
Abstract
Hemorrhagic transformation (HT) is a severe complication occurring in ischemic stroke patients undergoing tPA thrombolytic therapy, which significantly limits its clinical applicability. The mechanism and intervention of HT is still not fully understood. In our study, we found that an increased mobilization of circulating formyl peptide receptor 1 (FPR1) expressing leucocytes into the ischemic brain after tPA treatment in mice. In Fpr1−/− mice, neutrophil mobilization and HT occurrence after tPA thrombolysis decreased. Notably, pharmacological inhibition of FPR1 using a novel antagonist T0080 effectively mitigated tPA-associated HT, concurrently reducing neutrophil infiltration into the brain and preserving blood–brain barrier (BBB) integrity. We further revealed that brain infiltration neutrophils facilitate BBB leakage and neuron death by producing cytotoxic molecules such as reactive oxygen species (ROS), matrix metalloproteinase-9 (MMP9), and tumor necrosis factor-alpha (TNF-α). Neutrophil depletion and adoptive transfer experiments in vivo with FPR1+ neutrophils demonstrate that the essential role of FPR1+ neutrophils in mediating HT post-tPA administration in mice. Collectively, these findings identify neutrophils FPR1 activation as a key mechanistic driver exacerbating HT following tPA thrombolysis in ischemic stroke.
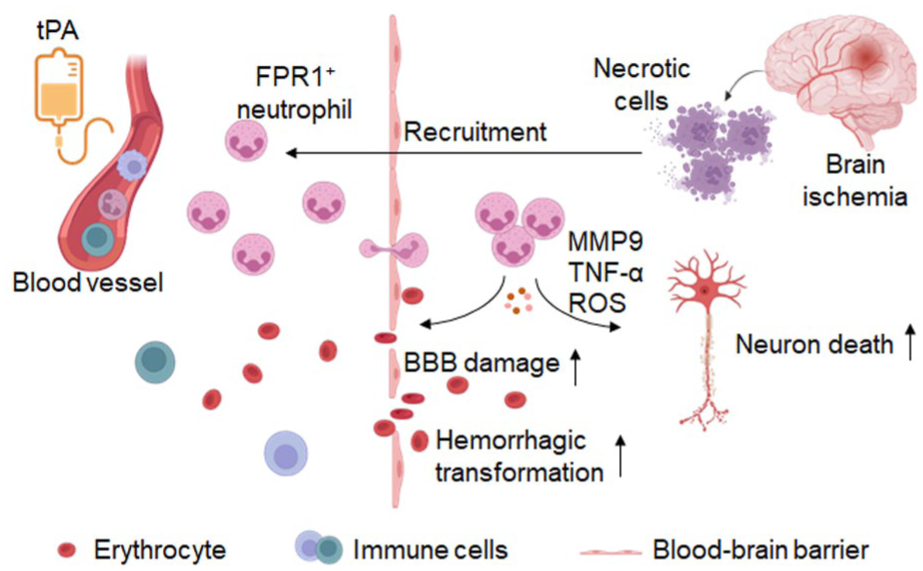
tPA triggers the mobilization of peripheral immune cells—particularly FPR1+ neutrophils—which migrate into the ischemic brain. These FPR1+ neutrophils amplify blood-brain barrier (BBB) disruption and neuronal injury through the release of cytotoxic factors, such as MMP-9, TNF-α, and reactive oxygen species (ROS). This detrimental cascade exacerbates BBB dysfunction and brain parenchymal damage, consequently elevating the risk of hemorrhagic transformation.
Keywords
Get full access to this article
View all access options for this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.