
Abstract
Objectives
Most diffuse gliomas are reported to harbor isocitrate dehydrogenase (IDH) mutations. However, when these mutations are tested in clinical practice, the results are often negative.
Methods
This study examined the frequency of IDH1 and IDH2 mutations in gliomas classified according to the revised 2016 World Health Organization classification, and investigated their prognostic relevance. We tested 87 gliomas for IDH1 and IDH2 mutations using the peptide nucleic acid clamp method.
Results
IDH1 mutations were observed in 42% of diffuse astrocytomas, 23% of anaplastic astrocytomas, all oligodendrogliomas and anaplastic oligodendrogliomas, and 17% of glioblastomas. An IDH2 mutation was identified in one case of diffuse astrocytoma. In the survival analysis of diffuse astrocytic tumors, patients with IDH1/2-wildtype anaplastic astrocytomas tended to have a poor prognosis, similar to that of glioblastomas.
Conclusions
IDH2 mutations were infrequent in gliomas. In anaplastic astrocytomas, the frequency of IDH1/2-wildtype was relatively high, and the prognosis of patients with this type of tumor was very similar to that of those with glioblastomas. It may therefore be necessary to reconsider the classification and treatment strategies for IDH1/2-wildtype anaplastic astrocytomas.
Introduction
In the revised 2016 edition of the World Health Organization (WHO) classification of tumors, molecular parameters, and in particular the genetic status of isocitrate dehydrogenase 1 (IDH1) and isocitrate dehydrogenase 2 (IDH2), became essential criteria for the diagnosis of diffuse astrocytic and oligodendroglial tumors. 1 Most diffuse gliomas are reported to harbor IDH mutations. 2 However, in our experience, when IDH mutations are tested using immunohistochemistry or molecular studies during routine diagnostic processes, the results are negative more often than expected, even in lower grade diffuse gliomas in adults. This raises the question of whether there is a difference in the clinical IDH mutation rate from that of the published literature. Regional or racial differences may contribute to this difference. It may be that, in Asia in particular, the distribution of IDH mutations differs from the frequency described in the WHO classification. Another possibility is that these differences arise because only IDH1 mutations are currently widely tested, while IDH2 mutations are not.
In the present study, we aimed to examine the status of IDH1 and IDH2 mutations in diagnosed cases of diffuse astrocytomas, anaplastic astrocytomas, oligodendrogliomas, and glioblastomas, and to estimate the frequencies of the two mutations in these tumors according to the revised 2016 WHO classification. Furthermore, we aimed to explore any new pathological and clinical features associated with these mutations.
Materials and methods
Patients
We included cases diagnosed as diffuse astrocytoma, anaplastic astrocytoma, oligodendroglioma, anaplastic oligodendroglioma, or glioblastoma at St. Vincent’s Hospital, Korea between 2001 and 2018. Pilocytic astrocytoma and pleomorphic xanthoastrocytoma, which were classified as other astrocytic tumors in the revised fourth edition of the WHO classification of tumors of the central nervous system, 1 were excluded from the study. The tumors were either excision specimens or stereotactic biopsy specimens.
Existing hematoxylin and eosin-stained slides, immunohistochemically stained slides, and molecular data were reviewed by two expert pathologists (C.Y.Y. and U.C.) using the new definition criteria from the WHO classification. 1 Hematoxylin and eosin staining was performed on 4 µm-thick formalin-fixed paraffin-embedded tissue sections. First, the tissue sections were dewaxed with xylene and hydrated through alcohol and water. Rehydrated sections were then stained with hematoxylin solution for 30 minutes, washed in tap water, and differentiated with 70% ethanol and 1% HCl solution for 5 s. The tissue sections were washed again in tap water. Next, the sections were stained by applying eosin for 10 minutes. The slides were then passed through changes of alcohol before being cleared in xylene baths.
Clinical information was collected from the medical records of the selected cases. Written informed consent was received from all patients. This study was approved by the St. Vincent’s Hospital Institutional Review Board (VC18SESI0160).
