
Abstract
Cribriform morular thyroid carcinoma is a rare thyroid malignancy with uncertain histogenesis. It predominantly affects young women and is strongly associated with familial adenomatous polyposis. This paper reports a rare case of sporadic cribriform morular thyroid carcinoma in a female patient in her early 50s, with somatic genetic testing revealing a KMT2C mutation. She presented with a solitary lesion confined to the right thyroid lobe and had no family history of familial adenomatous polyposis. Colonoscopy and germline genetic testing revealed no abnormalities. This finding suggests a potential link between KMT2C mutations and sporadic cribriform morular thyroid carcinoma. The clinical and imaging manifestations of this malignancy lack specificity, and the final diagnosis depends on routine pathological examination and immunohistochemical analysis. This report indicates the need for the clinical investigation of family history and genetic testing, thus contributing to the clinical realization of standardized follow-up monitoring and management.
Introduction
Cribriform morular thyroid carcinoma is a rare thyroid malignancy predominantly affecting young women, with an average age of onset of 24 years and a female-to-male ratio as high as 61:1. 1 It is strongly associated with familial adenomatous polyposis (FAP) but may also present sporadically. Harach et al. 2 first described this tumor type in 1994; they characterized thyroid carcinomas exhibiting follicular, cribriform, solid, and papillary structural features as a distinct entity and revealed their correlation with FAP. Subsequently, in 1999, Cameselle-Teijeiro et al. 3 introduced the cribriform morular variant of papillary thyroid carcinoma. In 2017, the World Health Organization (WHO) classified the cribriform morular variant as a rare subtype of papillary thyroid carcinoma (PTC) in the WHO categorization of endocrine organ tumors. 4 However, in the 2022 fifth edition of the WHO Classification of Endocrine and Neuroendocrine Tumors, cribriform morular thyroid carcinoma was reclassified from a PTC subtype to a tumor of uncertain cell lineage. 5
To the best of our knowledge, there are currently no documented cases linking KMT2C mutations to sporadic cribriform morular thyroid carcinoma. Herein, we reported a case of sporadic cribriform morular thyroid carcinoma in a female patient with KMT2C mutation and reviewed relevant literature to deepen awareness of this rare disease.
Case presentation
A female patient in her early 50s was admitted to our hospital on 29 January 2024, after a right thyroid nodule was detected during a routine check-up a year ago. She exhibited no symptoms and did not undergo any treatment. She had neither received neck radiation nor had a family history of thyroid cancer. Upon admission, physical examination revealed that the neck was soft, and the trachea was centrally located. In the right thyroid lobe, a mass measuring approximately 2.5 cm × 1.5 cm was detectable by touch. The mass was firm, nontender, and not fixed to surrounding tissues; moreover, it moved during swallowing. No significant lymph node enlargement was detected in the cervical region.
In the middle of the right thyroid lobe, a hypoechoic nodule measuring 17 mm ×17 mm × 26 mm was detected via color Doppler ultrasound (Figure 1(a) and (b)). The nodule had unclear borders and irregular margins, with an elastography score of 3 (Figure 1(c)). Abundant blood flow signals were detected within and around the nodule via color Doppler flow imaging (Figure 1(d)). Neck computed tomography revealed a slightly hypodense nodule in the right thyroid lobe, measuring 2.4 cm ×1.6 cm, with punctate calcifications (Figure 1(e)) and heterogeneous postcontrast enhancement (Figure 1(f)). There was no extrathyroidal tumor extension, and no significant bilateral lymphadenopathy was observed. On 1 February 2024, the patient underwent right thyroid lobectomy and isthmectomy. Based on intraoperative frozen section analysis, the tumor was diagnosed as minimally invasive follicular carcinoma or hyalinizing trabecular tumor, prompting right central lymph node dissection.
