
Abstract
Following ischemic stroke, substance P (SP)-mediated neurogenic inflammation is associated with profound blood-brain barrier (BBB) dysfunction, cerebral edema, and elevated intracranial pressure (ICP). SP elicits its effects by binding the neurokinin 1 tachykinin receptor (NK1-R), with administration of an NK1-R antagonist shown to ameliorate BBB dysfunction and cerebral edema in rodent and permanent ovine stroke models. Given the importance of reperfusion in clinical stroke, this study examined the efficacy of NK1-R antagonist treatment in reducing cerebral edema and ICP in an ovine model of transient middle cerebral artery occlusion (tMCAo). Anesthetized sheep (n = 24) were subject to 2-hours tMCAo and randomized (n = 6/group) to receive early NK1-R treatment (days 1–3 post-stroke), delayed NK1-R treatment (day 5 post-stroke), or saline vehicle. At 6-days post-stroke animals were re-anaesthetized and ICP measured, followed by MRI to evaluate infarction, edema and BBB dysfunction. Following both early and delayed NK1-R antagonist administration, ICP was significantly reduced on day 6 compared to vehicle animals (p < 0.05), accompanied by a reduction in cerebral edema, midline shift and BBB dysfunction (p < 0.05). This study demonstrates that NK1-R antagonist treatment is an effective novel therapy for cerebral edema and elevated ICP following stroke in an ovine model, warranting future clinical evaluation.
Introduction
Cerebral edema is associated with poor outcomes following ischemic stroke.1,2 The advent of reperfusion therapies has decreased the prevalence of malignant edema,3 –5 however, successfully reperfused patients with large infarct cores 4 may still develop cerebral edema with consequent poor outcomes. At present, pharmacological therapy includes hyperosmolar agents that have limited efficacy, 6 and may be associated with rebound edema.7 –9 Surgical decompressive hemicraniectomy (DC)10,11 is performed in selected patients with malignant middle cerebral artery infarction and improves survival and function, 12 though outcomes in patients >60 years are less favorable.12 –14 As such, identifying an efficacious pharmacotherapy that targets the molecular mechanisms underlying edema genesis and prevents its formation represents an unmet need in neurointensive medicine.
Neurogenic inflammation has been identified as a potential therapeutic target to attenuate cerebral edema.15 –19 In ischemic stroke, neurogenic inflammation is initiated due to noxious stimulation of primary afferent nerves present in high densities around cerebral blood vessels, leading to the aberrant release of neuropeptide substance P (SP).20,21 SP preferentially binds the neurokinin 1 tachykinin receptor (NK1-R), a G-protein coupled receptor expressed in endothelial cells of the blood-brain barrier (BBB). 22 NK1-R are abundant within endothelial caveolae, which facilitate transcellular transport across the barrier.23,24 Following stroke, SP binding to the NK1-R compromises BBB integrity by increasing transcellular transport of osmotically-active molecules, such as albumin, across the endothelium. 25 This drives an abnormal accumulation of water in the cerebral parenchyma and consequent rise in ICP. We have shown in both rodent and ovine species that increased perivascular SP is associated with BBB dysfunction, cerebral edema, elevated ICP and poor functional outcome post-stroke.15,18,26 Furthermore, administration of an NK1-R antagonist reduces BBB permeability, cerebral edema and functional deficits post-stroke in rodent models.15,17,27,28
Numerous putative stroke agents developed in rodents have failed to demonstrate clinical efficacy, 29 prompting more rigorous pre-clinical screening in accordance with the Stroke Therapy Academic and Industry Roundtable (STAIR) guidelines. 30 For promising therapeutics such as the NK1-R antagonist, after demonstration of efficacy in rodents, additional screening in a large animal species (i.e. ovine, porcine, non-human primate) is recommended to increase the likelihood of successful clinical translation. 30 Advances in acute stroke management have also highlighted the need for more reperfusion-centric pre-clinical stroke research, 31 particularly relevant given the extended window for reperfusion with intravenous thrombolysis32 –34 and endovascular clot retrieval.35 –38 Large animal models encompassing both permanent and transient occlusion thereby have utility in appropriately replicating the clinical scenario, establishing the development of edema (particularly given the higher proportion of white matter compared with rodent counterparts39,40; an important consideration given the propensity for vasogenic edema to develop in the white matter 41 ) and providing a window for therapeutic intervention with novel anti-edema agents.
In line with pre-clinical recommendations, we have previously determined the temporal profile of cerebral edema and elevated ICP following both permanent 42 and transient stroke 43 in an ovine model. Specifically, following permanent middle cerebral artery occlusion (MCAo), significant cerebral edema and elevated ICP was observed within 24-hours of stroke onset, 42 whereas following 2-hours transient MCAo (tMCAo) with reperfusion, ICP was shown to be significantly elevated at 5- and 6-days post-ictus, accompanied by profound midline shift. 43 We subsequently demonstrated that NK1-R antagonist treatment produced significant reductions in ICP, comparable to that with DC following permanent MCAo. 18 However, whether NK1-R antagonist treatment is also effective following stroke with reperfusion in a large animal model remains unclear. As such, this study sought to assess the efficacy of NK1-R antagonist treatment following tMCAo in an ovine model. Given the delayed nature of cerebral edema and elevated ICP following stroke with reperfusion both clinically (3–5 days) and in the ovine model (5–6 days) 43 two different treatment regimens were assessed to determine if the NK1-R antagonist could produce a clinically meaningful reduction in ICP when administered either early in the evolution of cerebral edema (days 1–3, before elevated ICP was established), or later in the evolution (day 5, when ICP was already elevated), post-stroke.
Materials and methods
Ethics statement
This study was approved by the South Australian Health and Medical Research Institute (SAHMRI) Animal Ethics Committee (104/11; SAM141, SAM3). All experiments were conducted in accordance with the Australian National Health and Medical Research Council code of care and use of animals for scientific purposes (8th Edition, 2013) and adhere with the Animal Research Reporting of In Vivo Experiments (ARRIVE 2.0) guidelines. 44
Experimental design
Twenty-four Merino sheep (Ovis aries) were used in this study (n = 12 wethers (M); n = 12 ewes (F), 61 ± 6 kg, 18–36-months old). The minimum number of animals were used per experimental group, based on previous studies (n = 6/group). Animals were sourced from a single farm in Gum Greek, South Australia, and transported to the SAHMRI Preclinical Imaging Research Laboratories, and acclimated for 2-weeks in conventional outdoor paddocks prior to experimental procedures. Animals were preoperatively randomized to one of the following treatment regimens: 1) early treatment (NK1-R antagonist administered consecutively on days 1–3 post-stroke, n = 6; 3 M; 3 F); 2) late treatment (NK1-R antagonist administered at 5-days post-stroke, n = 6; 3 M; 3 F); or 3) saline vehicle (n = 6; 3 M; 3 F). Stroke was induced on day 0, and ICP monitoring and magnetic resonance imaging (MRI) was performed on day 6 post-stroke following which animals were euthanized (Figure 1). A separate cohort of animals (n = 6; 3 M; 3 F) underwent late NK1-R antagonist treatment (administered at 5-days post-stroke) and repeat whole blood sample collection at 4 hourly intervals for assessment of pharmacokinetics (PK).
