
Abstract
Blood-brain barrier (BBB) dysfunction occurs in numerous central nervous system disorders. Unfortunately, a limited understanding of the mechanisms governing barrier function hinders the identification and assessment of BBB-targeted therapies. Previously, we found that non-muscle myosin light chain kinase (nmMLCK) negatively regulates the tight junction protein claudin-5 in brain microvascular endothelial cells (BMVECs) under inflammatory conditions. Here, we used complementary animal and primary cell co-culture models to further investigate nmMLCK and claudin-5 during neuroinflammation. We found that nmMLCK-knockout mice resisted experimental autoimmune encephalomyelitis (EAE), including paralysis, demyelination, neutrophil infiltration, and BBB dysfunction. However, transiently silencing claudin-5 culminated in a fulminant disease course. In parallel, we found that neutrophil-secreted factors triggered a biphasic loss in the barrier quality of wild-type BMVEC monolayers, plus pronounced neutrophil migration during the second phase. Conversely, nmMLCK-knockout monolayers resisted barrier dysfunction and neutrophil migration. Lastly, we found an inverse relationship between claudin-5 expression in BMVECs and neutrophil migration. Overall, our findings support a pathogenic role for nmMLCK in BMVECs during EAE that includes BBB dysfunction and neutrophil infiltration, reveal that claudin-5 contributes to the immune barrier properties of BMVECs, and underscore the harmful effects of claudin-5 loss during neuroinflammation.
Keywords
Introduction
The blood-brain barrier (BBB) is a vital defender of the central nervous system (CNS). 1 Unfortunately, BBB dysfunction occurs in numerous CNS disorders, including multiple sclerosis (MS), for which it may play a key role in pathogenesis.2 –4 Thus, a deeper understanding of the mechanisms controlling BBB function could offer new insight into CNS disorders and identify BBB-specific therapies to preserve or restore its integrity in patients.
Neutrophils are early responders to infection and injury. 5 Patients with MS have elevated neutrophil-to-lymphocyte ratios, 6 elevated serum levels of neutrophil chemoattractants,7 –13 and neutrophils with primed phenotypes.11,13 However, the precise role of neutrophils remains poorly understood.14,15 Mice induced with experimental autoimmune encephalomyelitis (EAE), a model of inflammatory demyelination, 16 exhibit neutrophil infiltration and CNS vascular leakage starting shortly before paralysis onset.12,17 –20 Although their depletion pre-onset attenuates leakage and paralysis,17,18 depletion post-onset has little effect, 17 implying that neutrophils contribute to early BBB dysfunction that permits later disease progression.
Neutrophils can modulate barrier integrity by releasing factors targeting tight junctions in endothelial cells (ECs).21,22 Tight junctions are multiprotein complexes formed between adjacent ECs to limit paracellular leak. 23 The claudin family establishes its backbone, 24 with claudin-5 as the main member in brain microvascular ECs (BMVECs) under basal conditions.25 –27 However, claudin-5 is suppressed in MS and EAE.4,28 Since its dysregulation is associated with cognitive dysfunction and inflammation, 29 strategies fostering its expression may prove widely beneficial. 28
Previously, we’ve shown that non-muscle myosin light chain kinase (nmMLCK) regulates cytokine-induced nuclear accumulation of transcriptional repressors for claudin-5 in BMVECs. 30 Here, we investigated the contributions of nmMLCK and claudin-5 to EAE disease progression and neutrophil-BMVEC interactions. We found that nmMLCK-knockout mice were resilient to EAE unless claudin-5 was transiently silenced, which caused a lethal disease course. Similarly, nmMLCK-knockout BMVEC monolayers resisted barrier dysfunction and neutrophil migration induced by conditioned media from activated neutrophils. Finally, modulating claudin-5 revealed an inverse relationship between its expression and neutrophil migration. Our findings support a pathogenic role for nmMLCK in BMVECs via BBB dysfunction and neutrophil infiltration in EAE. Additionally, they reveal that claudin-5 contributes to the immune barrier function of BMVECs, underscoring the importance of its preservation during neuroinflammation.
Methods
Reagents, antibodies, siRNAs, plasmids, and microscopy
Vendors, concentrations, and sequences for reagents, antibodies, siRNAs, and plasmids are listed in Supplementary Tables S1–S3. Microscopy details, including instruments, objectives, and magnifications, are listed in Supplementary Table S4.
Animal usage
All animal use was approved by the Institutional Animal Care and Use Committee at the University of South Florida, was performed per the Guide for the Care and Use of Laboratory Animals, 31 and is reported per the Animal Research Reporting In Vivo Experiments guidelines. 32 Mice were housed in individually ventilated cages in a specific pathogen-free facility under a twelve-hour light-dark cycle with food and water ad libitum. Constitutive knockout mice for non-muscle myosin light chain kinase (nmMLCK) on a C57BL/6 background (MGI: 2662927) were a kind gift from Dr. Daniel Martin Watterson (Northwestern University, IL).33,34 Homozygous knockouts (nmmlck−/−) and control littermates (nmmlck+/+) were generated from heterozygous knockout breeders and genotyped using a commercial Vendor (Transnetyx).
Experimental autoimmune encephalomyelitis (EAE) induction
EAE was induced in male nmmlck+/+ or nmmlck−/− mice (10–12 weeks) using a commercial kit. 19 Mice were injected subcutaneously twice with an emulsion of MOG35–55 peptide in complete Freund’s adjuvant, then injected intraperitoneally (i.p.) with pertussis toxin two and twenty-four hours later. Mice were evaluated daily via 5-point rubric: 0 = no clinical signs; 1 = tail paralysis; 2 = hindlimb weakness; 3 = hindlimb paralysis; 4 = hindlimb and forelimb weakness or paralysis; and 5 = moribund or found dead. 19 Water-softened chow and water gel cups were provided, and bladders were palpated to assist with urination. Mice scoring ≥4 were euthanized and received a score of 5 for the remainder of the experiment. Individuals blinded to group allocation handled mice at similar times of day. Also, cages were kept on the same shelf and housed a mixture of control and experimental mice. All mice except one nmmlck+/+ mouse (Figure 1), which died shortly post-induction, were included. Exact numbers are listed in Supplementary Table S5.
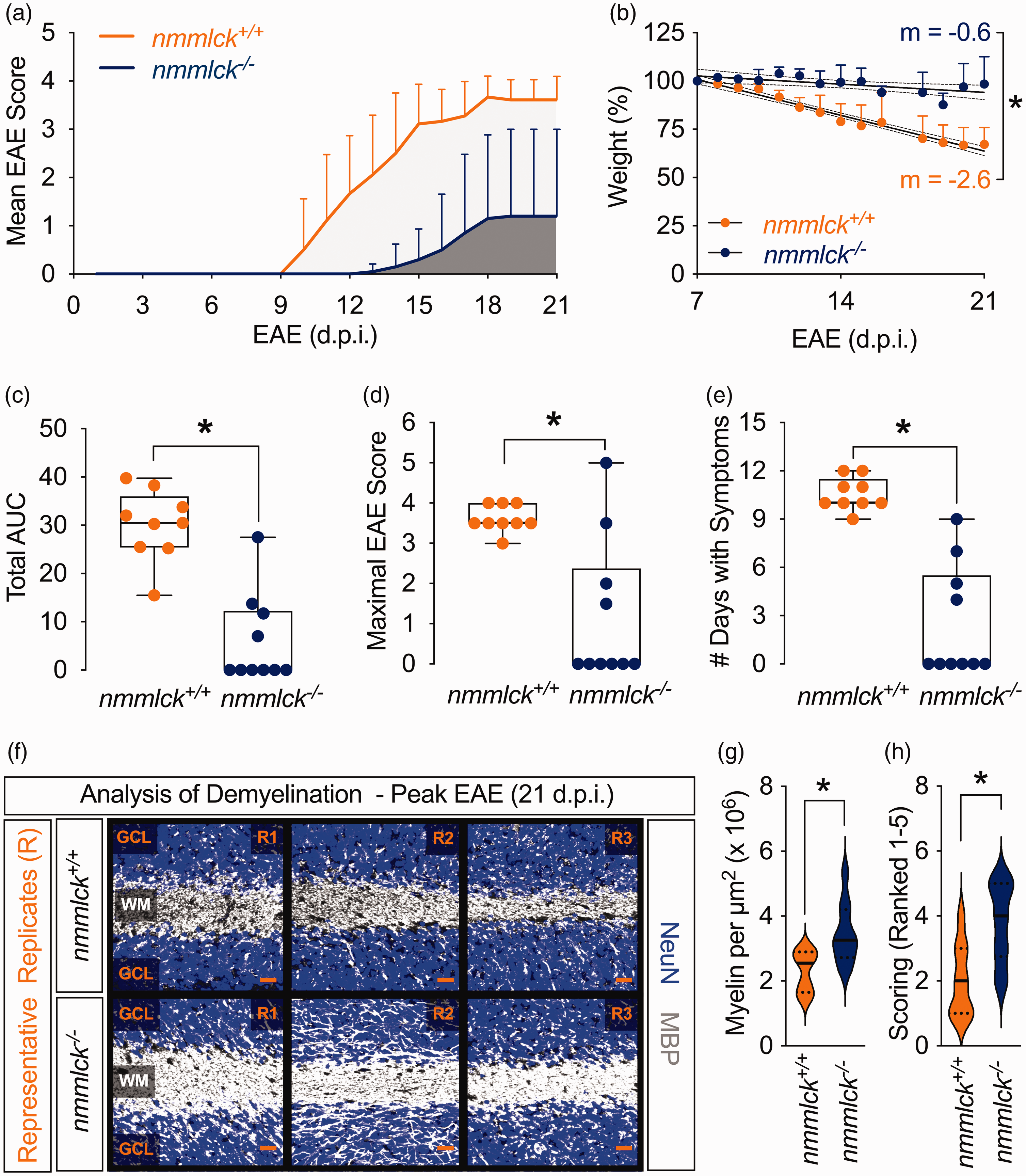
Lack of nmMLCK reduces EAE severity and demyelination. (a–e) EAE was induced in ten nmmlck−/− mice and nine nmmlck+/+ mice. (a) Disease was evaluated with a 5-point rubric through twenty-one days post-induction (d.p.i). Data are shown as the mean ± SD. (b) Weights were recorded daily starting at seven d.p.i. Data are shown as the mean ± SD. The lines of best fit (solid) are shown with 95% CIs (dotted). Slopes (m) were analyzed with F-tests; * indicates p < 0.05. (c–e) Based on (a), disease severity was evaluated for cumulative disease burden via total area under the curve (AUC) (c), maximal EAE score (d), and number of symptomatic days (e). Data are shown as the median and IQRs and analyzed with Mann-Whitney U tests; * indicates p < 0.05. Each dot reflects an individual mouse. (f–h) Cerebellums were harvested from the mice in (a–e) and immunolabeled for myelin basic protein (MBP, grey) and neuronal nuclear antigen (NeuN, blue). (f) Representative micrographs showing the granular cell layers (GCLs) and white matter (WM) tracts for nmmlck+/+ mice (upper) and nmmlck−/− mice (lower). Each replicate (R) shown is an individual mouse. Scale bar = 20 µm. (g–h) For each mouse, a single region in the cerebellar WM was collected for automated or manual evaluation of demyelination. Violin plots for the corrected integrated density for MBP in arbitrary units per µm2 (g) and histopathologic scoring (h) on a scale from 1–5, with 5 reflecting 81–100% of baseline in healthy mice. Data are shown as the median (solid) and IQRs (dotted) and analyzed with unpaired t-tests; * indicates p < 0.05.