IDH1 and IDH2 mutation analysis
The IDH1 and IDH2 mutation analysis was performed using peptide nucleic acid (PNA)-mediated real-time polymerase chain reaction (PCR). The PNAClamp IDH1 Mutation Detection Kit (Panagene, Inc., Daejeon, Korea) used for these experiments detects five hotspot mutations in codon 132 of the IDH1 gene. According to the manufacturer’s protocol, DNA was extracted from formalin-fixed paraffin-embedded tissue using a Maxwell® DNA Purification Kit (Promega, Madison, WI, USA). The target DNA samples were prepared at a concentration of 10 to 25 ng/μL in a total volume of 7 μL by adding distilled water. Each reaction contained template DNA, a primer and PNA probe set, and SYBR Green PCR Master Mix (Kapa Biosystems, Woburn, MA, USA), which were included in the kit. Real-time PCR was performed using a CFX96 (Bio-Rad, Hercules, CA, USA). Five minutes of pre-denaturation at 94°C was followed by 40 cycles of denaturation at 94°C for 30 s, PNA clamping at 70°C for 20 s, annealing at 63°C for 30 s, and extension at 72°C for 30 s. The PNA probe was complementary to the wildtype sequence and suppressed the amplification of the wildtype target. This suppression induced the preferential amplification of mutant sequences by competitively inhibiting the binding of DNA primers to the wildtype sequence. The test results were determined following the manufacturer’s protocol, which has been previously described. 3 The experimental protocol and the interpretation of the results were the same for the IDH2 mutation analysis using the PNAClamp IDH2 Mutation Detection Kit (Panagene, Inc.), which detects 11 hotspot mutations in codons 140 and 172 of the IDH2 gene.
Statistical analysis
Statistical analysis was performed using IBM SPSS Statistics for Windows, version 21.0 (IBM Corp., Armonk, NY, USA). A chi-squared test or Fisher’s exact test was applied to assess the possible associations between qualitative variables. Overall survival was defined as the time between diagnosis and death or the last follow-up visit. Kaplan–Meier survival analysis was performed, and the survival curves were compared using the log-rank test. A two-sided p-value of less than 0.05 was considered significant when comparing two survival curves. When multiple survival curves were compared, a p-value less than the Bonferroni-corrected threshold (i.e., 0.017) was considered significant.
Results
The study cohort included 87 patients with a male to female ratio of 0.98. Patient age ranged widely, from 8 to 79 years. However, gliomas occurred most frequently in individuals between the ages of 40 and 60 years, regardless of the histological type. Of the 87 cases, there were 24 diffuse astrocytomas, 22 anaplastic astrocytomas, four oligodendrogliomas, one anaplastic oligodendroglioma, and 36 glioblastomas (Table 1). Fifty-six tumors were excision specimens (13 diffuse astrocytomas, 14 anaplastic astrocytomas, three oligodendrogliomas, one anaplastic oligodendroglioma, and 25 glioblastomas), and 31 tumors were stereotactic biopsy specimens (11 diffuse astrocytomas, 8 anaplastic astrocytomas, 1 oligodendroglioma, and 11 glioblastomas). In terms of treatments, various treatment options were applied depending on each individual patient’s clinicopathological condition as well as their diagnosis. Many patients with glioblastomas received chemoradiation therapy after surgery, whereas patients with anaplastic astrocytomas received more diverse treatments, which depended on the completeness of the tumor removal, symptoms, and progression status (Table 1).
Clinical features of the patients and IDH1/2 mutations.

DA, diffuse astrocytoma; AA, anaplastic astrocytoma; OD, oligodendroglioma; AO, anaplastic oligodendroglioma; GB, glioblastoma.
1Percentage within each diagnosis.
2Chemotherapy included combination therapy (procarbazine, lomustine, and vincristine), carmustine, lomustine, and temozolomide.
Histologically, diffuse astrocytomas showed fibrillary astrocytic proliferation, and no mitosis or nuclear atypia was observed. In anaplastic astrocytomas, substantial astrocytic proliferation was observed, as were mitosis and cellular atypia. No necrosis or microvascular proliferation was observed in anaplastic astrocytomas. Glioblastomas had high tumor cell density, cellular pleomorphism, nuclear atypia, and frequent mitosis. Necrosis and microvascular proliferation were also observed in these tumors. In oligodendrogliomas, the proliferation of monomorphic cells with uniform round nuclei was characteristic; these cells often contained a perinuclear halo. Fine branching capillaries and calcification were also frequently observed histological features of oligodendrogliomas. In anaplastic oligodendrogliomas, nuclear atypia was notable and was accompanied by mitotic figures, necrosis, and microvascular proliferation. These histological findings were identical in both IDH-mutated and IDH-wildtype cases (Figure 1).