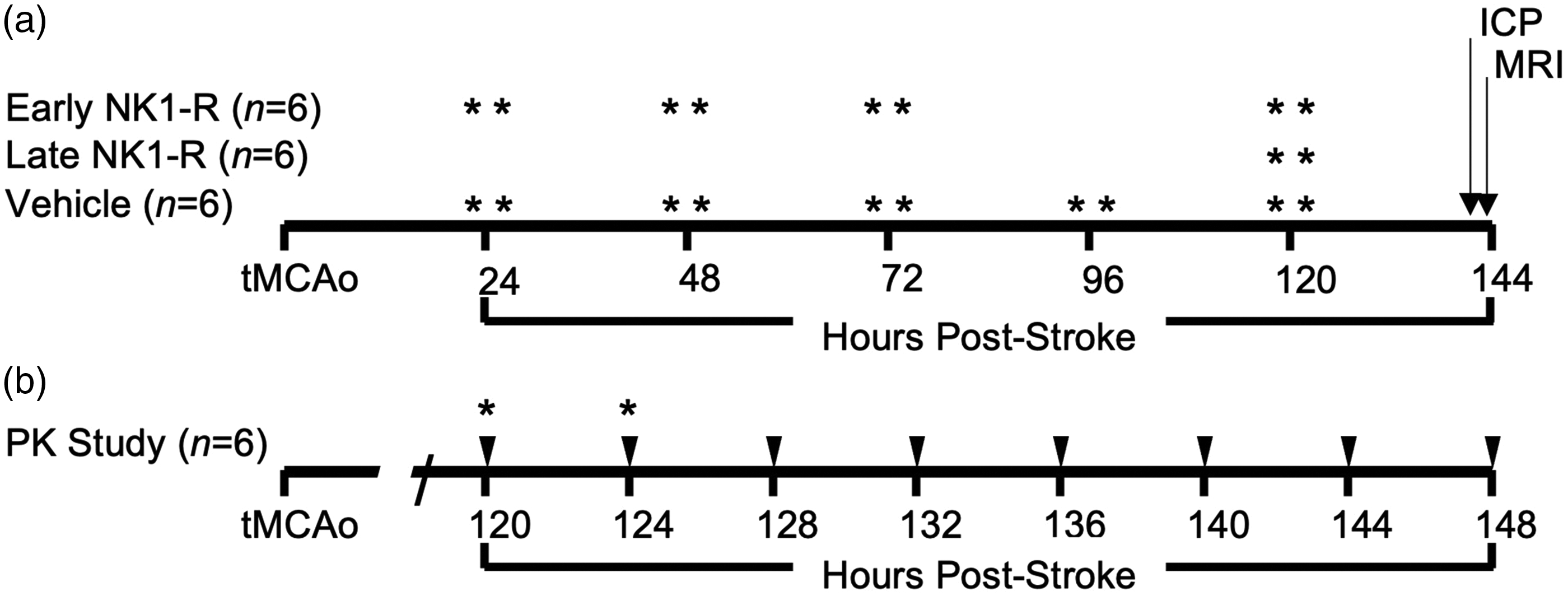
Experimental timelines. (a) Animals underwent 2-hour transient middle cerebral artery occlusion (tMCAo) followed by daily injections of either saline (vehicle), 2 bolus injections NK1-R antagonist (indicated by *) at 24–72 hours (early NK1-R group) or 2 bolus injections NK1-R antagonist on day 5 (120 hours; late NK1-R group) post-stroke. At 6 days (144 hours) post-stroke, animals underwent intracranial pressure (ICP) monitoring and magnetic resonance imaging (MRI) before euthanasia and brain collection and (b) animals underwent 2-hour tMCAo. On day 5 post-stroke animals received 2 bolus injections of the NK1-R antagonist (indicated by *). Blood was collected immediately prior to the first bolus and every 4 hours post-injection (indicated by the arrows).
Surgical approach
Preoperative preparation
Animals were fasted for 12-hours preoperatively to reduce intraoperative regurgitation. Anesthesia was induced via intramuscular (IM) administration of ketamine (0.05 mL/kg, 100 mg/kg injection, CEVA, Australia) and diazepam (0.08 mL/kg, 5 mg/mL injection, Pamlin, CEVA, Australia). Animals were intubated and a jugular catheter (18 G, Terumo SURFLO, Japan) inserted for delivery of intraoperative and postoperative (as required) crystalloid fluids (Hartmann’s solution, Baxter Health, Australia). Animals were placed on their left side on the operating table and anesthesia maintained throughout surgery with inhalational isoflurane (1.5–2.0%; mixed in 3 L air and 500 mL oxygen; Henry Schein, Australia) and continuous ketamine infusion via jugular line (2 mL/hour). An arterial catheter (20 G, Terumo SURFLO, Japan) was placed in the distal hindlimb for arterial blood gas sampling, with the catheter regularly flushed via a 500 mL bag of 0.9% sodium chloride (Baxter Health, Australia) attached to a pressurized bag pump (maintained at 300 mmHg). A pediatric blood pressure cuff (Easy Care Cuff, Phillips, Australia) was placed on the proximal forelimb for non-invasive measure of mean arterial blood pressure (MABP), recorded at 5-minute intervals.
Intraoperative procedures
tMCAo with reperfusion surgery was performed as previously described. 43 Briefly, a pterional craniotomy was performed, dura incised, MCA visualized, and an aneurysm clip (Aesculap YASARGIL Aneurysm Clip, Germany) applied to the proximal MCA and left in situ for 2-hours, then removed to achieve reperfusion. The dura was closed watertight with synthetic regeneration matrix (Durepair, Medtronic, USA) and a cranioplasty performed using autologous bone and polyacrylic cement (Sledgehammer, Keystone, Germany) to restore intracranial dynamics. The muscle was reopposed using a horizontal mattress suture technique (Vicryl, ETHICON, Australia), and the incision site treated with 1.0 mL 0.5% subcutaneous local anesthetic bupivacaine hydrochloride (Marcain, 5 mg/mL, AstraZeneca, Australia). Hourly arterial blood samples were analyzed (OPTI-3 CCA Electrolyte and Blood Gas Analyzer, Roche, USA) to ensure sodium, potassium, hematocrit, hemoglobin, bicarbonate, pO2 and pCO2 levels were maintained within normal ovine limits.
Postoperative recovery
Animals were removed from anesthesia, extubated, and transported to a postoperative recovery pen. Once lucid, subcutaneous non-steroidal anti-inflammatory (NSAID, 0.7 mg/kg, 50 mg/mL every 12-hours, Carprofen, Norbrook, Australia) and IM buprenorphine (Temgesic, 1.0 mL, 324 μg/mL buprenorphine hydrochloride, Reckitt Benckiser, Australia) were administered for pain relief, and IM procaine benzylpenicillin antibiotic (1 mL/25 kg every 12-hours, Depocillin, Intervet, Australia). NSAID and antibiotic treatment was continued for 3-days postoperatively, and as required thereafter. Clinical record sheets were maintained at least twice daily, including assessment of clinical signs of stroke and general wellbeing, including urine and fecal output, food and water intake, and signs of apathy. Animals remained in indoor housing for the entire postoperative period, monitored remotely via 24-hour closed-circuit television.