Cerebellar demyelination analysis
Twenty-one days post-induction (d.p.i.), anesthetized mice were transcardially perfused with AF488-tomato lectin (TL; 100 µL), 1X phosphate-buffered saline (PBS), and 2% (w/v) paraformaldehyde (PFA) in 1X PBS. Excised brains were post-fixed with 2% (w/v) PFA in 1X PBS overnight at 4°C. Cerebellar slices (100 µm) were blocked for one hour in 1X PBS with 0.1% (v/v) Triton X-100 and 2 mM calcium (PBSTC) plus 10% (v/v) normal donkey serum, then incubated overnight with primaries for myelin basic protein (MBP) and neuronal nuclear antigen (NeuN) in blocking solution at 4°C. Slices were washed six times with PBSTC and incubated overnight with fluorescent secondaries in blocking solution at 4°C. Slices were washed six times with PBSTC and mounted with non-setting media.
Blinded individuals imaged a single region within the cerebellar white matter (WM) tracts (lobules IV-VIII). For automated analysis, average-intensity Z-projections (AIPs) were processed in FIJI (v2.0.0-rc-69/1.52p) 35 to calculate the integrated density of MBP fluorescence in arbitrary units per area (AU/µm2). The mean grey value for each pixel was corrected by subtracting the background (mean of three spots), then multiplied by the AIP’s area. For manual analysis, the AIPs from EAE-induced mice were scored for MBP on a scale from 1–5 against those from healthy mice as a baseline, e.g., 5 = 81–100%.
Neutrophil labeling and cerebellar infiltration analysis
Fab fragments from an anti-Ly6G antibody (Clone: 1A8) were created and labeled with AF488 using commercial kits. Eight d.p.i., mice were injected via tail vein with 10 µL of eluent. Fifteen minutes later, anesthetized mice were transcardially perfused with AF594-TL (100 µL), 1X PBS, and 2% (w/v) PFA in 1X PBS. Excised brains were post-fixed with 2% (w/v) PFA in 1X PBS overnight at 4°C. Cerebellar slices (100 µm) were mounted with non-setting media.
For automated analysis, Z-stacks were acquired and stitched together for reconstruction with Zen (black edition; v2.0). In turn, Z-stacks were processed with Imaris (v9.5) with the Spots function to count Ly6G+ cells per volume (µm3) of the entire slice. For manual analysis, blinded individuals imaged four regions in the cerebellar WM tracts (lobules IV-VIII). Maximum-intensity Z-projections (MIPs) were created in FIJI (v2.0.0-rc-69/1.52p), 35 then Ly6G+ cells were tallied per area (µm2) and averaged, plus classified as intravascular or extravascular based on morphology and co-localization with TL-labeled microvessels.
BBB function analysis
Evans blue (960 Da), which binds to serum albumin (66 kDa), was used to assess large solute leakage.19,36 Eight d.p.i., mice were injected i.p. with a 2% (w/v) solution in 0.9% (w/v) saline (100 µL). Twenty-four hours later, anesthetized mice were transcardially perfused with 0.9% (w/v) saline and 4% (w/v) PFA in 1X PBS. Blinded individuals imaged serial cerebellar coronal sections (1 mm) or whole spinal cords in the 700 and 800 channels of an Odyssey CLx near-infrared (NIR) fluorescence imager to capture dye and tissue autofluorescence. 19 Image Studio (v5.2) was used to prepare images.
We adapted previous strategies that used fluorescent dextrans (3 and 10 kDa) to assess small solute leakage.37 –39 Eight d.p.i., mice were anesthetized and injected retro-orbitally with 100 µL of a cocktail containing 3 mg/mL each of TRITC-Dextran (3 kDa) and FITC-Dextran (10 kDa) in 0.9% (w/v) saline. Thirty minutes later, whole blood was drawn via cardiac puncture and transferred to serum separation tubes to clot. Meanwhile, mice were transcardially perfused with 0.9% (w/v) saline and decapitated, then their excised brains were weighed after being cleaned of meninges and pial vessels. Sera and brains were flash-frozen in liquid nitrogen and kept at −80°C until ready. Thawed brains were ground in 1X PBS (0.4 mL) using a 7-mL Dounce tissue grinder. Afterward, homogenates were centrifuged at 15,000 × g for 20 minutes at 4°C. In turn, 100 µL of brain supernatant (diluted threefold with 1X PBS) or serum (diluted tenfold with 1X PBS) was loaded in triplicate into a black 96-well plate and read on a SpectraMax M5 with filters for FITC (490/520 nm) and TRITC (557/576 nm). Also, diluted sera and brain supernatants from non-injected mice were loaded in triplicate as-is or spiked with dextrans to ensure the samples were above the background but within a linear range. To calculate the brain permeability index (PI) for each dextran, the raw mean of each mouse’s fluorescence values (in RFU) for brain supernatant and serum was corrected by subtracting the mean fluorescence values (in RFU) of their respective blanks, then normalized by dividing the brain values by weight (in g) and serum values by volume (in mL). 38 Finally, each mouse’s PIs were divided by the mean PI of the respective dextran in the control group.
Cerebellar perivenular inflammatory lesion (PIL) analysis
We adapted previous strategies with biotinylation reagents to locate sites with vascular leakage.40 –42 Eight d.p.i., a 0.3 mg/mL solution of sulfo-NHS-LC-biotin (556 Da) was prepared in 1X PBS, then anesthetized mice were transcardially perfused with AF488-TL (100 µL), biotinylation solution (20 mL), and 1% (w/v) PFA in 1X PBS. Excised brains were post-fixed with 1% (w/v) PFA in 1X PBS overnight at 4°C. Cerebellar slices (100 µm) were blocked for one hour in PBSTC plus 10% (v/v) normal donkey serum, then incubated overnight with primaries for NeuN in the blocking solution at 4°C. Slices were washed six times with PBSTC, then incubated with fluorescent secondaries and Texas Red-streptavidin (TR-SA) in the blocking solution overnight at 4°C. Slices were washed six times with PBSTC, then mounted with non-setting media. Z-stacks were acquired and stitched together for reconstruction with Zen (black edition; v2.0). Blinded individuals identified PILs based on characteristic extravascular biotinylation by TR-SA reactivity and non-endothelial labeling by AF488-TL, then tallied PILs per entire slice.
Microvessel isolation for cell culture
Primary mouse brain microvascular endothelial cells (BMVECs) were isolated from nmmlck+/+ or nmmlck−/− pups (7–10 days).19,30 Excised brains were cleaned of meninges and pial vessels, then ground in phenol red-free DMEM plus 2% (v/v) fetal bovine serum (DMEM-S) using a 40-mL Dounce tissue grinder (5 strokes with loose pestle). Homogenates were mixed equally with a 36% (w/v) solution of 70-kDa dextran prepared in DMEM-S, then centrifuged at 10,000 × g for 10 minutes at 4°C. Pellets were suspended in DMEM-S and passed through a 70-µm filter, then digested in 1X HBSS containing Nα-tosyl-L-lysine chloromethyl ketone (1 µg/mL), DNase I (10 U/µL), and collagenase/dispase (1 mg/mL) for 40 minutes at 37°C. Digests were suspended in Complete Endothelial Cell Medium, plated on mouse collagen type IV (COL4)-coated cultureware (10 µg/mL), and cultured at 37°C in a humidified, 5% CO2 incubator for 5–7 days before use. Cells were used at passages 0 or 1.
Gene silencing and transfer in cell culture
BMVECs were transfected with the Nucleofector II/2b system and kit for Primary Mammalian Endothelial Cells.19,30 Commercial siRNAs targeting mouse Cldn5 were used, with non-targeting siRNAs as controls. Similarly, a commercial pCMV6-Myc-DDK vector containing the full-length cDNA for mouse Cldn5 was used, with the empty vector backbone as the control. A complete description is listed in the Supplementary Methods. Western blotting (Figures 6(a) and 7(a)) or microscopy (Figure 6(e)) were used to confirm silencing or transfer.
Neutrophil isolation and generation of conditioned media (NCM)
Neutrophils were isolated from whole blood or bone marrow depending on the number required via negative selection using commercial magnetic-activated cell sorting kits. To create NCM, bone marrow-derived nmmlck+/+ neutrophils were plated in Complete Endothelial Culture Medium at 1 × 106 cells/mL, then incubated with N-Formylmethionine-leucyl-phenylalanine (fMLF, 100 nM) or vehicle for 30 minutes at 37°C in a humidified 5% CO2 incubator. Afterward, NCM were centrifuged at 400 × g for 5 minutes to remove cells and debris before their immediate use.