Micrographs of representative IDH-mutant and -wildtype gliomas. The histological findings of IDH-mutant (a, c, e) and IDH-wildtype (b, d, f) tumors were not different in diffuse astrocytomas (a, b), anaplastic astrocytomas (c, d), or glioblastomas (e, f). IDH-wildtype anaplastic astrocytomas exhibited many mitoses and nuclear pleomorphisms but lacked microvascular proliferation and necrosis in the whole field (hematoxylin and eosin stain, original magnification ×200).
IDH1 mutations were observed in 10 out of 24 (42%) diffuse astrocytomas. Among the 22 anaplastic astrocytomas, IDH1 mutations were observed in only 5 (23%) cases, while 17 (77%) cases were wildtype. IDH1 mutations were observed in all oligodendroglioma cases (4/4, 100%) and in the single case of anaplastic oligodendroglioma (1/1, 100%). Thirty out of 36 (83%) glioblastomas were IDH1-wildtype, while the other 6 cases (17%) harbored IDH1 mutations. When the IDH2 mutation test was performed, only 1 case of diffuse astrocytoma harbored an IDH2 mutation, while the other 86 tumor cases had no such mutations (Table 2). All pediatric patients (patients under the age of 20 years) had IDH1/2-wildtype tumors (Table 1). When the pediatric patients were excluded, 33% (27/82) of the patients with gliomas had IDH1/2-mutated tumors, and 26% (5/19) of the patients with anaplastic astrocytomas had IDH1/2-mutated tumors. Overall, 32% (18/56) of all excision specimens and 28% (9/31) of all biopsy specimens had IDH1/2 mutations. Specimen type did not correlate with mutation status. Among the IDH1/2-wildtype diffuse and anaplastic astrocytomas, 54% (7/13) and 71% (12/17), respectively, were biopsied samples. The specimen type and IDH1/2 mutation had no significant correlations in either type of tumor.
Frequencies of IDH1 and IDH2 mutations in glioma.

DA, diffuse astrocytoma; AA, anaplastic astrocytoma; OD, oligodendroglioma; AO, anaplastic oligodendroglioma; GB, glioblastoma.
We performed an overall survival analysis according to the IDH1/2 mutation status. The median follow-up duration of the study cohort was 21.7 months (range, 1–233.9 months), and 26 (30%) patients died during the follow-up period. The 2-year and 5-year overall survival rates were 71% and 59%, respectively. The 5-year overall survival rate was 73% in diffuse astrocytoma patients, 65% in anaplastic astrocytoma patients, and 34% in glioblastoma patients. All patients with oligodendrogliomas survived during the follow-up period (range, 11.7–146.6 months). The patient with anaplastic oligodendroglioma died 122.7 months after the initial diagnosis.
We next compared the survival curves between the different groups of patients with diffuse astrocytic tumors. According to the survival analysis, patients with anaplastic astrocytomas exhibited no significant differences in overall survival compared with glioblastoma patients (5-year survival rate 65% vs. 34%) (Figure 2a). Patients with IDH1/2-mutated diffuse astrocytomas had a more prolonged overall survival than patients with IDH1/2-wildtype diffuse astrocytomas (5-year survival rate 86% vs. 41%, p = 0.042) (Figure 2b). In lower-grade astrocytic tumors (i.e., diffuse or anaplastic astrocytomas; grades 2 and 3), IDH1/2-mutated cases had better overall survival than IDH1/2-wildtype cases (5-year survival rate 88% vs. 57%, p = 0.016) (Figure 2c). There was no significant difference in overall survival between IDH1/2-mutated and -wildtype cases in the combined group of anaplastic astrocytomas and glioblastomas (grades 3 and 4) (5-year survival rate 45% vs. 50%). There was also no significant difference in survival curves between the IDH1/2-mutated and -wildtype glioblastoma cases.