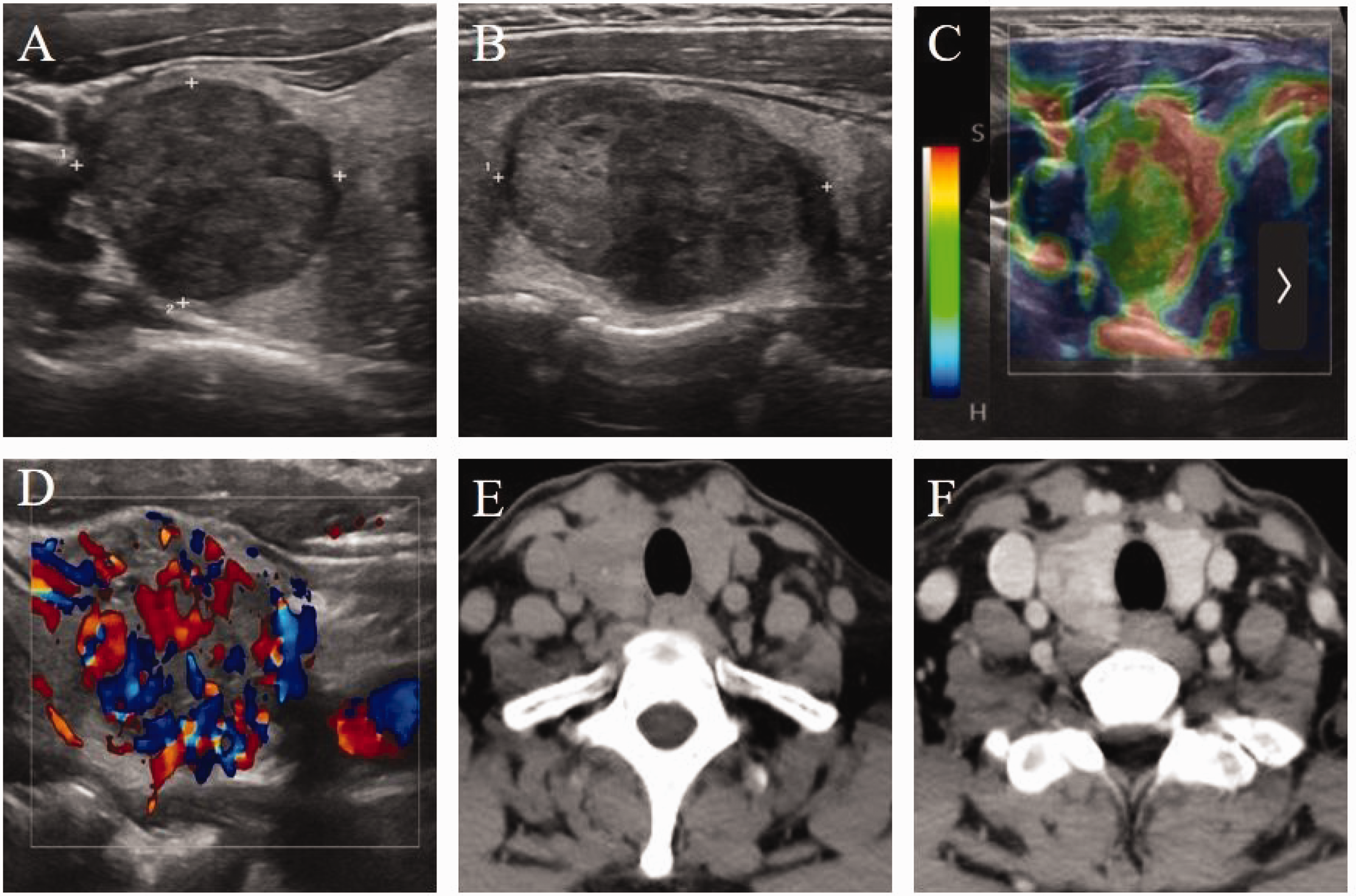
Ultrasound and computed tomography (CT) images. The right lobe contains a hypoechoic mass measuring 17 mm × 17 mm × 26 mm (a = transverse, b = sagittal). Strain elastography score = 3 (c). The hypoechoic mass indicated abundant blood flow signals in the right lobe (d). Neck CT in the right thyroid lobe revealed a slightly hypodense nodule, measuring 2.4 cm × 1.6 cm, with punctate calcifications (e) and heterogeneous postcontrast enhancement (f).