Neutrophil adhesion and transendothelial migration (TEM)
For adhesion, BMVECs were seeded at confluence on mouse COL4-coated (10 µg/cm2) 96-well plates. When ready, 10% of the media was replaced with NCM. Six hours later, 2.5 × 105 neutrophils loaded with NucSpot Live 488 dye were added to each well. After thirty minutes, non-adherent neutrophils were rinsed away, then wells were fixed with 4% PFA in 1X PBS for 10 minutes at room temperature. Three regions were imaged on an Axiovert 5/7 equipped with a GFP filter set for each well. Images were opened in FIJI (v2.0.0-rc-69/1.52p), 35 then the mean number of cells per field of view (FoV) was converted to surface area (cm2).
For TEM, BMVECs were seeded at confluence in mouse COL4-coated (10 µg/cm2) 24-well transwell inserts (3-µm pores). Before starting, the transendothelial electrical resistance (TEER) of the inserts was confirmed to be >100 Ω⋅cm2 (voltohmmeter), then 10% of the media in the upper chambers was replaced with NCM. When ready, 1 × 105 neutrophils loaded with CellTracker Green CMFDA or Red CMTPX dyes were added to the upper chambers, and fMLF (100 nM) was added to the lower chamber. In Figures 4 and 5, live-cell imaging was done with an Axiovert 5/7 equipped with a Texas red filter set or EVOS FL imager equipped with GFP light cube. To calculate the fraction migrated, the mean cell number per FoV of three non-continuous regions was scaled to the total surface area of the plate, then divided by the input number. In Figures 6 and 7, media from the lower chambers was collected and the cell density determined via flow cytometry. To calculate the fraction migrated, cell density was scaled to the total volume of the lower chamber, then divided by the input number.
Small solute permeability
Sodium fluorescein (376 Da) was used to assess the permeability of BMVECs to small solutes.19,30 A complete description is listed in the Supplementary Methods. The apparent permeability coefficient for sodium fluorescein (PS) was calculated thus: PS = [L]/t × 1/A × V/[U], where [L] is the lower chamber concentration; t is time (in seconds); A is the membrane area (in cm2); V is the lower chamber volume (in mL); and [U] is the upper chamber concentration.
Transendothelial electrical resistance (TEER)
TEER was monitored in real-time with an electrical cell-substrate impedance sensing (ECIS) ZƟ platform.19,30 BMVECs were seeded at confluence in mouse COL4-coated (10 µg/mL) 8W10E+ arrays. When ready, 10% of the media was replaced with NCM, and TEER was measured continuously at 4000 Hertz for 24 hours. Raw values were normalized to the onset (t = 0), and peak changes in TEER were compared to onset values.
Gene silencing In vivo
Silencing was performed as per Campbell et al. 43 Seven d.p.i., EAE-induced nmmlck−/− mice were administered 20 µg of 2′-OMe-modified siRNAs targeting mouse Cldn5 or luciferase prepared using the in vivo-jetPEI delivery system via tail vein. Western blotting (Figure 7(d)) was used to confirm silencing 24 hours post-injection in brain microvessels from a separate cohort of naïve nmmlck−/− mice.
Immunofluorescence analysis
Expression of endogenous and DDK-tagged claudin-5 in BMVECs was assessed with immunofluorescence.19,30 A complete description is listed in the Supplementary Methods.
Two-color NIR Western blot (NIR-WB) analysis
Protein expression in brain microvessels, leukocytes, and BMVECs was assessed with two-color NIR-WB.19,30 A complete description is listed in the Supplementary Methods. Blots were imaged on an Odyssey CLx NIR fluorescence imager. Image Studio (v5.2) was used to prepare images and perform densitometry. Relative expression was determined by dividing the target protein signal (800 channel) by the β-actin signal (700 channel).
Statistical analysis
For animal studies, a priori power analyses were performed with power = 0.8 and α = 0.05. In Figures 1 and 7, an effect size of d = 1.2 was chosen based on the resilience of nmmlck−/− mice to injury and vascular dysfunction33,34,44,45 and the compromised vascular integrity of Cldn5-null mice, 40 respectively. In Figures 2 and 3, a larger effect size of d = 1.8 was chosen based on Figure 1. Where appropriate, Shapiro-Wilk tests were used to assess normality. Data are presented as the mean and the standard deviation (SD), or the median and the interquartile ranges (IQRs). Lines of best fit are shown with 95% confidence intervals (CIs). Unpaired t-tests, Mann-Whitney U tests, F-tests, or one-way Analysis of variance (ANOVA) tests with post-hoc Tukey’s test were used and are listed in Supplementary Table S6. Analysis was performed using GraphPad Prism (v10.3.0), and p < 0.05 was considered significant.
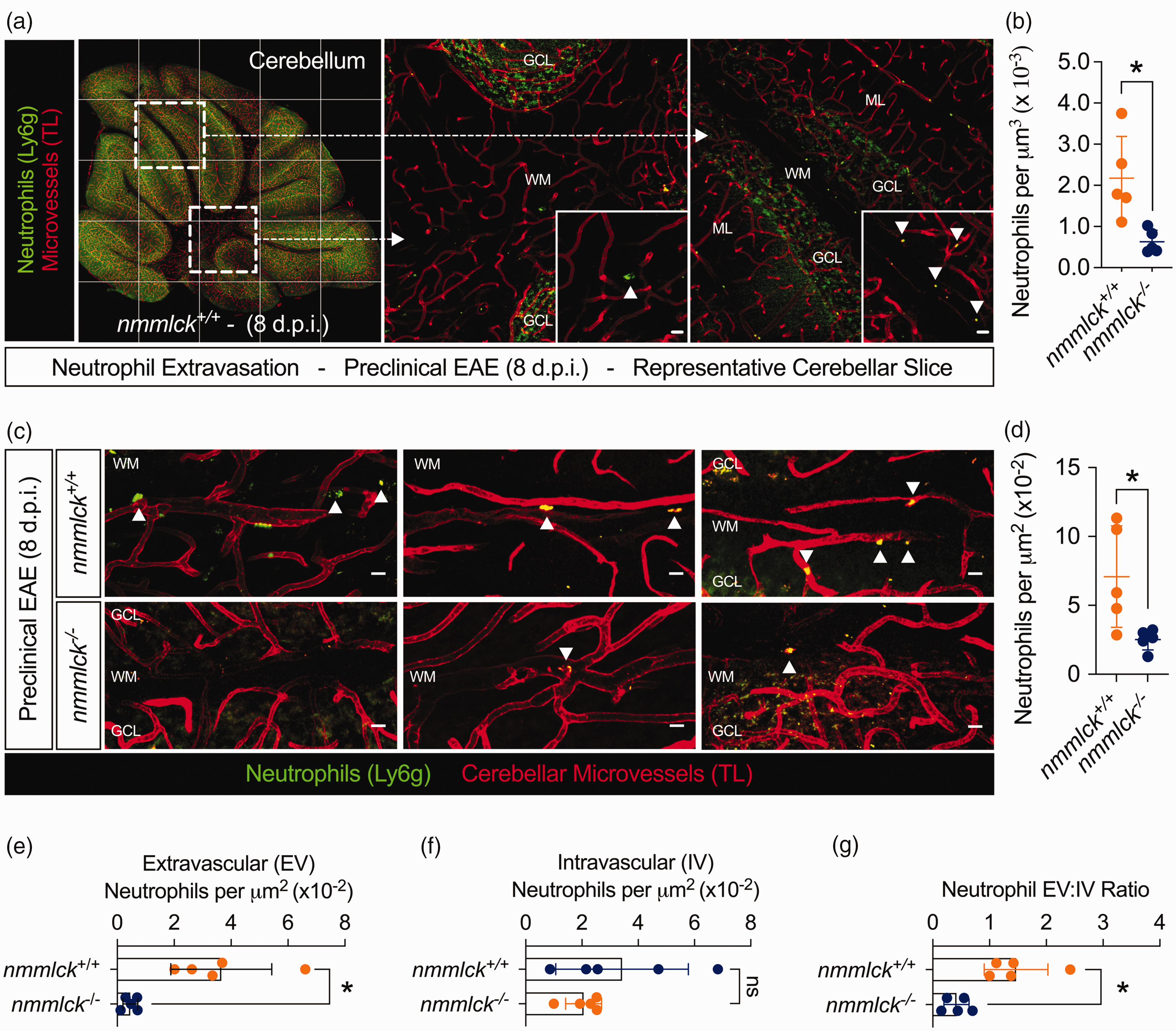
Lack of nmMLCK reduces neutrophil infiltration in pre-clinical EAE. (a–g) EAE was induced in five nmmlck−/− mice and five nmmlck+/+ mice. Eight d.p.i., mice were injected with fluorescent Fab fragments from an anti-Ly6G antibody to label neutrophils (green) and perfused with fluorescent tomato lectin (TL, red) to label microvessels. Intravascular and extravascular neutrophils are marked by up and down arrowheads, respectively. Scale bars = 10 µm. (a) Representative cerebellar reconstruction from an nmmlck+/+ mouse (left) stitched from maximum intensity z-projections (MIPs, right). (b) Using Imaris, the Spots function quantified neutrophils per µm3 in cerebellar reconstructions. Data are shown as the mean ± SD and analyzed with unpaired t-tests; *indicates p < 0.05. Each dot reflects an individual mouse. (c) Representative cerebellar MIPs from a second data set for nmmlck+/+ mice (upper) and nmmlck−/− mice (lower). (d–g) Blinded individuals identified and counted neutrophils to quantify the neutrophils per µm2 (d), extravascular (EV) neutrophils per µm2 (e), and the intravascular (IV) neutrophils per µm2 (f) to calculate the ratio of EV-to-IV neutrophils (g). Data are shown as the mean ± SD and analyzed with unpaired t-tests; * indicates p < 0.05 and n.s. indicates p > 0.05. Each dot reflects an individual mouse (average of four micrographs per mouse).