NK1-R antagonist administration
Animals received intravenous (IV) boluses of either the NK1-R antagonist (1 mg/kg EU-C-001; Australian Patent AU2002328837B2; donated by PresSura Pharmaceuticals) 45 or saline vehicle (1 mL/kg 0.9% sodium chloride, Baxter Health, Australia) administered over the course of 10-minutes via jugular catheter (Figure 1). In the early treatment group, animals received two boluses of treatment daily on days 1–3 post-stroke (treatment administered at 24-, 29-, 48-, 53-, 72- and 77-hours post-stroke). In the late treatment group, animals received two boluses of treatment on day 5 alone (120- and 124-hours post-stroke). Vehicle animals received two boluses of saline daily on days 1–5 post-stroke. Treatment timing was informed by our previous studies characterizing cerebral edema and elevated ICP evolution in the transient ovine stroke model 43 and dosing based on our previous studies of NK1-R treatment in the ovine permanent MCAo model. 18
NK1-R antagonist pharmacokinetic assessment
An additional cohort of sheep underwent blood collection for determination of PK parameters. Following tMCAo (as above), animals were administered 2 IV boluses of NK1-R antagonist (1 mg/kg EU-C-001) at 5-days (120- and 124-hours) post-stroke. Blood samples were collected immediately prior to administration of the first bolus and at 4 (immediately prior to the second), 8-, 12-, 16-, 24- and 28-hours thereafter. Whole blood was collected via jugular catheter in potassium-EDTA anticoagulant tubes (BD Vacutainer, USA) with 12 mL of blood collected per animal, per time point. Tubes were gently inverted to ensure mixing of anticoagulant to avoid microclotting. Samples were then placed on ice for 20 minutes and centrifuged at 1500 G for 10-minutes at 18–25°C. Plasma was aliquoted into 2 mL polypropylene tubes (LoBind, PCR clean, Eppendorf, Australia) and samples snap frozen in liquid nitrogen and transferred to a −80°C freezer until analysis.
NK1-R antagonist (EU-C-001) plasma concentrations were determined by CPR Pharma Pty Ltd Services (Australia). Briefly, EU-C-001 and deuterated EU-C-001 (d4-EU-C-001, internal standard (IS)) were extracted from 50 µL plasma samples via protein precipitation extraction with 0.1% formic acid (MS grade, Honeywell Fluka, Germany) in 100% methanol (LiChrosolv, Merck, Germany). Analytes were then separated using high-performance liquid chromatography on an ACE C18-AR column and quantified using mass spectrometry (MS) on an API4000 MS/MS (SCIEX, USA) in positive multiple reaction monitoring mode. The sample run time was 5-minutes using varying concentrations of mobile phases consisting of 0.1% formic acid in 10% acetonitrile (LiChrosolv, Merck, Germany), and 0.1% formic acid in 50% acetonitrile/50% methanol (LiChrosolv, Merck, Germany). Data acquisition and processing was performed via Analyst software (SCIEX) and processed in Watson LIMS (Thermo Scientific) software. Peak area ratios (versus IS) were calculated and linear regression with 1/x2 weighting used to construct calibration curves that ranged from 0.2 to 200 ng/mL. Assay performance was assessed against quality control samples of 0.6, 15 and 150 ng/mL.
NK1-R antagonist concentrations were used to calculate PK parameters. Firstly, the volume of distribution (V) and total clearance (CL) were estimated using a one compartment model in Phoenix 64 WinNonlin (v8.3 software, Certara, USA). These were then used to calculate the elimination rate constant (k), half-life (t1/2), concentration at time 0 hours (C0) and area under the concentration-time curve from time 0 to infinity (AUC0-inf) using standard PK equations (Supplementary equations 1–4). 46
Intracranial pressure monitoring
At 6-days post-stroke, animals were re-anesthetized with IM ketamine and diazepam (as above), maintained with isoflurane (1.5–1.75%), and placed in the sphinx position on an operating table. Bilateral burr holes (5 mm) were drilled using a Codman Cranial Hand Drill (Codman & Shurtleff Inc., USA), ∼1 cm lateral to the sagittal suture and 1 cm posterior to the horn buds. The underlying dura was incised, and plastic bolts (Microsensor Skull Bolt Kit for Intraparenchymal Procedures, Codman & Shurtleff Inc., USA) secured into the burr holes. A Millar strain gauge Codman Microsensor transducer (Codman & Shurtleff Inc., USA) was used to measure ICP in each hemisphere. The nylon lead of the Microsensor was attached to a bridge amplifier and PowerLab data acquisition device (ADInstruments, Australia). LabChart software (v8, ADInstruments, Australia) installed on a computer to which the PowerLab and bridge amplifier were connected allowed for the analogue signal from the Codman microsensor to be continually recorded at frequencies of approximately 1KHz.
Transducers underwent two-point calibration checks (0 and 100 mmHg) to ensure accuracy, following which they were advanced through skull bolts ∼15 mm into the cerebral parenchyma. ICP was continually recorded for 3 hours, then transducers were removed, and two-point checks re-performed to determine the degree of probe drift during monitoring, if any. Physiological variables were recorded hourly. ICP recordings from the right (ipsilateral) hemisphere were preferentially used for analysis, with the recordings from the left (contralateral) hemisphere collected as a backup in the case of equipment failure.
Magnetic resonance imaging
Image acquisition
Following ICP recording, animals were transported to an MRI suite and maintained under anesthesia (3% isoflurane) for neuroimaging. The facility MRI was upgraded during the study, such that n = 12 animals were scanned on a 1.5 Tesla (T) Siemens Syngo2004A MRI (Siemens AG, Germany) and n = 6 animals were scanned on a 3 T Siemens Magnetom Skyra MRI (Siemens AG, Germany). The following sequences were acquired: time-of-flight magnetic resonance angiography (MRA), diffusion weighted imaging (DWI; b0 and b1000 s/mm2), T2 fluid attenuated inversion recovery (FLAIR), T1 magnetization-prepared rapid gradient-echo (T1 MP-RAGE) and T2-weighted imaging (T2w). T1 MP-RAGE images were also reacquired following IV contrast administration of 1 mL/kg gadolinium-diethylene-triamine-pentaacetic acid (Magnevist, Bayer HealthCare, Germany). Supplementary Table 1 details sequence parameters for each of the machines.
Image analysis
All data was converted to NIfTI format using MRIcroGL (https://www.nitrc.org/projects/mricrogl). MRA images were qualitatively assessed to confirm successful MCA reperfusion. 43 Infarct volume on b1000 DWI and cerebral edema volume on FLAIR was calculated using semi-automated three-dimensional (3D) segmentation tools in ITK-SNAP (v4.0.2).43,47 Midline shift was used as a surrogate marker of cerebral edema on T2w scans, measured as the distance in mm from the septum pellucidum at the level of the foramen of Monro (Horos, v3.1.1).