Postoperative pathology revealed the presence of a thyroid tissue specimen measuring 2.5 cm × 1.5 cm × 1.5 cm. The cut surface was gray-white to gray-red, solid, and firm, with a visible capsule and well-defined borders (Figure 2(a)). Microscopically, the tumor displayed follicular, tubular, cribriform, and solid nest growth patterns, with prominent morular structures and areas of hyaline degeneration (Figure 2(b) and (c)). Tumor cells showed mild-to-moderate atypia with nuclear crowding (Figure 2(d)). Vascular invasion (less than four foci) was observed; however, no capsular or extracapsular invasion, intraglandular dissemination, nerve invasion, or necrosis was identified. No metastasis was detected in the right central lymph nodes.
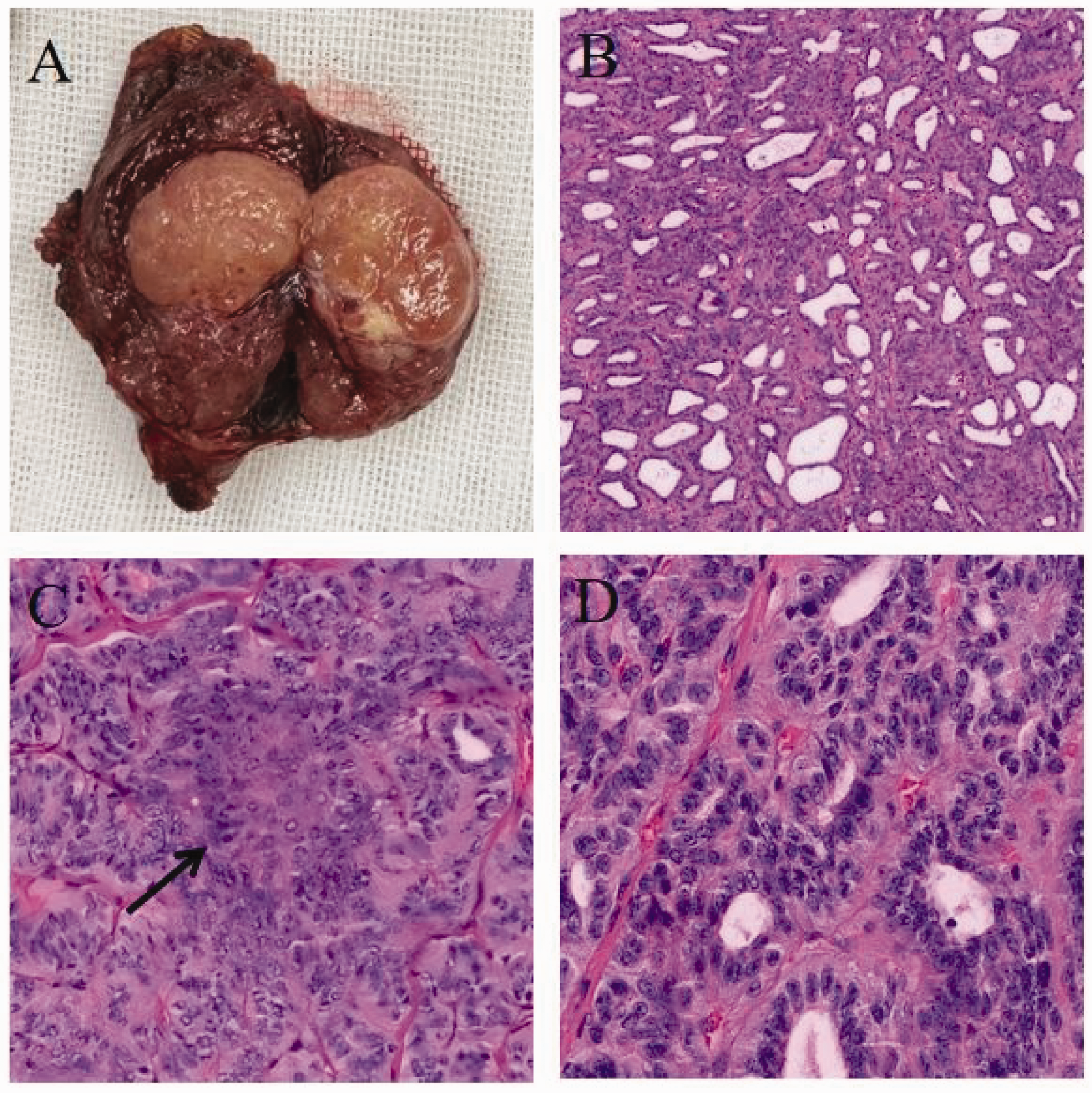
Postoperative specimen and pathology. Thyroid specimen measuring 2.5 cm × 1.5 cm × 1.5 cm. The cut surface was gray-white to gray-red, solid, and firm, with a visible capsule and well-defined borders (a). The tumor displayed follicular, tubular, cribriform, and solid nest growth patterns, with prominent morular structures and areas of hyaline degeneration (b). Morular structure (c). Tumor cells showed mild-to-moderate atypia with nuclear crowding (d).
Immunohistochemical staining revealed the following findings: β-catenin was positive in both nuclear and cytoplasmic regions (Figure 3(a)). PAX8 showed partial weak positivity. CgA, Sy, thyroglobulin (TG), CT, and parathyroid hormone (PTH) were negative. Ki-67 was positive (2%–3%). Thyroid transcription factor 1 (TTF1) (Figure 3(b)), estrogen receptor (ER) (Figure 3(c)), progesterone receptor (PR) (Figure 3(d)), and CK19 were positive in the cribriform area but negative in the morular area. CD56 was positive in a few cells. CDX2 (Figure 3(e)) was positive in the morular area but negative in the cribriform area. CD10 (Figure 3(f)) was focally positive in the cribriform area and positive in the morular area.