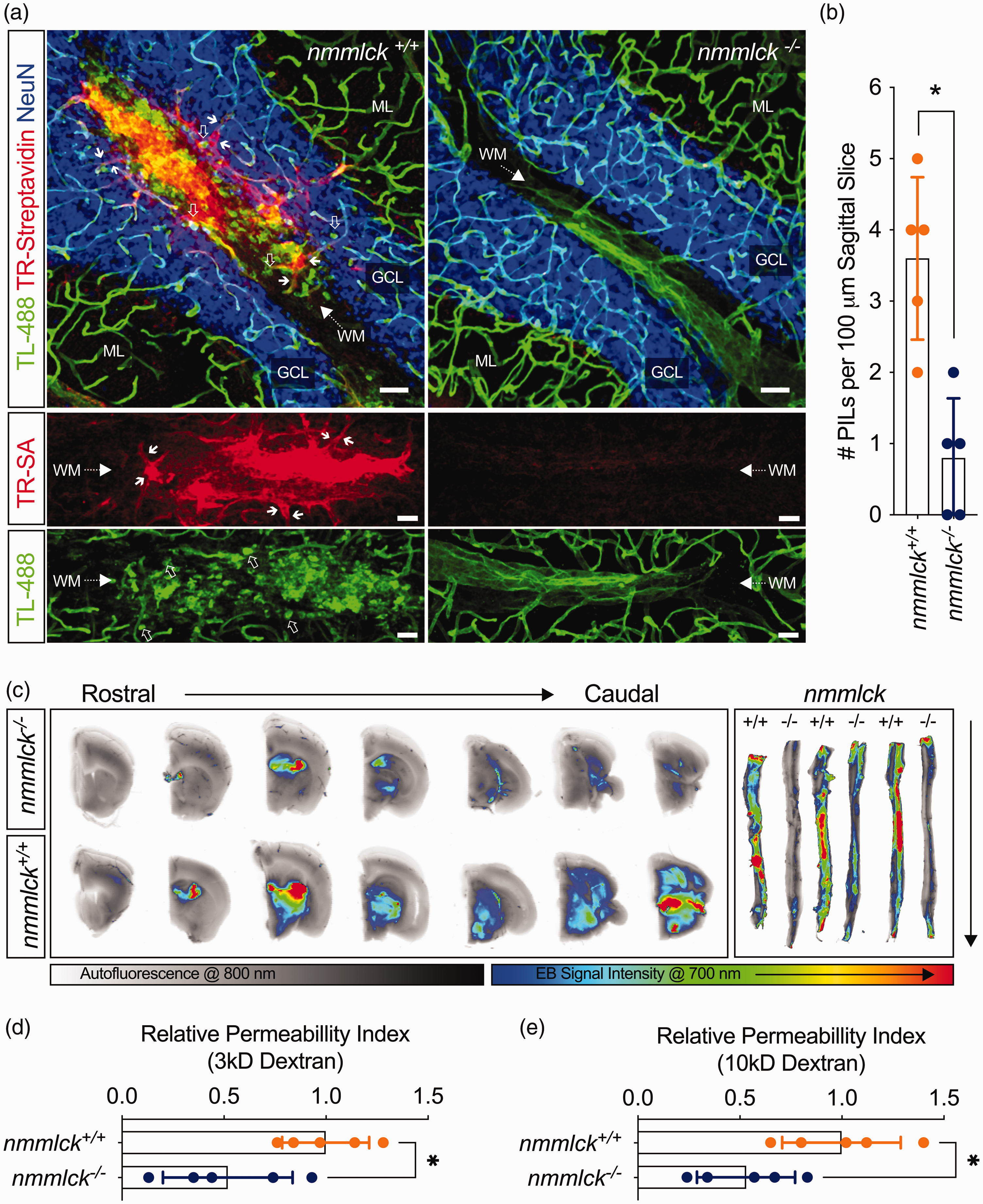
Lack of nmMLCK reduces BBB dysfunction in pre-clinical EAE. (a–e) EAE was induced in five nmmlck−/− and five nmmlck+/+ mice (a,b,d,e), or three nmmlck−/− and three nmmlck+/+ mice (c). For (b), (d), and (e), data are shown as the mean ± SD and analyzed with an unpaired t-test; * indicates p < 0.05. Each dot reflects an individual mouse. (a) Eight d.p.i., mice were perfused with fluorescent tomato lectin (TL) and a biotinylation reagent. Confocal micrographs centered over cerebellar white matter (WM) tracts show sites with vascular leakage in an nmmlck+/+ mouse (left) or vascular integrity in an nmmlck−/− mouse (right) based on the presence or absence of non-endothelial labeling by TL (green) and extravascular biotinylation by Texas Red-streptavidin (TR-SA, red). NeuN (blue) was immunolabeled to mark the granular cell layers (GCLs). Scale bars = 40 µm. (b) Quantification of perivenular inflammatory lesions (PILs) per slice for data shown in (a). (c) Eight d.p.i., mice were injected intraperitoneally with Evans blue (EB) to assess large solute leakage. Cerebellums and spines were imaged in the 700 (rainbow) and 800 (grey) channels of an Odyssey CLx NIR imager to detect EB and tissue autofluorescence. For the cerebellum, serial coronal sections of an nmmlck−/− mouse (upper) and nmmlck+/+ mouse (lower) are shown. (d,e) Eight d.p.i., mice were injected retro-orbitally with a cocktail of 3 kDa TRITC-dextran (d) and 10 kDa FITC-dextran (e) to assess small solute leakage. For intergroup comparison, the calculated permeability index (PI) of each dextran for each mouse was divided by the mean PI of the respective dextran in the nmmlck+/+ group.
Results
Lack of nmMLCK attenuates disease severity and cerebellar demyelination in EAE
As a first step, we induced MOG35–55 peptide-based EAE in nmmlck+/+ and nmmlck−/− mice and monitored them for three weeks. Compared to their counterparts, nmmlck−/− mice had mild disease (Figure 1(a)) with minimal weight loss (Figure 1(b)). In line, cumulative disease burden based on area under the curve (AUC; Figure 1(c)), maximal scores (Figure 1(d)), and days with symptoms (Figure 1(e)) were all significantly lower in nmmlck−/− mice. These results indicate that nmMLCK deficiency protects against EAE-induced disease severity.
In the MOG35–55 model, lesions form throughout the CNS. 46 However, we focused on the cerebellum as it is commonly affected in patients with MS, 47 plus neutrophils are more involved with brain inflammation in EAE. 48 After three weeks, we observed a greater presence of myelin basic protein (MBP) in the cerebellar white matter (WM) regions of nmmlck−/− mice (Figure 1(f)), and this difference was significant when assessed by automated image processing (Figure 1(g)) or manual histopathological scoring (Figure 1(h)). These results indicate that nmMLCK deficiency protects against EAE-induced cerebellar demyelination.
CD4+ T cells isolated from healthy mice 49 or EAE-induced mice do not express nmMLCK (Supplementary Figure S1(c)). Nevertheless, we assessed their presence and abundance in the cerebellum since the MOG35–55 model is CD4+ T cell-driven. 50 In nmmlck−/− mice, CD4+ T cells were readily seen (Supplementary Figure S1(a)), and there was no significant difference in the total number by genotype (Supplementary Figure S1(b)). Separately, no differences in leukocyte types were found in the blood differential tests of EAE-induced nmmlck−/− mice (Supplementary Table S7). These results suggest that the protective phenotype in nmmlck−/− mice during EAE is not due to a defective induction phase.
Lack of nmMLCK limits pre-onset neutrophil infiltration and BBB dysfunction in EAE
Neutrophil infiltration and CNS vascular leakage precede paralysis onset.12,17 –20 As such, we narrowed our focus to 8 days post-induction (d.p.i.), which reflects a time point ∼1 day before onset in nmmlck+/+ mice (Figure 1(a)). To track neutrophils, we injected mice with fluorescent Fab fragments from an anti-Ly6G antibody shortly before sacrifice. Afterward, we counted Ly6G+ cells in the cerebellum (Figures 2(a) and 2(c)). Compared to their counterparts, nmmlck−/− mice had significantly fewer neutrophils per volume (Figure 2(b)) by automated image processing or per area (Figure 2(d)) by manual counting. Furthermore, the number of intravascular neutrophils in nmmlck−/− mice was non-significantly different (Figure 2(f)), but the number of extravascular neutrophils was significantly lower (Figure 2(e)). Accordingly, their extravascular-to-intravascular ratio was significantly lower (Figure 2(g)). These results indicate that nmMLCK deficiency limits EAE-induced neutrophil infiltration in the cerebellum.
To find sites of vascular leakage in situ, we transcardially perfused mice with fluorescent TL (71 kDa), which binds N-acetyl-glucosamine on glycoproteins, 51 and sulfo-NHS-LC-Biotin (556 Da), which reacts with primary amines. 40 In the cerebellar WM regions of nmmlck−/− mice, TL is restricted to the luminal surface, and minimal biotinylation is detected via fluorescent streptavidin (Figure 3(a)). Conversely, non-endothelial labeling and extravascular biotinylation occur over putative venules in nmmlck+/+ mice (Figure 3(a)). Accordingly, we have termed these sites as perivenular inflammatory lesions (PILs). Compared to their counterparts, nmmlck−/− mice had significantly fewer PILs per tissue slice (Figure 3(b)). In parallel, we conducted permeability assays with sodium fluorescein (376 Da), fluorescent dextrans (3 and 10 kDa), or Evans blue (960 Da), which binds serum albumin (66 kDa). Compared to their counterparts, nmmlck−/− mice had significantly lower leakage of sodium fluorescein (Supplementary Figure S2) and both dextrans (Figures 3(d) and 3(e)). Similarly, they had qualitatively less Evans blue leakage via near-infrared (NIR) imaging of their brains and spines (Figure 3(c)). These results indicate that nmMLCK deficiency limits EAE-induced CNS vascular leakage of small and large solutes.