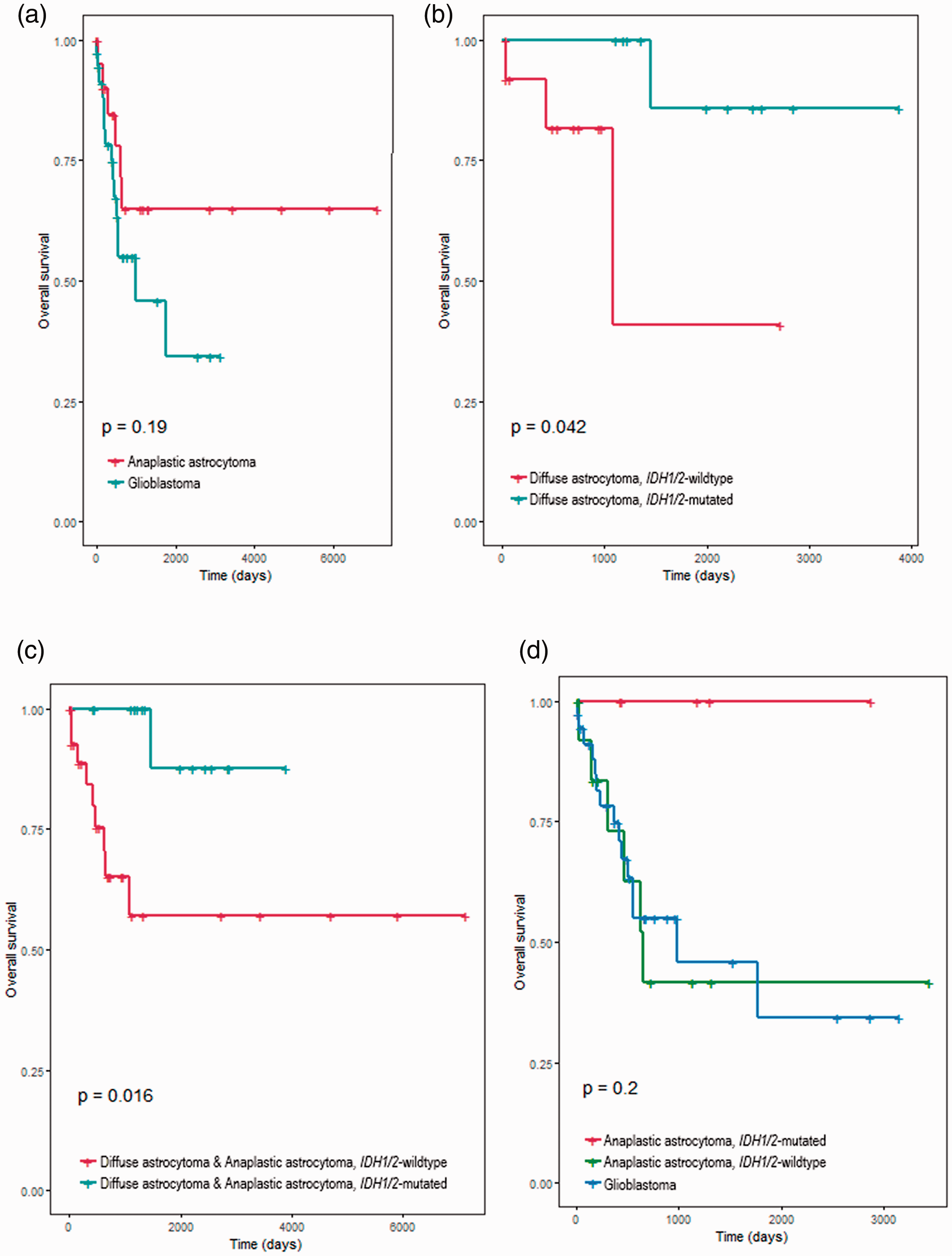
Kaplan–Meier curves comparing the overall survival of the diffuse astrocytic tumors when the different groups were compared according to their histological type and IDH mutation status. (a) Patients with anaplastic astrocytomas did not have significantly different overall survival compared with those with glioblastomas. (b) In patients with grade 2 diffuse astrocytomas, those with tumors harboring IDH1 or IDH2 mutations had better overall survival than those with wildtype tumors (p = 0.042). (c) IDH1/2 mutation status also had a prognostic impact on lower-grade diffuse astrocytic tumors; IDH1/2-mutated tumors had better survival (p = 0.016). (d) We performed multiple comparisons between the survival curves of adult patients with IDH1/2-mutated anaplastic astrocytomas, IDH1/2-wildtype anaplastic astrocytomas, and glioblastomas. The survival curves of the three groups showed no significant differences, although patients with IDH1/2-mutated anaplastic astrocytomas seemed to have a better prognosis than the other patients.