To determine parenchymal water content, quantification of the relaxation signal was performed on T2w images at the level of the foramen of Monro. Eight regions of interest (ROIs) measuring 10 mm2 were drawn in the ipsilateral and contralateral hemispheres. The mean signal intensity for each ROI was summed, and the average intensity for all ROIs reported for each hemisphere. Signal intensity ratios were then calculated by normalizing the signal intensity within the stroke hemisphere to the signal intensity of the contralateral hemisphere. The normalized values in the stroke affected hemisphere were exported for final interpretation.
BBB integrity was quantified on pre- and post-contrast T1 MP-RAGE images using the FMRIB Software library (FSL) (www.fmrib.ox.ac.uk/fsl) (Supplementary Figure 1).48,49 Brain masks were created with tools in ITK-SNAP to facilitate linear affine registration to an ovine atlas.50,51 Once aligned, the region of contrast extravasation in the stroke hemisphere was segmented (ITK-SNAP) and mirrored to the contralateral hemisphere. The atlas mask, hemispheric masks, and enhancement and mirrored enhancement masks were registered back to native T1 space. The mean number of voxels and total volume of the voxels was measured and expressed as a percentage of the ischemic hemisphere. The signal to noise ratio (SNR) was estimated from the hemispheres, region of enhancement and mirrored region of enhancement and pre-contrast values were subtracted from post-contrast.
Immunohistochemistry
Following MRI, animals were euthanized via jugular exsanguination under general anesthesia (3% isoflurane) and common carotid perfusion with tris-buffered saline (Sigma-Aldrich, Australia) following IV heparin administration (5000 IU/5 mL, Pfizer, USA). Brains were removed and sliced into 10 mm coronal sections using a custom matrix, then immerse-fixed in 10% neutral-buffered formalin for at least 14 days prior to being processed, embedded in paraffin wax, and sectioned coronally at 5 µm intervals. Immunohistochemical (IHC) examination was performed via albumin (1:2000, Dako Pty Ltd, USA; A0001), caveolin-1 (1:1000 EDTA retrieval, Cell Signalling Technology Pty Ltd, USA; 3238) and claudin-5 (1:500 EDTA retrieval, Invitrogen Pty Ltd, Australia; 35-2500) to examine vasogenic edema, transcellular and paracellular permeability, respectively. Sections were mounted and scanned at 40 × magnification (NanoZoomer 2.0-RS, Hamamatsu Photonics, Japan).
IHC quantification
Cumulative thresholding analysis 52 of IHC was performed in the peri-infarct area and corresponding region in the contralateral hemisphere using custom QuPath (v0.5.0) script. For each antibody the 3,3′-Diaminobenzidine stain (Sigma-Aldrich, D12384, Australia) was isolated and the number of pixels at each of the pixel intensities (0–2; 0.01 increments) determined. For albumin analysis, 2 ROIs were placed in the peri-infarct area measuring 1500 × 1500 µm2 and mirrored to the contralateral hemisphere. For caveolin-1 and claudin-5, 10 arterioles in the peri-infarct and corresponding area in the contralateral hemisphere were isolated. Values were converted into a percentage of thresholded material across each pixel intensity level, and the area under the curve (AUC) quantified for each treatment group.
Statistical analysis
Data are expressed as mean±SD. Data were tested for normality using the Shapiro-Wilk normality test, or by assessing Q-Q plots of residuals. Physiological data (MABP, pH, pO2, pCO2) were analyzed using one-way analysis of variance (ANOVA) and Tukey’s post-hoc tests (Prism v.9.4.0, GraphPad, USA). ICP measurements underwent logarithmic exponential transformation as previously described42,53 with values reported at hourly intervals for 3 hours and analyzed by two-way ANOVA with Tukey’s post-hoc tests. Cerebral perfusion pressure (CPP) was calculated by subtracting hourly ICP measurements from MABP at the corresponding time and analyzed via two-way ANOVA and Tukey’s post-hoc. MRI parameters (infarct volume, cerebral edema, midline shift, and T2w prolongation) were analyzed by one-way ANOVA followed by Tukey’s post-hoc, and contrast extravasation on T1 MP-RAGE by paired t-test or one-way ANOVA with Tukey’s post-hoc. To determine the effect of sex on all outcome measures collected, two-way ANOVAs with Sidak’s post-hoc tests were performed. Spearman rank correlations were run between cerebral edema and midline shift, edema and T2w prolongation, ICP and cerebral edema, contrast extravasation and midline shift and contrast extravasation and cerebral edema to determine if a relationship was present. IHC analysis was performed by one-way ANOVA with Sidak’s post-hoc. A p-value <0.05 and r value >0.5 were considered significant.
Results
Surgery, mortality and postoperative course
No premature mortality was observed, with all animals surviving to terminal experimental endpoints and included in analyses. Reperfusion of the MCA was achieved in all animals, confirmed by MRA (data not shown).
Physiological parameters
Physiological parameters (Table 1) for pCO2 (F2, 15 =2.75), pO2 (F2, 15 = 3.05), pH (F2, 15 = 1.61) and MABP (F2, 15 = 1.93) were comparable between all groups throughout surgery (p > 0.05), irrespective of sex (p > 0.05). During ICP monitoring, pH (F2, 15 = 1.99) and pO2 (F2, 15 = 0.10) were normal between groups and comparable between sexes (p > 0.05). However, MABP in vehicle animals was significantly higher than both the late (p = 0.0012) and early (p = 0.031) NK1-R antagonist treatment groups (F2, 15 = 10.36), although no differences were observed between males and females (p > 0.05). pCO2 was also higher in vehicle animals compared with early (p = 0.0016) and late (p < 0.0001) NK1-R antagonist treatment groups (F2, 15 =18.86), but no effect of sex was observed (p > 0.05).
Physiological variables (mean ± SD).