Lack of nmMLCK does not prevent endothelial activation or neutrophil diapedesis
Inducible nmMLCK-knockout mice did not exist when we conducted our animal studies, Instead, we used co-culture models with brain microvascular endothelial cells (BMVECs) and neutrophils isolated from nmmlck+/+ or nmmlck−/− mice because both cell types express nmMLCK (Supplementary Figure S1(c)).30,52 Adopting a holistic approach, we created conditioned media from neutrophils stimulated with N-formylmethionine-leucyl-phenylalanine (NCM-fMLF) or the vehicle (NCM-VEH) to serve as our inflammatory stimulus. Regardless of genotype, an increase in total intercellular adhesion molecule 1 (ICAM-1) protein (Figure 4(a)) and a decrease in total platelet and endothelial cell adhesion molecule-1 (PECAM-1) protein (Figure 4(b)) was seen in the total protein lysates of BMVECs incubated with NCM-fMLF for six hours, suggesting that our in vivo findings are not the result of defective BMVEC activation.
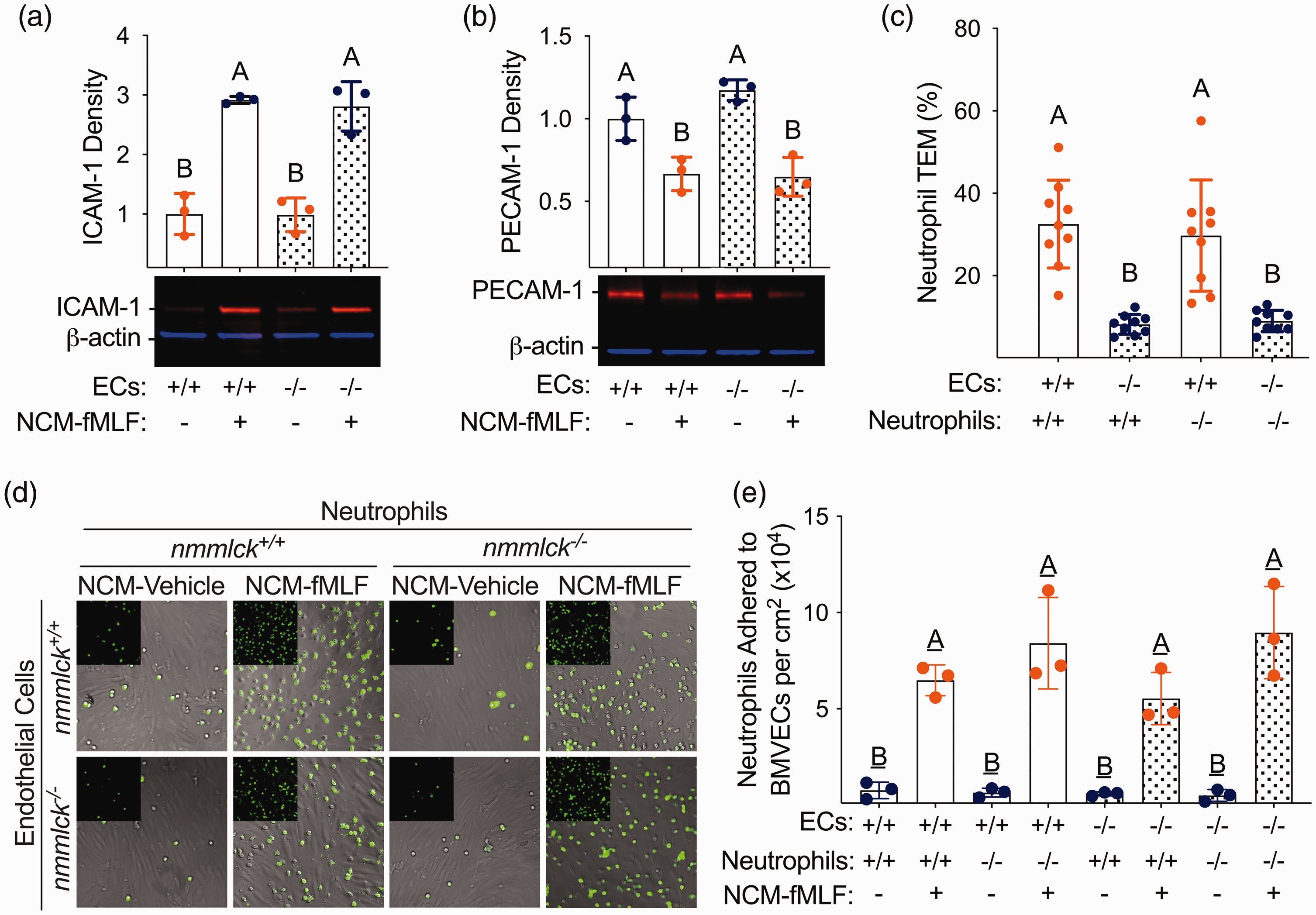
Lack of nmMLCK does not prevent BMVEC activation or neutrophil diapedesis. (a,b) Densitometry and representative two-color NIR Western blots for ICAM-1 (a) and PECAM-1 (b). Primary nmmlck+/+ or nmMLCK−/− BMVECs were incubated with conditioned media created from neutrophils stimulated with N-formylmethionine-leucyl-phenylalanine (NCM-fMLF) or vehicle (NCM-VEH). Six hours later, BMVECs were lysed and probed for target proteins (orange) and β-actin (blue). For normalization, target protein signals were divided by β-actin signals. Data are shown as the mean ± SD and analyzed with one-way ANOVA; different letters indicate p < 0.05 (Tukey’s). Each dot reflects an individual experiment. (c) Transendothelial migration (TEM) of naïve nmmlck+/+ or nmmlck−/− neutrophils across nmmlck+/+ (orange) or nmmlck−/− (blue) BMVEC monolayers after six hours. Beforehand, BMVECs were incubated with NCM-fMLF or NCM-VEH for six hours. Data are shown as the mean ± SD and analyzed with one-way ANOVA; different letters indicate p < 0.05 (Tukey’s). Each dot represents a single insert from three individual experiments (three inserts per experiment). (d,e) Adhesion of naïve nmmlck+/+ or nmmlck−/− neutrophils to nmmlck+/+ or nmmlck−/− BMVEC monolayers after twenty minutes. Beforehand, BMVECs were incubated with NCM-fMLF (orange) or NCM-VEH (blue) for six hours. (d) Representative micrographs of NucSpot Live 488-laden neutrophils (green) adherent to BMVEC monolayers (grey - brightfield). (e) Quantification of the data shown in (d) to determine the number of adherent neutrophils per surface area (cm2). Data are shown as the mean ± SD and analyzed with one-way ANOVA; different letters indicate p < 0.05 (Tukey’s). Each dot represents the mean of three fields of view for three separate experiments.
The results of adhesion and transendothelial migration (TEM) assays further support this position. Naïve neutrophils readily adhered to BMVECs pre-incubated with NCM-fMLF for six hours (Figure 4(d)), with no significant difference in the number of adherent cells based on genotype after twenty minutes (Figure 4(e)). Similarly, naïve neutrophils readily migrated across BMVEC monolayers pre-incubated with NCM-fMLF for six hours, with no significant difference in the fraction migrated based on genotype after six hours (Figure 4(c)). These results suggest that our in vivo findings are not the result of defective neutrophil diapedesis.
Lack of nmMLCK limits barrier loss and neutrophil migration in BMVECs
Encouraged by these results, we continued forward with our co-culture models to further study nmMLCK in BMVECs. For barrier function, we used transendothelial electrical (TEER) and transwell-based permeability assays (TPAs) to monitor changes after incubating BVMECs with NCM-fMLF. A biphasic loss in TEER was observed over twenty-four hours for nmmlck+/+ monolayers (Figure 5(a)). The first phase was transient and resolved within two hours, but the second phase was a gradual and persistent loss starting at four hours. For both phases, the peak changes in TEER were significantly lower versus the starting time point (Figure 5(b)). Conversely, no significant changes in TEER were seen for either phase in nmmlck−/− monolayers (Figures 5(a) and 5(b)). In line, the apparent permeability coefficient (PS) for sodium fluorescein was greater for nmmlck+/+ monolayers after 24 hours (Figure 5(c)). These results indicate that neutrophil-secreted factors trigger a biphasic loss in the barrier quality of BMVECs and that nmMLCK deficiency in BMVECs limits both phases of neutrophil-induced barrier dysfunction.