Finally, we excluded all pediatric gliomas and performed a subgroup analysis of the anaplastic astrocytomas according to IDH mutation status. These were compared with glioblastomas as the control group. All 11 patients with IDH1/2-mutated anaplastic astrocytomas survived for more than 5 years, whereas only 42% of patients with IDH1/2-wildtype anaplastic astrocytomas survived for this period. However, there was no significant difference between the survival curves of the two groups. Patients with IDH1/2-mutated anaplastic astrocytomas tended to have better overall survival than patients with glioblastomas (5-year survival rate 100% vs. 34%), but this difference was not significant (Figure 2d). When we compared patients with IDH1/2-wildtype anaplastic astrocytomas with patients with glioblastomas, there was also no significant difference between the two groups (5-year survival rate 42% vs. 34%) (Figure 2d).
Discussion
The classification of central nervous system tumors was based primarily on histological findings until the publication of the 2007 WHO classification. 4 The revised WHO classification (2016) 1 introduced a modified classification system for diffuse astrocytic and oligodendroglial tumors. In the modified system, molecular parameters, and especially IDH1 and IDH2 mutation status, became an essential foundation for the classification of such tumors. IDH-mutant and IDH-wildtype gliomas have different tumor development processes and therefore respond differently to different therapies. 2 According to the revised taxonomy, tumors that are histologically classified as astrocytomas, oligodendrogliomas, and glioblastomas can then be divided into IDH-mutant and IDH-wildtype subgroups according to their IDH mutation status. If an IDH-mutant glioma has a 1p/19q co-deletion, they are further classified as oligodendroglioma or anaplastic oligodendroglioma, IDH-mutant, and 1p/19q co-deleted. 1
Studies on the frequency of IDH1 and IDH2 mutations have reported that most diffuse gliomas are of the IDH-mutated type. 1 It is generally considered that approximately 80% of low-grade gliomas and secondary glioblastomas have IDH mutations. 5 According to a report in an Asian population, however, IDH1 mutations account for 55.3% of all diffuse glioma cases, IDH2 mutations account for 2.2% of such cases, and wildtype IDH1/2 accounts for 42.6% of such cases. 6 IDH has three forms; IDH1 plays a leading role in the development of gliomas, 6 but IDH2 may also be associated with glioma development.5,7 These two mutations are mostly exclusive and very rarely coincide. In most cases with IDH1 mutations, amino acid substitutions occur at R132H, and this alteration accounts for most of the IDH alterations that occur in gliomas. In IDH2, amino acid substitutions occur at R172K, and the frequency of IDH2 mutations is variable but generally low (0.3%–5.2%) in all cases of glioma,8–14 if not absent. 15
In our experience, however, IDH mutation-negative tumors are often encountered in routine pathology in clinical practice, when the presence or absence of these mutations is tested by immunohistochemical or molecular studies. This experience is somewhat different from that of published findings. Although the way in which the testing is performed may cause this difference, it may also be because only IDH1 mutations are currently examined, whereas IDH2 mutations are not routinely tested. Another possibility involves regional and/or racial differences in the IDH mutation rate. For example, in the Asian population, the mutational landscape may differ from the mutation frequencies that are presented in the WHO classification. In the present study, the frequency of IDH mutations was markedly different from that reported in the revised 2016 WHO classification. 1 Notably, the frequency of IDH1/2-wildtype anaplastic astrocytomas (77%) was much higher than the 20% described in the 2016 WHO classification. 1 The frequency (74%) remained higher than expected even when pediatric patients were excluded. A few previous studies performed in Asian populations have reported the frequency of IDH1/2-wildtype anaplastic astrocytomas, and these frequencies were the same or higher than those of IDH1/2-mutated anaplastic astrocytomas: 64.1% (25/39, Chinese), 6 50% (5/10, Korean), 16 57.1% (8/14, Chinese), 12 and 72% (21/29, Japanese). 17 The underdiagnosis or false-negative mutation results caused by undersampling may have affected these numbers. The inclusion of pediatric gliomas in the present study might have raised the rate of IDH1/2-wildtype anaplastic astrocytomas. Nevertheless, our data and those of previous studies demonstrate that IDH1/2-wildtype anaplastic astrocytomas may be much more prevalent than what is conventionally reported, and more attention and emphasis on this point are needed to improve the treatment of patients.