Immunohistochemical staining. β-Catenin was positive in both the nuclear and cytoplasmic regions (a). TTF1 (b), ER (c), and PR (d) were positive in the cribriform area but negative in the morular area. CDX2 was negative in the cribriform area but positive in the morular area (e). CD10 was focally positive in the cribriform area and positive in the morular area (f).
Germline genetic testing revealed no relevant mutations. Somatic genetic testing identified a TERT promoter mutation (c.-124C > T, C228T, mutation frequency: 12.06%) and a KMT2C exon-32 mutation (c.4664A > G, p.D1554G, mutation frequency: 53.36%). No gene fusions were detected in AFF3, AGGF1, AGK, AKAP13, APAP9, ALK, AP3B1, ATIC, BAIAP2L1, BCL2L11, BICC1, BRAF, CARS, CASP7, CAAR2, CCDC30, CCDC6, CCNY, CD74, CLTC, CREB3L2, DCTN1, EGFR, EML4, ERC1, ETV6, EZR, FAM114A2, FGFR1, RGFR2, FGFR3, FKBP15, FN1, GFPT1, GLIS1, GLTS3, GOLGA5, GOPC, HIP1, HOOK3, IGF2BP3, IRF2BP2, KIAA1217, KIAA1549, KIF5B, KLH17, KTN1, LMNA, LOC389473, LRIG3, LTK, MACF1, MET, MKRN1, MSN, NACC2, NCOA4, NPM1, NTRK1, NTRK2, NTRK3, OFD1, PAX8, PCM1, PLAG1, POR, PPARG, PPFIBP1, PPKAR1A, QKI, RAD51, RAF1, RANBP2, RBMS, RBPMS, RELCH, RET, RMDN3, RNF213, ROS1, SDC4, SEC31A, SEPTIN14, SHTN1, SLC26A11, SLC34A2, SND1, SPECCIL, SQSTM1, SSBP2, STRN, SYN2, TACC1, TACC3, TANK, UACA, TBL1XR1, TFG, THADA, TPM3, TRIM33, TPR, TRIM24, TRIM27, VCL, ZC3HAV1, and ZNF703. No mutations were detected in AKT1, ALK, APC, ATM, BANP, BRAF, CDK12, CDKN2A, CHEK2, CTNNB1, DICER1, FGFR, EIF1AX, EP300, ERBB4, EZH1, FAM193A, FARSB, FGFR1, FGFR2, FGFR3, FGFR4, FLT3, GLIS3, GNAQ, GNAS, HRAS, IDH1, IDH2, KIT, KLK1, KMT2D, KRAS, LRP1B, MEN1, MET, MTOR, NCOR2, NF1, NF2, NOTCH1, NRAS, NTRKS, PIK3CA, PLEKHS1, POR, PPARG, PTEN, PTH, RB1, RBM10, RET, ROS1, SMAD4, SPOP, STK11, SUGCT, TP53, TRIM61, TSC2, TSHR, VHL, and ZNF148.