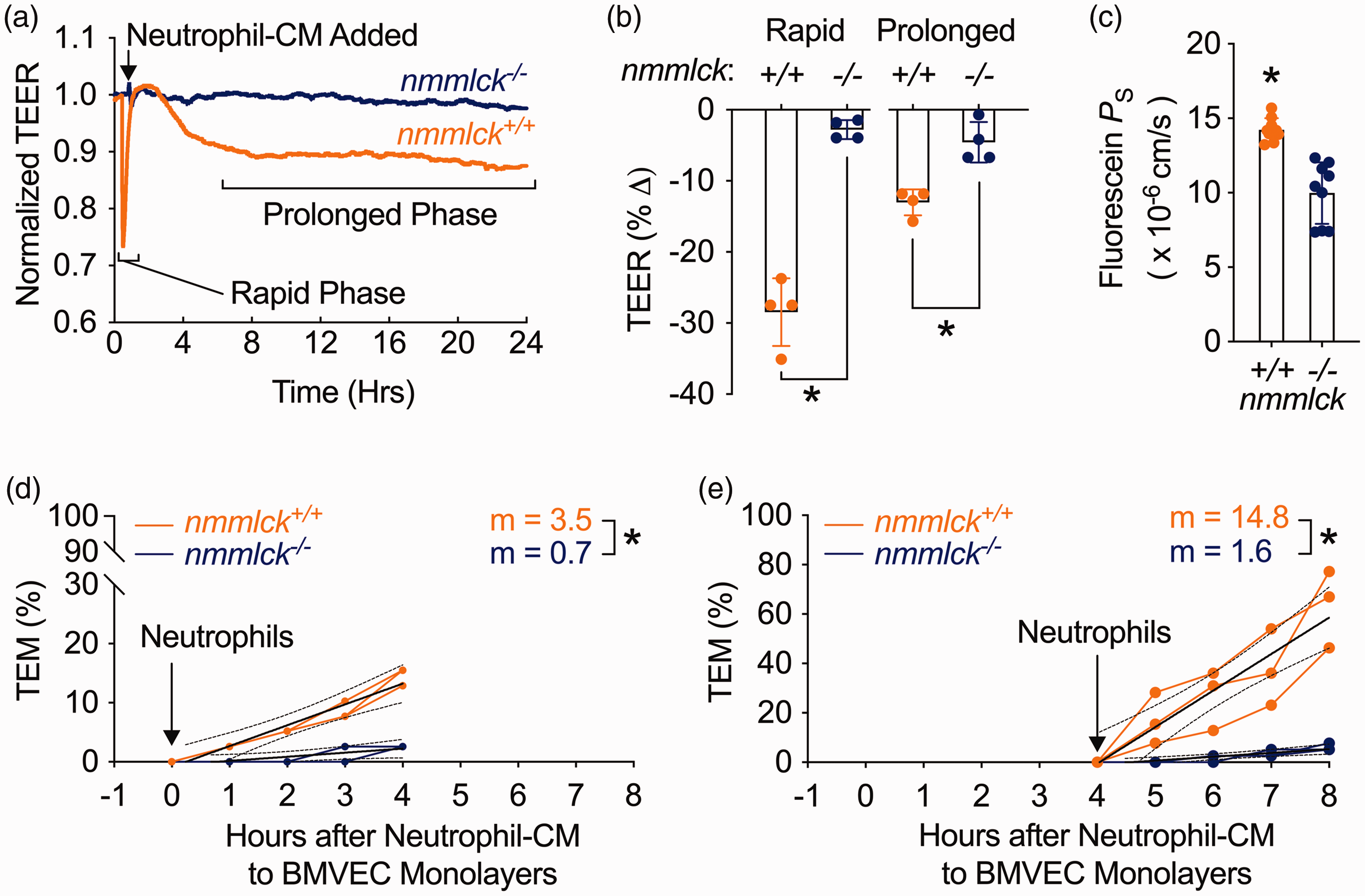
Endothelial nmMLCK mediates neutrophil-induced biphasic loss of barrier quality. (a) Representative transendothelial electrical resistance (TEER) tracings of nmmlck+/+ (orange) and nmmlck−/− (blue) BMVECs for twenty-four hours post-addition of conditioned media (CM) from fMLF-stimulated nmmlck+/+ neutrophils. Data are shown as the mean (solid) of four individual wells. (b) Quantification of maximal TEER changes during the rapid (left) and prolonged (right) phases for (a). Data are shown as the mean ± SD and analyzed with unpaired t-tests; * indicates p < 0.05. (c) The apparent permeability coefficient for sodium fluorescein (PS) across nmmlck+/+ (orange) and nmmlck−/− (blue) BMVEC monolayers twenty-four hours post-addition of neutrophil CM from fMLF-stimulated nmmlck+/+ neutrophils. Data are shown as the mean ± SD and analyzed with an unpaired t-test; * indicates p < 0.05. Each dot represents a single insert from three individual experiments (three inserts per experiment). (d,e) Transendothelial migration (TEM) for naïve nmmlck+/+ neutrophils across nmmlck+/+ (orange) or nmmlck−/− (blue) BMVEC monolayers during the rapid (d) or prolonged (e) phases of barrier dysfunction was monitored after adding CM from fMLF-stimulated nmmlck+/+ neutrophils, then adding neutrophils and fMLF as a chemoattractant zero (d) or four (e) hours later. Values from three individual experiments are shown as connected dots. The lines of best fit (solid) are shown with 95% CIs (dotted). Slopes (m) were analyzed with F-tests; * indicates p < 0.05.
For TEM, we explored the possibility that neutrophil migration might differ during the two phases of barrier dysfunction (Figure 5(a)). To test this, we added naïve nmmlck+/+ neutrophils at zero or four hours after the addition of NCM-fMLF, then tracked migration for four hours. Based on slope analysis, the TEM rate was significantly greater across nmmlck+/+ monolayers for both phases (Figures 5(d) and 5(e)), with a fivefold increase during the first phase and a tenfold increase during the second. These results indicate that neutrophils secrete factors that make BMVECs more receptive to neutrophil TEM, particularly during the second phase of barrier dysfunction, all of which require endothelial nmMLCK.
Silencing claudin-5 undoes the protective effects of nmMLCK deficiency in BMVECs and EAE
We’ve shown that nmMLCK mediates cytokine-induced barrier dysfunction in BMVECs via the downregulation of the tight junction protein claudin-5. 30 Here, we transfected nmmlck+/+ BMVECs with small, interfering RNAs (siRNAs) or expression vectors for mouse Cldn5 to study how its expression influences the physical and immune barrier functions of BMVECs. Claudin-5 knockdown alone (Figures 6(a) and 6(b)) significantly increased the PS for sodium fluorescein (Figure 6(d)) and the fraction of migrated neutrophils after six hours (Figure 5(c)). Conversely, the expression of Myc-DDK-tagged claudin-5 alone (Figure 6(e)) significantly decreased the PS for sodium fluorescein (Figure 6(g)) and the fraction of migrated neutrophils after six hours (Figure 6(f)). These results reveal an inverse relationship between basal claudin-5 expression and successful neutrophil migration, implying that claudin-5 contributes not only to the physical barrier function of BMVECs, but also to their immune barrier function.
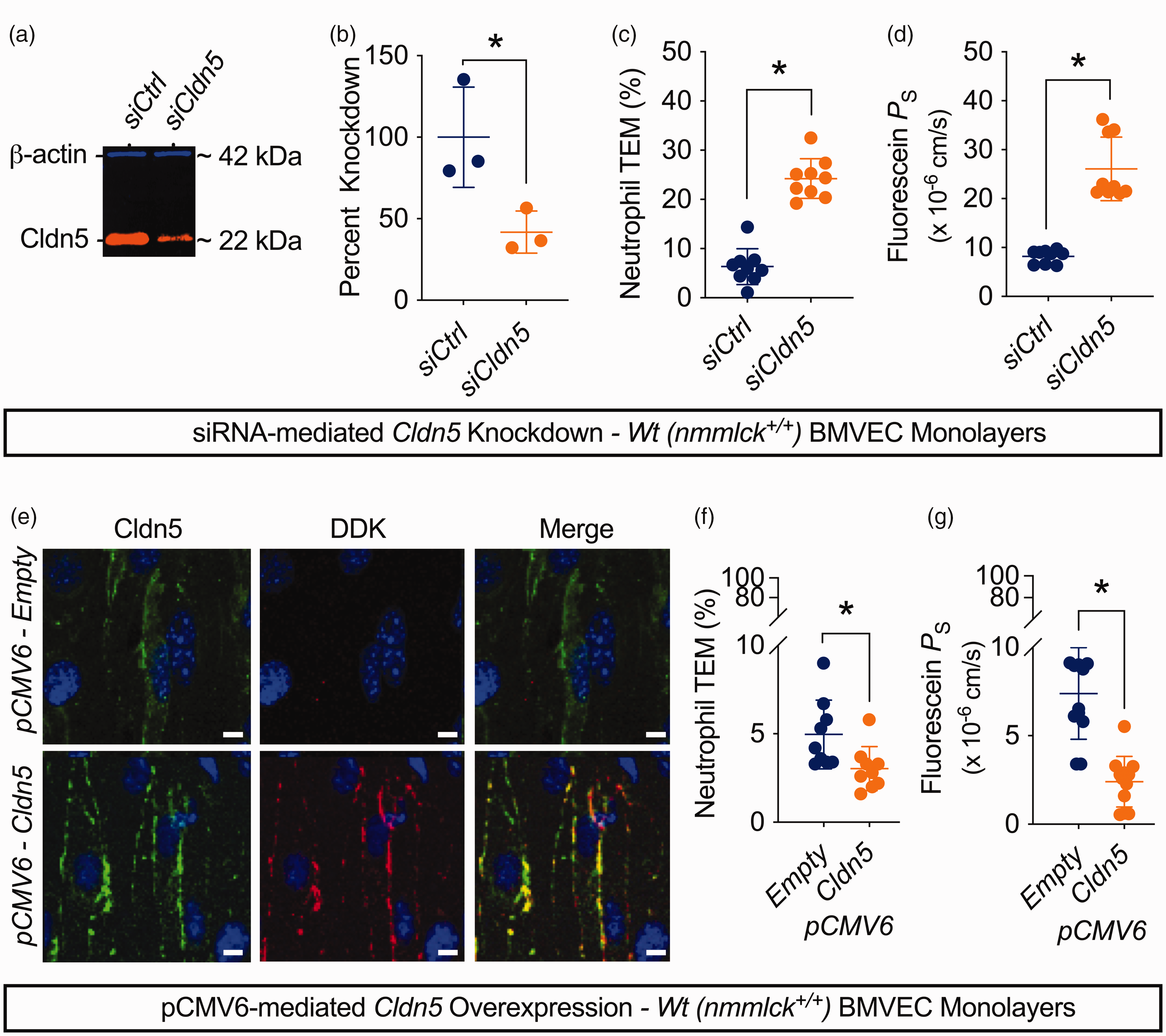
Claudin-5 acts as a physical and immune barrier in BMVECs. (a–d) nmmlck+/+ BMVECs were transfected with siRNAs targeting mouse claudin-5 (siCldn5) or non-targeting siRNAs as a control (siCtrl), then used after a twenty-four-hour recovery period. Data are shown as the mean ± SD for three independent experiments (three inserts per experiment in (c) and (d)) and analyzed by unpaired t-tests; * indicates p < 0.05. (a) Representative two-color, NIR Western blot for claudin-5 (CLDN5, orange) and β-actin (blue) to confirm knockdown. (b) Quantification of knockdown efficiency by normalizing claudin-5 signal to the β-actin signal in the siCtrl and siCldn5 groups. (c) Fraction of naïve nmmlck+/+ neutrophils that migrated across siCtrl- or siCldn5-transfected monolayers after six hours using fMLF as a chemoattractant. (d) The apparent permeability coefficient for fluorescein (PS) across siCtrl- or siCldn5-transfected monolayers. (e–g) nmmlck+/+ BMVECs were transfected with a pCMV6-Myc-DDK vector for mouse claudin-5 (Cldn5) or the empty vector backbone as a control (empty), then used after a twenty-four-hour recovery period. Data are shown as the mean ± SD for three independent experiments (three and four inserts per experiment in (f) and (g), respectively), and analyzed by unpaired t-tests; * indicates p < 0.05. (e) Representative confocal micrographs for endogenous (green) and DDK-tagged claudin-5 (red) to confirm plasmid functionality. (f) Fraction of naïve nmmlck+/+ neutrophils that migrated across empty- or Cldn5-transfected monolayers after six hours using fMLF as a chemoattractant. (g) The apparent permeability coefficient for fluorescein (PS) across empty- or Cldn5-transfected monolayers.