In contrast to the results from the anaplastic astrocytomas, all oligodendrogliomas harbored IDH1 mutations, while IDH1/2-wildtype glioblastomas were more common than IDH1/2-mutant glioblastomas (69% vs. 31%) in our study, which is in agreement with known findings. 1 Contrary to our expectations, we identified an IDH2 mutation in only one case of diffuse astrocytoma; interestingly, this patient was the only non-Asian white patient in our cohort. A few studies have reported relatively high frequencies of IDH2 mutations in gliomas: 5.2% (7/134) 13 and 4.5% (3/67). 14 However, our experimental results are more in line with other previous papers that have reported lower frequencies: 0.3%, 6 0.9%, 9 2.8%, 10 3.1%, 11 and 3.7%. 12 Reliable figures for IDH2 mutation frequency in gliomas are expected to be revealed in future studies containing more clinical cases or through a meta-analysis.
In the present study, patients with IDH1/2-wildtype anaplastic astrocytomas had a similar prognosis to that of patients with glioblastomas. Although the prognostic difference between IDH1/2-mutated and IDH1/2-wildtype anaplastic astrocytomas and glioblastomas was not significant, the number of patients in this study was relatively small, with low statistical power. However, a similar result was published in a previous study. At an institution where patients with anaplastic astrocytomas and glioblastomas received the same therapy, patients with IDH-wildtype anaplastic astrocytomas did not exhibit better prognosis than patients with glioblastoma. 18 These authors suggested that the histological grading criteria of “necrosis” and “vascular proliferation” lose their prognostic significance in the absence of IDH mutations. Therefore, if tumors that have been histologically classified as anaplastic gliomas, and especially anaplastic astrocytomas, are IDH-wildtype, it may be worth reconsidering whether these tumors should be diagnosed as IDH-wildtype anaplastic astrocytomas or as IDH-wildtype glioblastomas through meticulous microscopic examination. The treatment strategy for IDH-wildtype astrocytomas should also be reconsidered.
The 2016 WHO classification of CNS tumors is a “provisional” classification, and IDH1/2-wildtype gliomas have remained a heterogeneous group of tumors that need to be further studied for classification. Although not as much is known about them compared with IDH-mutant gliomas, the molecular and pathological characteristics of IDH-wildtype lower grade gliomas are being discovered. Not all IDH1/2-wildtype lower grade gliomas exhibit poor prognosis. Studies have shown that epidermal growth factor receptor (EGFR) amplification, telomerase reverse transcriptase (TERT) promoter mutation, the gain of chromosome 7, and the loss of chromosome 10q are all associated with the prognosis of these tumors.19,20 As more knowledge is gained about the molecular types with poor prognosis among IDH1/2-wildtype tumors, the cIMPACT-NOW (Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy) has suggested diagnostic criteria to classify them as “diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV.” 21 Our results support the need for a new molecular classification of IDH1/2-wildtype gliomas, and especially anaplastic astrocytomas, and suggests that there may be more of these tumors than the known prevalence, depending on the population.
Small patient numbers and patient heterogeneity are the major limitations of the present study, and it is hard to draw a strong conclusion with such limitations. A greater number of cases and an in-depth analysis to search for molecular signatures are needed.
In conclusion, the clinical frequency of IDH1 mutations may differ from that reported in the literature and may need to be reviewed in each country or region. IDH2 mutations do not appear to be frequently observed in glioma. However, because the IDH2 mutation frequency is low or not observed and the IDH gene mutation profile is clinically critical in glioma, it is necessary to test IDH2 along with IDH1 in glioma. Anaplastic astrocytomas had a higher frequency of IDH1/2-wildtype than was expected, and the prognosis of patients with this tumor type was similar to that of patients with glioblastoma. Therefore, a new IDH1/2-wildtype anaplastic astrocytoma classification is needed, and further study is required to better understand this entity.