guangzhou (Nos. 2023A04J0568, 2024A03J0868); the Health Science and Technology Project of Guangzhou (No. 20231A011047).