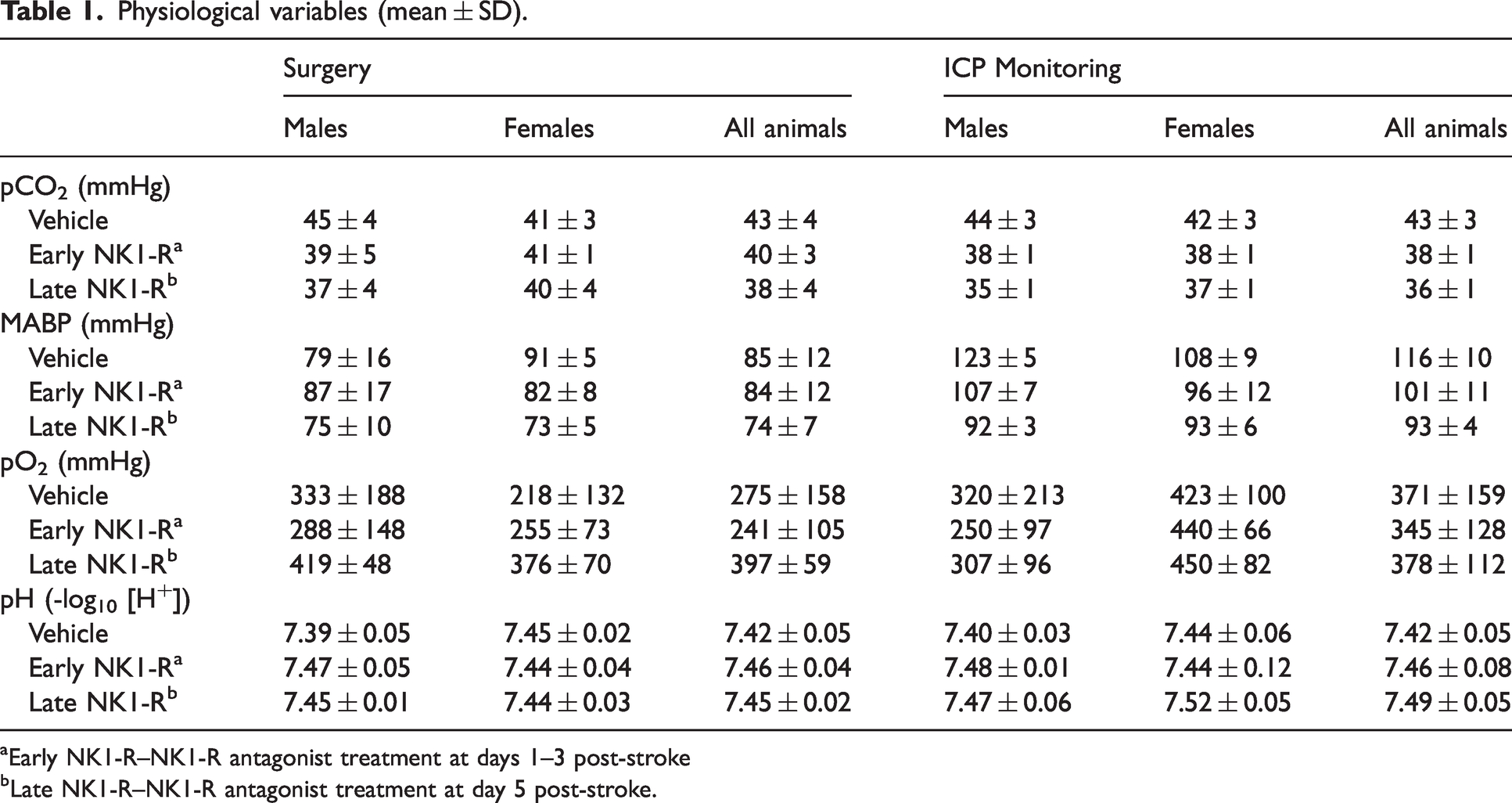
Early NK1-R–NK1-R antagonist treatment at days 1–3 post-stroke
Late NK1-R–NK1-R antagonist treatment at day 5 post-stroke.
Intracranial pressure
At 6-days post-stroke, ICP in vehicle animals was 17.74 ± 6.71 mmHg during the first hour of monitoring (Figure 2(a)), higher than ICP recorded in sham animals (9 ± 3 mmHg) in our previous studies. 42 Elevated ICP in vehicle animals was sustained throughout the second (20.01 ± 5.41 mmHg) and final hour (24.51 ± 5.45 mmHg) of ICP recording. ICP in the early NK1-R antagonist treatment group was less than that observed in vehicle animals (F2, 44 = 28.06). This was evident during the second (ICP 13.06 ± 3.35 mmHg, p = 0.022), and final (ICP 13.56 ± 2.76 mmHg, p = 0.0002) hour of ICP monitoring (Figure 2(a)). ICP in the late NK1-R antagonist treatment group was also less than vehicle animals, evident throughout the first (9.21 ± 4.10 mmHg, p = 0.007), second (9.64 ± 4.12 mmHg, p = 0.0005), and final hour (11.43 ± 1.97 mmHg, p < 0.0001) of ICP recording (Figure 2(a)). There were no differences in ICP between the early and late NK1-R antagonist treatment groups (p > 0.05). Despite differences in ICP between vehicle and treatment animals, no differences in CPP were observed between any of the treatment groups at any time point (F2, 44 = 0.93, all p > 0.05, Figure 2(b)). CPP and ICP was comparable between sexes irrespective of treatment group (p > 0.05, data not shown).
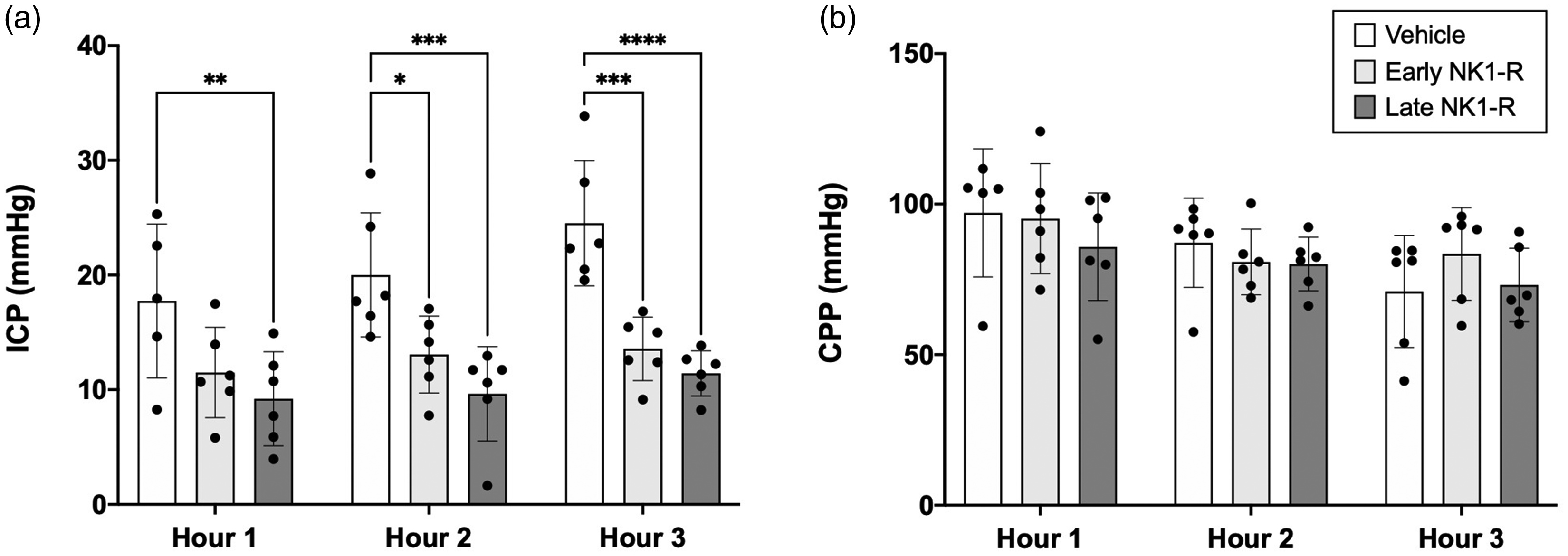
Intracranial pressure (ICP) and cerebral perfusion pressure (CPP). (a) Early NK1-R antagonist treatment administered from 1–3 days post-stroke brought about a reduction in ICP compared with vehicle, which was evident in the second (p < 0.05) and third (p < 0.001) hour of ICP recording. Delayed NK1-R antagonist treatment at day 5 post-stroke also brought about a reduction in ICP, which was evident throughout the first (p < 0.01), second (p < 0.001) and third (p < 0.0001) hour of monitoring compared with vehicle. (b) no difference in CPP was observed between treatment groups at any of the time points (p > 0.05). Data presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 compared with vehicle.