Finally, to connect the protective effects of nmMLCK deficiency to claudin-5 expression, we performed siRNA-mediated knockdown in nmmlck−/− BMVECs and in EAE-induced nmmlck−/− mice. Claudin-5 knockdown alone (Figure 7(a)) in nmmlck−/− BMVECs significantly increased the PS for sodium fluorescein (Figure 7(c)) and the fraction of migrated neutrophils after six hours (Figure 7(b)), which aligns with the results from nmmlck+/+ BMVECs (Figures 6(c) and 6(d)). As for animal studies, we injected nmmlck−/− mice with siRNAs at 7 d.p.i. to transiently knockdown claudin-5 and monitored them for another two weeks. Mice injected with siRNAs targeting the luciferase gene (siLuc) had mild disease (Figure 7(d)) akin to the non-injected nmmlck−/− mice in Figure 1(a). Conversely, mice injected with siRNAs targeting claudin-5 (siCldn5) had aggressive disease (Figure 7(d)) worse than the nmmlck+/+ mice in Figure 1(a) that culminated in complete mortality (Figure 7(e)). Accordingly, the cumulative disease burden based on AUC (Figure 7(f)), maximal scores (Figure 7(g)), and days with symptoms (Figure 7(h)) were all significantly greater in the siCldn5 group. Overall, these results show that the protective effects of nmMLCK deficiency in EAE are undone when claudin-5 is transiently suppressed shortly before paralysis onset, underscoring the harmful effects of its loss during neuroinflammation.
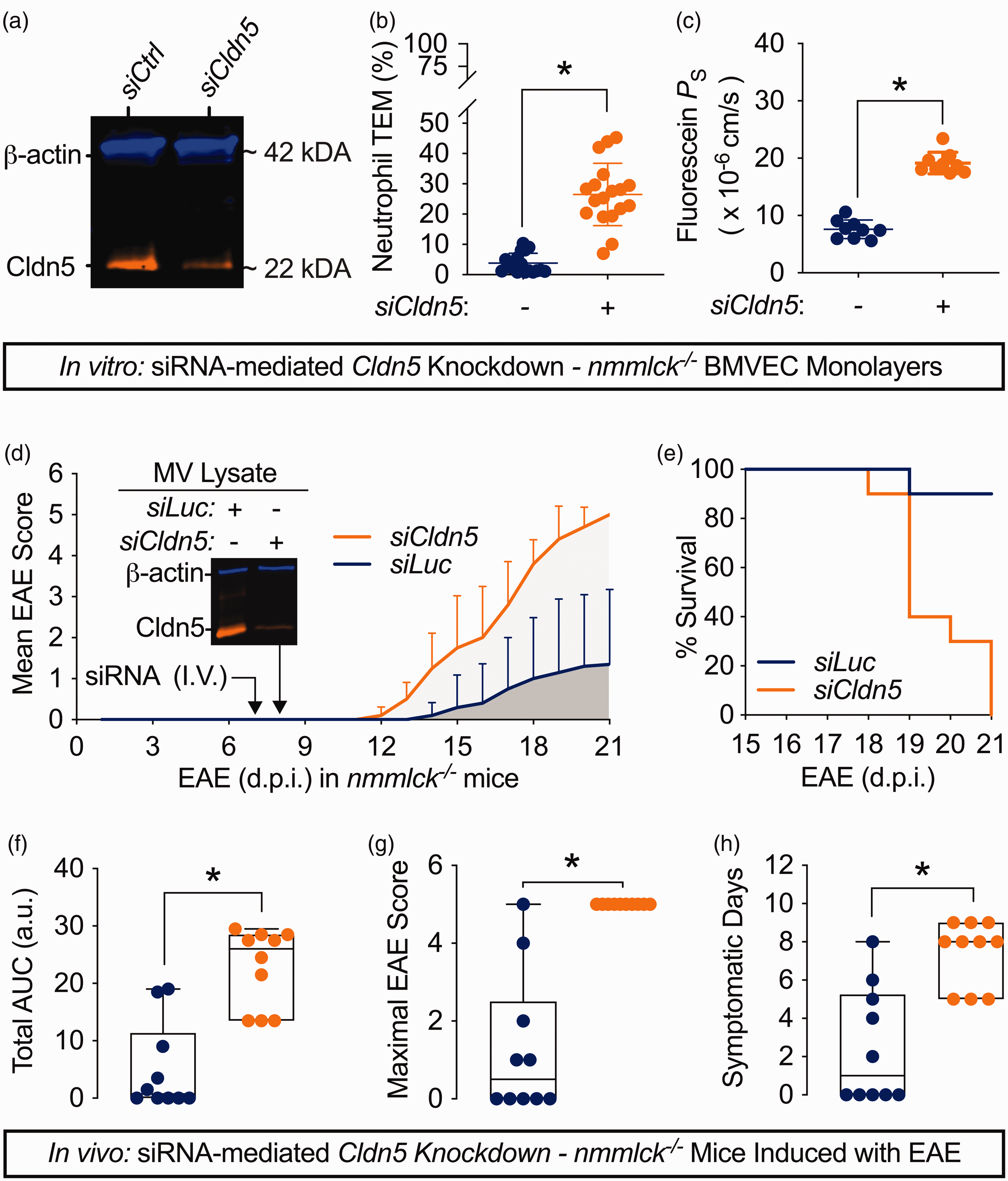
Silencing claudin-5 undoes the protective effects of nmMLCK loss in BMVECs and EAE. (a–c) nmmlck−/− BMVECs were transfected with siRNAs targeting mouse claudin-5 (siCldn5) or non-targeting siRNAs as a control (siCtrl), then used after a twenty-four-hour recovery period. Data are shown as the mean ± SD for three independent experiments (six or three inserts per experiment in (b) and (c), respectively), and analyzed by unpaired t-tests; * indicates p < 0.05. (a) Representative two-color, NIR Western blot for claudin-5 (CLDN5, orange) and β-actin (blue) to confirm knockdown. (b) The fraction of naïve nmmlck+/+ neutrophils that migrated across siCtrl- or siCldn5-transfected monolayers after six hours using fMLF as a chemoattractant. (c) The apparent permeability coefficient for fluorescein (PS) across siCtrl- or siCldn5-transfected monolayers. (d–h) EAE was induced in twenty nmmlck−/− mice. Seven days post-induction (d.p.i.), half of the mice received siRNAs targeting Cldn5 (siCldn5, orange), while the other half received siRNAs targeting luciferase (siLuc, blue). (d) Disease was evaluated with a 5-point rubric through twenty-one d.p.i. Data are shown as the mean ± SD and analyzed with a Mann-Whitney U Test; * indicates p < 0.05. Also, a representative two-color, NIR Western blot for claudin-5 (CLDN5, orange) and β-actin (blue) in microvessel (MV) lysates from a separate cohort of naïve nmmlck−/− mice is provided to show knockdown efficiency after twenty-four hours. (e) Kaplan-Meier curve for siCldn5-injected (orange) or siLuc-injected (blue) nmmlck−/− mice between 15–21 d.p.i. (f–h) Based on (d), disease severity was evaluated for cumulative disease burden via total area under the curve (AUC) (f), maximal EAE score (g), and days with symptoms (h). Data are shown as the median and IQRS and analyzed with a Mann-Whitney U Test; * indicates p < 0.05. Each dot reflects an individual mouse.
Discussion
BBB dysfunction is a quintessential hallmark of MS and its animal models.2,53,54 However, the mechanisms governing BBB function and the contributions of neutrophils in disease remain poorly understood. Here, we used nmMLCK-knockout mice and primary co-cultures with mouse BMVECs and neutrophils to explore both topics.