The final diagnosis, based on pathological and immunohistochemical results, was cribriform morular thyroid carcinoma, staged as pT2N0M0. The patient was well-informed about her condition, and she actively participated in the treatment plan. She also engaged in the follow-up schedule, attending regular assessments and monitoring the treatment outcomes. After surgery, she received thyroid stimulating hormone (TSH)–suppressive levothyroxine therapy, maintaining serum TSH levels within the normal or low range. Given her negative thyroglobulin status and absence of lymph node metastasis, radioactive iodine therapy was not administered. The patient and her son were screened for FAP and underwent colonoscopy, which yielded negative results. Over the past 11 months, she has been regularly monitored using thyroid function tests, serum thyroglobulin, anti-thyroglobulin antibodies, and neck ultrasound, all of which showed no signs of recurrence or metastasis. Currently, the patient is being followed up.
This case has been reported following the Case Report (CARE) guidelines. 6 Publication of this case report was approved by the Ethics Committee of Zhejiang Cancer Hospital. As all patient data were anonymized, informed consent was deemed unnecessary.
Discussion
Cribriform morular thyroid carcinoma is an extremely rare thyroid malignancy, often presenting as painless thyroid enlargement or being incidentally detected during routine physical examinations. Most patients have normal preoperative thyroid function with nonspecific clinical symptoms.7,8 Ultrasound typically shows benign features, such as well-defined, oval, hypoechoic solid nodules, often without a hypoechoic halo or calcifications, 9 leading to frequent misdiagnosis as follicular thyroid tumor or nodular goiter.4,10 Definitive diagnosis relies on routine pathological examination and immunohistochemical analysis.
On gross pathological examination, cribriform morular thyroid carcinoma typically appears as a well-circumscribed, gray-white, firm, tough tumor with a capsule, usually without necrosis or hemorrhage but occasionally showing cystic areas. 1 Histologically, the tumor is enveloped by a thick fibrous capsule, with the possibility of invading the capsule and vasculature. It exhibits papillary, follicular, cribriform, trabecular, and solid growth patterns, along with concentric morular structures resembling squamous metaplasia but lacking keratin pearls or intercellular bridges. Papillae are lined by columnar cells, whereas cribriform regions comprise tall columnar cells arranged in a pseudostratified pattern, with lumina lacking colloid. Although mitotic figures are absent and psammoma bodies are rare, tumor cell nuclei may exhibit features of papillary carcinoma, including nuclear crowding, overlapping, grooves, and intranuclear pseudoinclusions.1,7 In this case, the tumor exhibited the classical gross features of cribriform morular thyroid carcinoma. However, intraoperative frozen section analysis suggested a diagnosis of minimally invasive follicular carcinoma or hyalinizing trabecular tumor, leading to an underestimated diagnosis. This highlights the need for clinicians and pathologists to enhance their understanding of this rare tumor.