Magnetic resonance imaging
All animals displayed evidence of infarction in the territory of the MCA on MRI at 6-days post-stroke (Figure 3). Infarct volume measured on DWI was comparable in both vehicle and NK1-R antagonist-treated animals (F2, 15 = 3.21, p > 0.05; Figure 4(a)). Cerebral edema measured on FLAIR was reduced following both early (p = 0.009) and late (p = 0.022) NK1-R antagonist treatment compared with vehicle (F2, 15 =7.26; Figure 3; Figure 4(b)). This was accompanied by a significant reduction in midline shift on T2w images (F2, 15 = 12.99) in both early (0.89 ± 0.43 mm, p = 0.001) and late (1.13 ± 0.65 mm, p = 0.001) NK1-R antagonist treatment groups compared with vehicle (3.08 ± 1.09 mm; Figure 4(c)). Despite a reduction in edema and midline shift, the amount of water in the stroke hemisphere, as measured by T2w prolongation, was only reduced following late (p = 0.043) NK1-R antagonist treatment (F2, 15 = 3.68; Figure 4(d)).
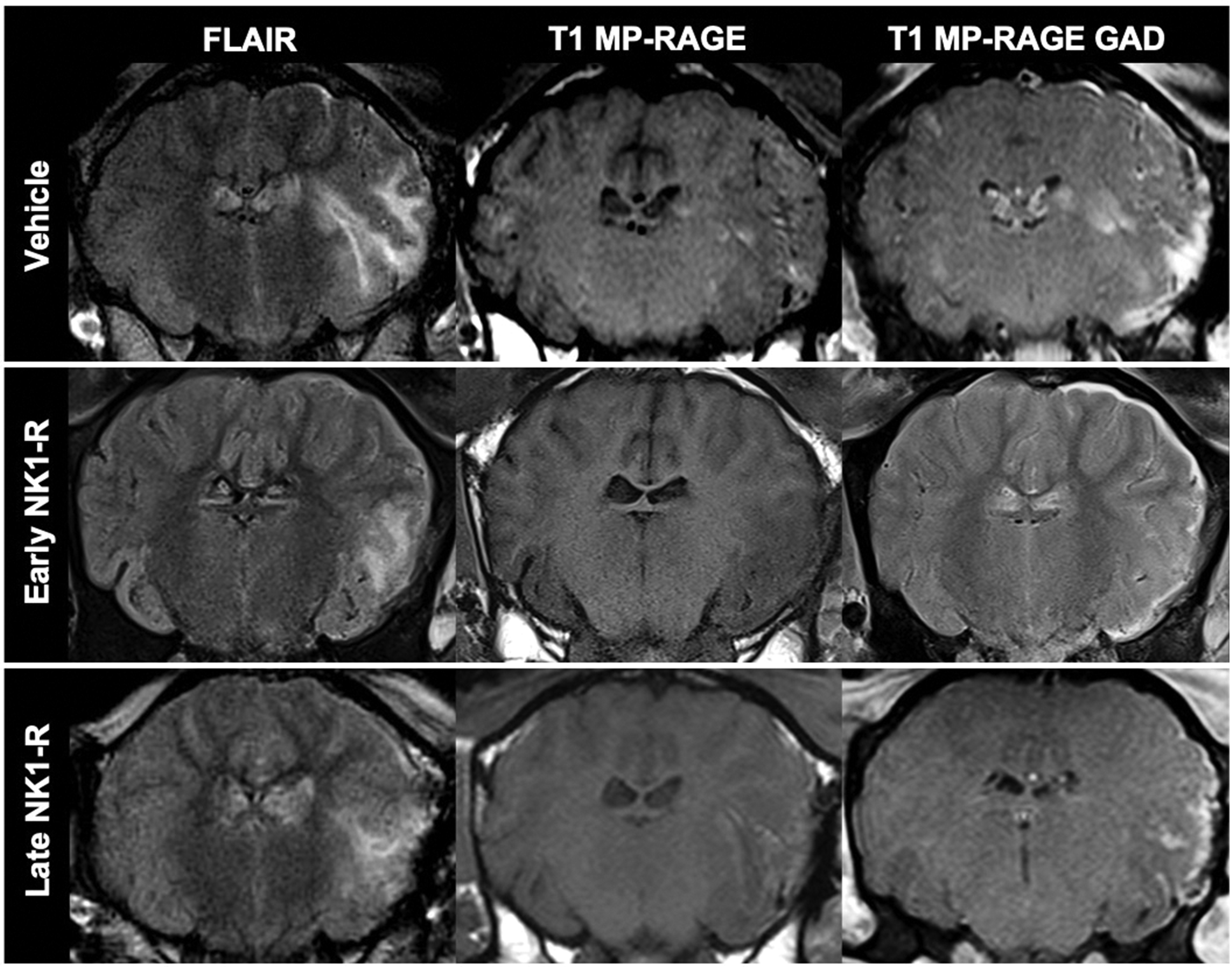
MRI findings following stroke. FLAIR images show a visible reduction in cerebral edema following both early and late NK1-R antagonist administration compared to vehicle. T1 MP-RAGE images were comparable between treatment groups prior to contrast (gadolinium; GAD) administration. Following administration, contrast in the territory of the MCA in the stroke affected hemisphere was observed on T1 MP-RAGE in all animals, although extravasation into parenchymal tissue appeared greater in vehicle compared with NK1-R treatment animals. Images shown are from the same animal/treatment group.