Claudin-5
While the barrier-forming properties of claudin-5 have been known since its discovery,40,55 recent studies underscore its vital importance to CNS health. Genetic disorders with mutations in its first extracellular loop, a conserved region responsible for charge and size selectivity, 24 are associated with developmental delay, seizures, and hemiplegia.56,57 Similarly, its dysregulation has been reported for psychiatric disorders,58 –60 with microdeletion of 22q11.2, in which CLDN5 resides, a known major genetic risk factor for schizophrenia. 59 Thus, claudin-5 is an attractive therapeutic target in neuroinflammatory disorders.4,29 For instance, Greene et al. reported that RepSox, a TGF-β signaling inhibitor, 61 enhances the barrier of hCMEC/D3 monolayers via claudin-5 and attenuates BBB disruption and neuroinflammation in mice induced with the kainic acid model of temporal lobe epilepsy. 62 Similarly, we showed that DMAQ-B1, an insulin receptor agonist, 63 enhances the barrier of primary mouse BMVEC monolayers via claudin-5 and preserves BBB integrity in pre-onset EAE. 19
Here, to connect the protective effects of nmMLCK deficiency to claudin‑5 expression, we modulated claudin-5 in vivo and in primary BMVECs. First and foremost, its transient silencing in EAE-induced nmmlck−/− mice ended in complete mortality (Figure 7(e)). Long-term claudin-5 loss is lethal in endothelial-restricted inducible knockdown mice 59 or knockout mice. 64 However, transient knockdown with the siRNAs and delivery system reported by Campbell et al. was non-lethal in healthy mice and caused no overt differences in histopathological or blood chemistry analyses. 43 Thus, we attribute the deaths in the siCldn5 group to transient silencing specifically during neuroinflammation. Additionally, we found an inverse relationship between the barrier quality of nmmlck+/+ BMVECs and neutrophil migration. Silencing enhanced migration (Figure 6(c)) at the cost of barrier integrity (Figure 6(d)). Conversely, overexpression suppressed migration (Figure 6(f)) and bolstered barrier integrity (Figure 6(g)). Our findings highlight the therapeutic potential of claudin-5 for preserving or restoring BBB integrity and limiting neutrophil infiltration in patients with neuroinflammatory disorders.
nmMLCK
In the periphery, nmMLCK modulates EC function via multiple pathways.65,66 However, little is known about its roles in BBB function or CNS disorders. While the literature is replete with studies using the pan-MLCK inhibitor ML-7,67 –77 which also affects smooth muscle MLCK, 78 only two nmMLCK-specific reports exist. Previously, we showed that nmMLCK is required for the occupancy of the transcription factors β-catenin and FoxO1 at a repressor region of Cldn5 in primary mouse BMVECs stimulated with the cytokine interleukin-1β (IL-1β). 30 Separately, Braun et al. reported that nmmlck−/− mice resisted cerebral microbleeds in a dietary model of hyperhomocysteinemia, but no BMVEC-focused analyses were mentioned. 79
Here, we expanded the current body of knowledge on several fronts. First, EAE-induced nmmlck−/− mice had mild disease (Figure 1(a)), which agrees with a previous EAE study that administered ML-7. 69 This was accompanied by myelin sparing in the cerebellum based on MBP (Figure 1(f)). Next, nmmlck−/− mice had less neutrophil infiltration (Figures 2(b) and 2(d)), less leakage of small or large solutes (Supplementary Figure S2, Figures 3 (c–e)), and fewer areas with vascular leakage (Figure 3(b)). In line, nmmlck−/− BMVEC monolayers resisted loss of barrier quality (Figures 5(a) and 5(c)) and neutrophil TEM (Figures 5(d) and 5(e)), the latter of which parallels studies with ML-7 to impede monocyte TEM across BMVECs.73,74,80 Finally, the protective effects of nmMLCK deficiency were undone with claudin-5 silencing in EAE-induced nmmlck−/− mice (Figure 7(d)) or nmmlck−/− BMVECs (Figures 7(b) and 7(c)). These results support a pathogenic role for nmMLCK in BMVECs during EAE that involves BBB dysfunction and neutrophil infiltration. Future studies that develop nmMLCK-specific inhibitors are warranted to assess their potential during neuroinflammation.
Neutrophils
Since most patient specimens come from autopsy and reflect an end stage of disease, 81 the direct study of neutrophils in chronic CNS disorders like MS is difficult. 15 Their presence in lesions is rarely caught except in severe types like opticospinal MS or fulminant MS,82 –84 or in patients experiencing rebound.18,85 Consequently, the current body of knowledge relies on animal models. Neutrophil-focused studies report that their presence in the CNS starts shortly before onset and persists through peak disease in EAE.12,17,18,86,87 In line, we observed neutrophils in the cerebellum shortly before onset (Figures 2(a) and (c)). This early infiltration seems to be a critical step as neutrophil depletion shortly before onset attenuates CNS vascular leakage and paralysis, but depletion post-onset has limited effect.12,17,18
Once activated, neutrophils produce and release various factors, including inflammatory cytokines, proteases, and reactive oxygen species.21,22 Here, we adopted a holistic approach by incubating nmmlck+/+ and nmmlck−/− BMVECs with conditioned media from fMLF-stimulated neutrophils as the inflammatory stimulus. We observed a biphasic response in barrier disruption and permissiveness to neutrophil migration in nmmlck+/+ BMVEC monolayers. The first phase had rapid loss in barrier quality (Figure 5(a)) and modest neutrophil TEM (Figure 5(d)). In contrast, the second phase had sustained loss in barrier quality (Figure 5(a)) and aggressive neutrophil TEM (Figure 5(e)). Conversely, nmmlck−/− BMVECs resisted barrier loss (Figure 5(a)) and neutrophil TEM (Figures 5(d) and 5(e)) during both phases. These findings indicate that activated neutrophils secrete factors that target nmMLCK in BMVECs to disrupt barrier integrity and prime them for further neutrophil TEM. This agrees with our previous work showing that pan-MLCK inhibitors stop rapid-onset barrier hyperpermeability and cytoskeletal rearrangement induced by complement component 5a-activated neutrophils in peripheral porcine ECs. 88
One possible neutrophil-secreted factor is IL-1β. We’ve shown that nmMLCK mediates IL-1β-induced claudin-5 loss in BMVECs. 30 Separately, Lévesque et al. reported that IL-1β was upregulated in CNS-infiltrating neutrophils in EAE-induced mice and produced a cytokine profile in BMVECs favoring neutrophils. 89 Future studies that identify the responsible factors and their intracellular signaling pathways are required to assess their therapeutic potential.
Limitations
Our study is limited in two main ways. First, the MYLK gene encodes five distinct splice variants for nmMLCK. 90 However, our mice lack all variants. 33 Future studies that employ variant-specific genetic manipulation, like transgenic mice with EC-restricted expression of splice variants, 91 are required. Second, other cells express nmMLCK.34,52 To ensure that the protective phenotype in nmmlck−/− mice during EAE was not due to an impaired CD4+ T cell response, we stained cerebellums (Supplementary Figure S1(a)) and found similar abundances (Supplementary Figure S1(b)). Additionally, to exclude impaired BMVEC activation or neutrophil diapedesis, we monitored ICAM-1 and PECAM-1 in nmmlck−/− BMVECs and found similar responses (Figures 4(a) and 4(b)), plus conducted adhesion and TEM assays with nmmlck−/− BMVECs and neutrophils and found similar responses (Figures 4(e) and 4(c)). Therefore, we attribute the in vivo findings of attenuated BBB dysfunction and neutrophil infiltration to the lack of nmMLCK in BMVECs which were resilient to neutrophil-mediated barrier dysfunction (Figures 5(a) and 5(c)) and neutrophil migration (Figures 5(d) and 5(e)). In support of this, Kempf et al. reported that endothelial-restricted reconstitution of nmMLCK in nmmlck−/− mice is sufficient to restore neutrophil infiltration and lung barrier dysfunction in a dual endotoxic shock-mechanical injury model. 92 Regardless, future studies with cell-specific knockout mice are required to define the contribution of nmMLCK in BMVECs to BBB dysfunction and EAE disease progression.
Conclusion
Our primary cell co-culture models support a pathogenic role for nmMLCK in BMVECs during EAE that involves BBB dysfunction and neutrophil infiltration, suggesting that its antagonism during neuroinflammation could benefit patients. Further support for this would come from studies that use BMVEC-restricted conditional knockout mice to define its relative contribution to disease progression. Also, our findings reveal that claudin-5 contributes not only to the physical barrier properties of BMVECs by limiting small solute leakage, which is well-known, but also to its immune barrier properties by serving as a barricade to neutrophil migration. Future studies that define the molecular underpinnings of these neutrophil-endothelial interactions are warranted to assess their therapeutic potential to mitigate BBB dysfunction and neutrophil infiltration during neuroinflammation.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X251318620 - Supplemental material for Progression of experimental autoimmune encephalomyelitis in mice and neutrophil-mediated blood-brain barrier dysfunction requires non-muscle myosin light chain kinase
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X251318620 for Progression of experimental autoimmune encephalomyelitis in mice and neutrophil-mediated blood-brain barrier dysfunction requires non-muscle myosin light chain kinase by Richard S Beard Jr, Brian A Hoettels, Jessica M McAllister, Jamie E Meegan, Travis S Wertz, Desiree A Self, Dylan E Hrkach, Daniel Greiner, Kristina Chapman, Nuria Villalba, Xiaoyuan Yang, Byeong J Cha, Cheryl L Jorcyk, Julia T Oxford, Mack H Wu and Sarah Y Yuan in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Data availability
All research data are available from the corresponding author upon reasonable request.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by funding from the National Institutes of Health grants HL150732 (S.Y.Y.), GM142110 (S.Y.Y. and M.H.W.), NS110934 (R.S.B.J.), GM143138 (M.H.W.), GM109095 and GM103408 (R.S.B.J. and J.T.O.).
Acknowledgements
The authors acknowledge Ofeira Faapouli, Raquel Brown, Cindy Keller-Peck, Lindsey Swaby, and the late Jonathan Overstreet (in memoriam) for their excellent technical support, plus the Advanced Microscopy and Cell Imaging core at University of South Florida and the Biomedical Research Institute at Boise State University for their services.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
S.Y.Y. and R.S.B.J. conceived and designed the study. R.S.B.J. performed, analyzed, and interpreted most of the experiments. B.A.H. contributed to drafting the manuscript, data analysis, and figure preparation. J.E.M., D.A.S., and X.Y. participated in neutrophil isolation, plus the design and analysis of transendothelial migration assays. D.E.H. performed and analyzed solute permeability assays. J.M.M., D.G., K.C. and N.V. participated in image acquisition and analyses. T.S.W. and D.A.S. assisted in histological preparations and contributed to the data analysis of all animal experiments. B.J.C. assisted with confocal imaging and analyses. C.L.J. and J.T.O. contributed to cell culture data analysis and manuscript preparations. M.H.W. provided the necessary reagents and participated in designing the in vivo knockdown studies. S.Y.Y. initiated, directed, and sponsored the work through all levels of development. All the authors critically discussed the results and approved the manuscript.
Supplementary material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.