Immunohistochemical staining plays a critical role in diagnosing cribriform morular thyroid carcinoma, with strong nuclear and cytoplasmic β-catenin expression being a hallmark. 1 Tumor cells typically show positive expression for TTF1, ER, and PR, whereas TG is partially positive or negative, calcitonin is negative, and the Ki-67 proliferation index is usually <5%. 11 Morular structures exhibit positive expression for CD5, CD10, CDX2, CK5, galectin-3, E-cadherin, and B-cell lymphoma 2, but are negative for TTF1, TG, ER, and PR. CDX2 and CD10 are key markers for identifying morular structures.4,7,11–14 In this case, β-catenin was strongly positive in the nucleus and cytoplasm, TG was negative, and the Ki-67 index was 2%–3%. Nonmorular tumor cells were positive for TTF1, ER, PR, and CK19, whereas morular areas showed positive expression for CDX2 and CD10. These findings are consistent with the literature.
Cribriform morular thyroid carcinoma is characterized by distinct molecular and genetic features. 15 It is associated with activation of the WNT/β-catenin signaling pathway, driven by germline or somatic APC mutations, or biallelic inactivation of genes such as CTNNB1 or AXIN1. 1 Unlike PTC and follicular-derived tumors, cribriform morular thyroid carcinoma primarily affects young women, likely due to interactions between ER, PR, and the WNT/β-catenin pathway, 16 suggesting a potential avenue for endocrine therapy that warrants further investigation. Mutations in the mitogen-activated protein kinase pathway (e.g. BRAF and RAS), which are common in PTC, are rare in cribriform morular thyroid carcinoma,11,17 highlighting its molecular distinction from PTC and justifying its reclassification as a separate entity. In this case, considering that this patient in her 50s has been postmenopausal for 10 years, female hormones may not have played a role in the progression of her cancer. Genetic analysis revealed no mutations in BRAF, RAS, or PIK3CA, further confirming the unique molecular characteristics of cribriform morular thyroid carcinoma compared with other thyroid cancers. Meanwhile, no APC and CTNNB1 mutations were detected in the genetic test. However, the patient did not undergo AXIN1 testing. Given the association of AXIN1 with the WNT/β-catenin pathway, we cannot rule out the potential impact of mutations in this gene on the patient's condition.
Differentiating familial from sporadic cribriform morular thyroid carcinoma is essential for effective clinical monitoring. Familial cribriform morular thyroid carcinoma typically presents as multifocal tumors involving both thyroid lobes, whereas sporadic cribriform morular thyroid carcinoma usually manifests as a solitary nodule confined to a single lobe. 18 Patients suspected of familial cribriform morular thyroid carcinoma should undergo early screening and proactive follow-up for FAP. FAP, an autosomal dominant condition caused by mutations in the APC gene, leads to activation of the WNT/β-catenin signaling pathway, promoting tumorigenesis. 1 Approximately 50% of cribriform morular thyroid carcinoma cases are associated with FAP, with most presenting initially as cribriform morular thyroid carcinoma, although sporadic cases also occur. 3 Studies by Cameselle-Teijeiro et al. 19 and Aydemirli et al. 20 have reported APC mutations in sporadic cribriform morular thyroid carcinoma. Therefore, both familial and sporadic cribriform morular thyroid carcinomas are known to commonly exhibit mutations in APC. The distinction is that in FAP and familial cribriform morular thyroid carcinoma, APC mutations are germline, leading to loss or dysfunction of the APC protein. In contrast, sporadic cribriform morular thyroid carcinoma results from somatic mutations in APC, CTNNB1, AXIN1, and/or KMT2D. 11 Consequently, germline APC mutation testing is considered the gold standard for diagnosing familial cribriform morular thyroid carcinoma. In the current case, the patient presented with a solitary lesion confined to the right thyroid lobe and had no family history of FAP. Colonoscopy and germline genetic testing revealed no abnormalities, effectively ruling out FAP and indicating the presence of sporadic cribriform morular thyroid carcinoma.