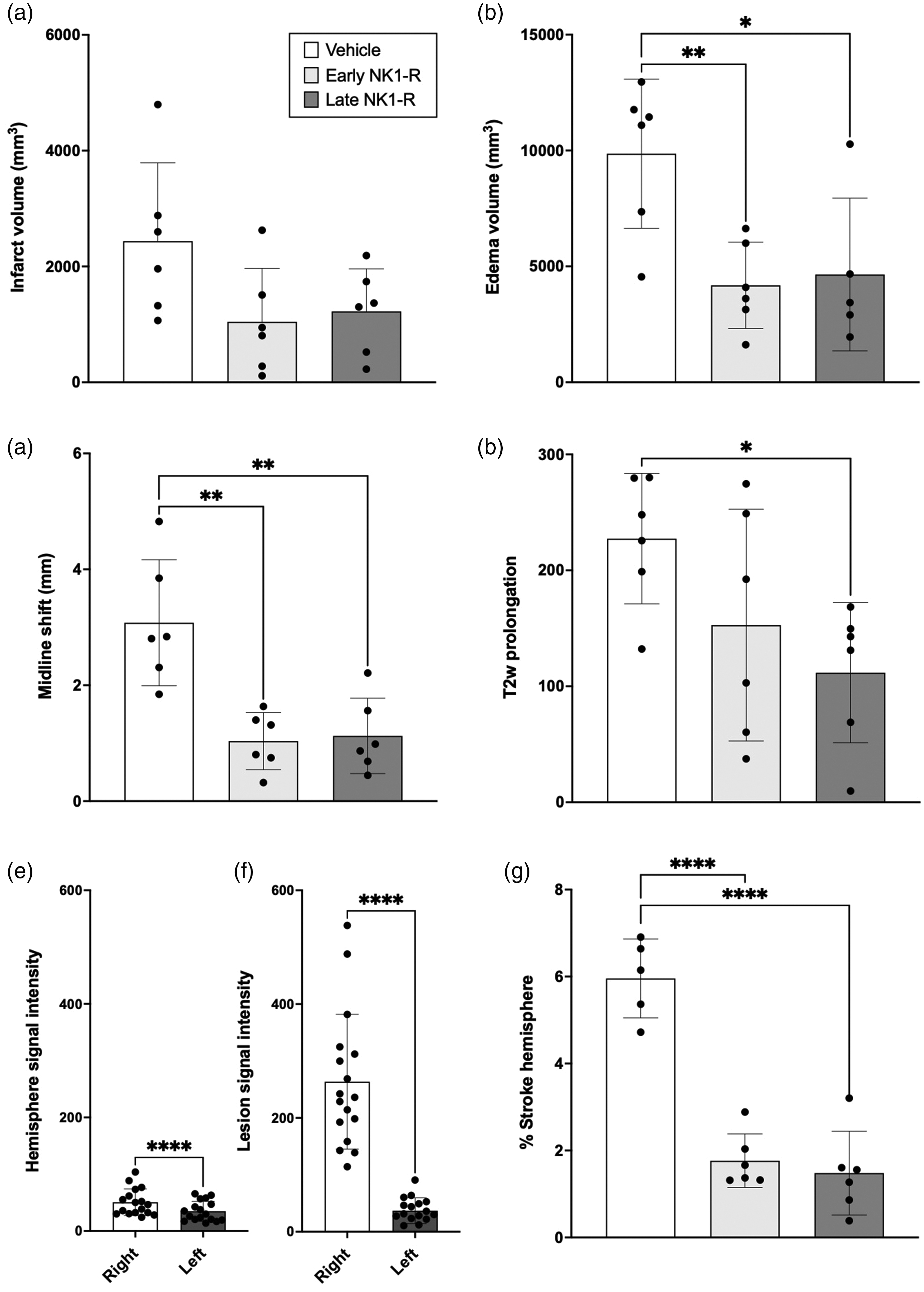
MRI assessment of infarct volume, cerebral edema, midline shift, T2w prolongation and blood-brain barrier permeability. (a) No difference in infarct volume was observed between vehicle and NK1-R antagonist treatment groups. (b) Cerebral edema was reduced following early and late NK1-R antagonist treatment, (c) accompanied by a reduction in midline shift seen in early and late treatment groups. (d) Despite a reduction in edema volume, water content in the stroke affected hemisphere was only reduced in late NK1-R antagonist treatment compared with vehicle animals. On T1 MP-RAGE images, there was significant signal enhancement in the ipsi- versus contra- lateral hemisphere (e) and lesion versus mirrored lesion (f) masks following contrast administration. The amount of contrast extravasation was greater in vehicle animals compared with both early and late NK1-R treatment groups (g). There were no differences observed between NK1-R treatment groups for edema volume, midline shift, T2w prolongation, or T1 MP-RAGE contrast extravasation (all p > 0.05). Data presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 compared with vehicle.
Analysis of T1 MP-RAGE images revealed significant contrast enhancement in the ipsilateral (50.64 ± 23.32) relative to the contralateral hemisphere (34.92 ± 17.57, p < 0.0001; Figure 4(e)), with further enhancement within the stroke lesion (263.5 ± 118.6) compared with the mirrored mask (36.87 ± 22.55, p < 0.0001, Figure 4(f)). The area of contrast extravasation was significantly greater in vehicle animals (5.96 ± 0.91%) compared with both early (1.77 ± 0.62%, p = 0.0001) and late (1.48 ± 0.96%, p = 0.0001) NK1-R treatment groups (F2, 14 = 47.5; Figures 3 and 4(g)).
No sex differences were observed for infarct volume, edema, T2w prolongation or contrast extravasation on T1 MP-RAGE (p > 0.05, data not shown). However, there was a significant sex difference in midline shift within the vehicle group, with males having a greater shift from the midline compared with females (p = 0.039, data not shown). No sex differences in midline shift were observed for either NK1-R antagonist treatment group (p > 0.05).
A strong positive correlation was observed between cerebral edema and midline shift (r = 0.83, 95% CI: 0.57–0.94, p < 0.0001), cerebral edema and T2w prolongation (r = 0.80, 95% CI: 0.52–0.93, p = 0.0002), cerebral edema and contrast extravasation (r = 0.70, 95% CI: 0.29–0.90, p < 0.004) and midline shift and contrast extravasation (r = 0.81, 95% CI: 0.54–0.93, p < 0.0001). The relationship between ICP and cerebral edema was only moderate (r = 0.64, 95% CI: 0.22–0.86, p = 0.007) (correlation data not shown).
IHC quantification
Following stroke, cumulative thresholding analysis revealed a significant effect when investigating albumin reactivity (F5, 25 = 12.02; p < 0.0001; Figure 5(a) and (b)). Post-hoc testing showed significantly higher albumin immunoreactivity in the ipsilateral peri-infarct area compared with the contralateral hemisphere (vehicle, p < 0.0001; and late NK1-R, p = 0.0063; Figure 5(a) and (b)). There was also a similar trend in the early NK1-R group (p = 0.052). However, no differences were observed between treatment groups, irrespective of hemisphere (p > 0.05). When investigating caveolin-1, no statistical differences were observed between treatment groups or hemispheres (F5, 24 = 0.35; p = 0.87; Figure 5(c) and (d)). There was a significant effect observed regarding claudin-5 immunoreactivity (F5, 28 = 6.05; p = 0.0006), with significantly less claudin-5 immunoreactivity in late NK1-R treatment animals compared with both vehicle (p = 0.0106) and early NK1-R animals (p = 0.026) in the ipsilateral hemisphere, as well as compared with early NK1-R in the contralateral hemisphere (p = 0.0125) (Figure 5(e) and (f)).