KMT2C is a member of the KMT2 gene family, and mutations in this gene are closely associated with various tumors. In Table 1, we summarized the potential mechanisms of KMT2C mutations in common cancers, including breast cancer, 21 lung cancer, 22 colorectal cancer, 23 and nervous system tumors.24–26 Additionally, Nieminen et al. 27 investigated the coexistence of somatic mutations in KMT2D and KMT2C in cases of cribriform morular thyroid carcinoma associated with FAP. They proposed that these mutations act as drivers of thyroid cancer by promoting tumorigenesis through the WNT signaling pathway. In our case of sporadic cribriform morular thyroid carcinoma, we tested several genes, including APC and KMT2D. Although both genes were negative, a somatic mutation in KMT2C was identified. This report represents the first description of a somatic mutation in KMT2C in a sporadic case of cribriform morular thyroid carcinoma. The detection of a somatic KMT2C mutation in this patient suggests that it may be linked to sporadic cribriform morular thyroid carcinoma, with a potential mechanism involving the WNT/β-catenin pathway. 27 Although the detection of a single case has limitations, this finding provides valuable insights for further investigation into the role of KMT2C in the pathogenesis of sporadic cribriform morular thyroid carcinoma. However, further research is needed to confirm its widespread applicability.
KMT2C mutation associations with other cancers.

Current treatment for cribriform morular thyroid carcinoma largely follows the guidelines for differentiated thyroid carcinoma. 28 In cases of sporadic cribriform morular thyroid carcinoma without high-risk factors, hemithyroidectomy is sufficient and acceptable. However, high-risk cases, including high-grade or extensively invasive tumors, extensive lymphovascular invasion, or extrathyroidal extension, require total thyroidectomy. For FAP-associated cribriform morular thyroid carcinoma, total thyroidectomy is mandatory due to the frequent presence of multicentricity and bilaterality. Lymph node dissection is performed when necessary, and radioactive iodine therapy may be used to address residual lesions.11,29 Cribriform morular thyroid carcinoma has an initial lymph node metastasis rate of 12%, 4 with distant metastases being rare, although lung metastases have been reported. 30 Overall, the prognosis is generally favorable, with low recurrence and mortality rates after surgical resection.4,7 The patient in the current case presented with a solitary lesion in a single thyroid lobe, staged as T2N0M0. She underwent right lobectomy, isthmectomy, and right central lymph node dissection. Postoperative follow-up over 11 months revealed no recurrence or metastasis. However, the overall follow-up period remains relatively short compared with the median survival time for thyroid cancer, necessitating attention to long-term follow-up outcomes.
In conclusion, we present a rare case of sporadic cribriform morular thyroid carcinoma in a middle-aged female patient, with somatic testing revealing a KMT2C mutation. This finding suggests a potential link between KMT2C mutation and sporadic cribriform morular thyroid carcinoma, highlighting the importance of genetic analysis in the diagnosis of this rare malignancy. For the diagnosis and management of cribriform morular thyroid carcinoma, especially in cases with nonspecific clinical and imaging characteristics, pathological morphology and immunohistochemical staining remain crucial to prevent underestimation by preoperative ultrasound or intraoperative frozen section analysis. However, relying solely on these diagnostic methods may be insufficient for accurate diagnosis and prognosis, particularly as genetic mutations increasingly play a key role in predicting cancer progression. This case underscores the need for clinicians and pathologists to enhance their understanding of the clinicopathological features, treatment, and prognosis of this rare malignancy, as outlined in the updated WHO classification, and to consider genetic testing as part of a comprehensive diagnostic and management strategy. We recommend incorporating family history evaluation and genetic testing in routine clinical practice to guide follow-up and management of patients with cribriform morular thyroid carcinoma.
Footnotes
Acknowledgement
None.
Author contributions
YZG collected and analyzed clinical data and drafted the manuscript. JJX and JWD contributed to surgical management, data collection, and critical revisions. JHC contributed to drafting and editing the manuscript. JWD and WDD diagnosed the case, reviewed the first draft, and prepared the final draft of the manuscript. All authors reviewed and approved the final version.
Data availability
Data are available from the corresponding author upon reasonable request.
Declaration of conflicting interests
The authors declare no financial or nonfinancial conflicts of interest.
Funding
No funding, grants, or other support were received for the preparation of this manuscript.