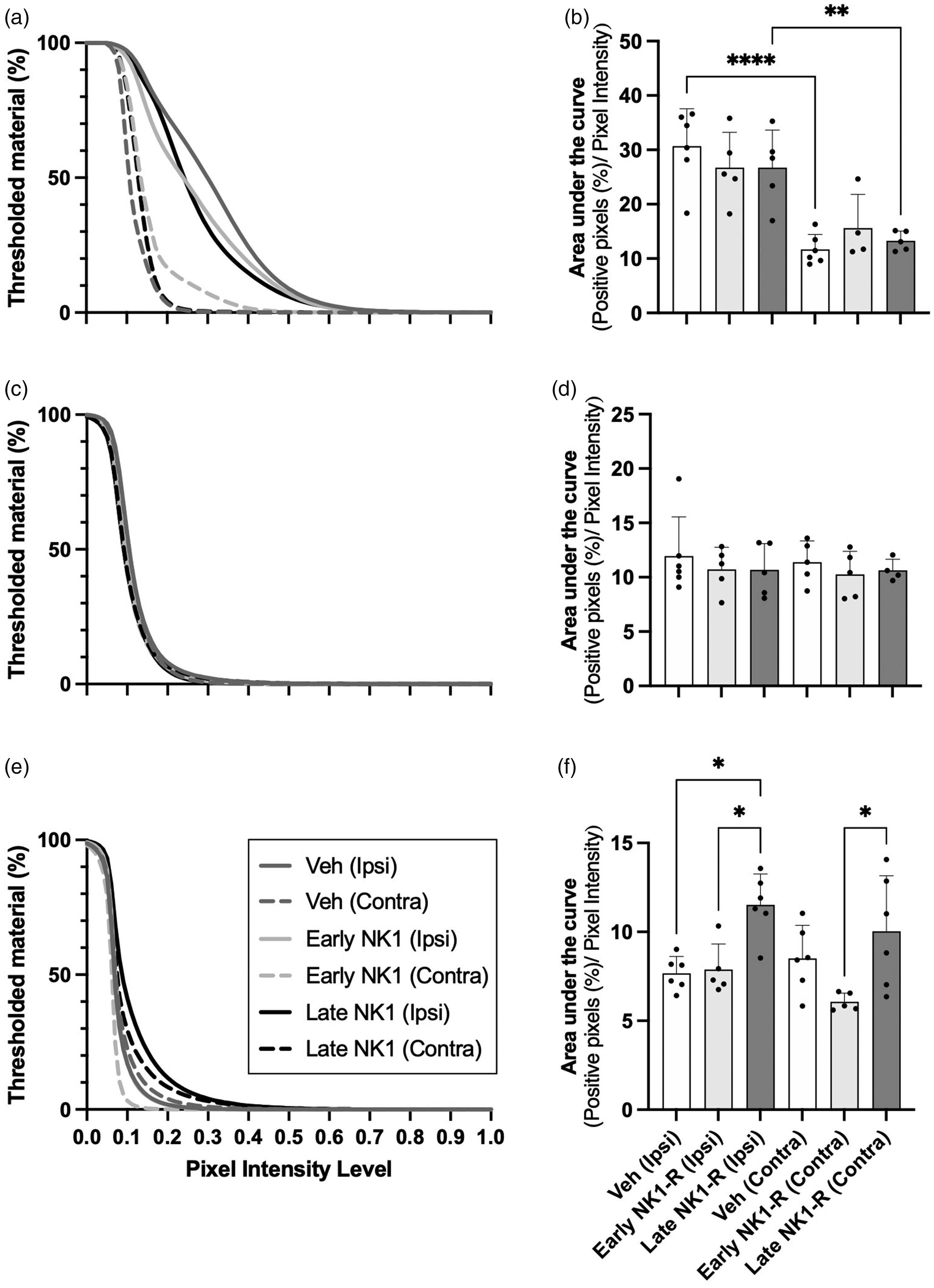
Immunohistochemical pixel intensity analysis of albumin, caveolin-1 and claudin-5. Albumin analysis revealed significant pixel intensity enhancement in the stroke versus contralateral hemisphere in the vehicle and late NK1-R treatment groups, although no other significant differences were observed (a, b). Caveolin-1 revealed no differences in pixel intensity between either hemispheres or treatment groups (c, d). Claudin-5 analysis revealed significant pixel intensity enhancement of the late NK1-R antagonist treatment group in the ipsilateral hemisphere compared with early treatment and vehicle, and contralateral hemisphere compared with early treatment, although no other differences were observed between groups or hemispheres (e, f). Data presented as mean ± SD. *p < 0.05, **p < 0.01 and ****p < 0.0001 compared with vehicle.
NK1-R antagonist pharmacokinetic parameters
PK parameters were determined from the plasma concentrations of 5 animals (n = 3M; 2 F). All data were included in the PK calculation, except for one animal where concentrations were 3.8- and 3-fold lower at the 4- and 8-hours post-administration time-points, respectively. Table 2 shows the summary PK parameters obtained of which V, CL, C0 and AUC0-inf were highly variable (2.6–4-fold) and k and t1/2 less variable (1.7-fold).
Summary of NK1-R antagonist pharmacokinetic (PK) parameters (n = 5).
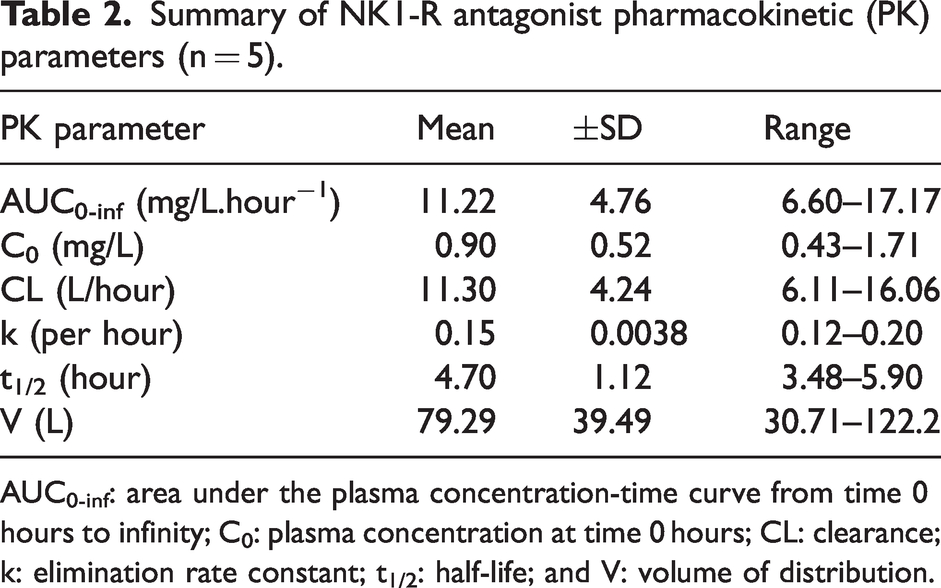
AUC0-inf: area under the plasma concentration-time curve from time 0 hours to infinity; C0: plasma concentration at time 0 hours; CL: clearance; k: elimination rate constant; t1/2: half-life; and V: volume of distribution.
Discussion
This study demonstrates the efficacy of NK1-R antagonist treatment in reducing cerebral edema and ICP following stroke with reperfusion in a large animal model. A significant and sustained reduction in ICP was observed following administration of the NK1-R antagonist either early (days 1–3 post-stroke), before the onset of cerebral edema and elevated ICP, or in a delayed fashion (5-days post-stroke), when significant cerebral edema and elevated ICP are established, accompanied by a decrease in BBB dysfunction, cerebral edema and midline shift when measured at 6-days post-stroke.
In supine, human adults, ICP ranges from 7–15 mmHg54,55 with values >20 mmHg generally considered elevated in the neurointensive care unit. 56 Although there is a paucity of disease specific literature, current guidelines recommend ICP be maintained at <22 mmHg in patients with evolving brain injury.57,58 In this study, ICP in vehicles exceeded 24 mmHg during the final hour of monitoring on day 6 post-stroke; a significant elevation compared with ICP observed in sham animals in previous studies (6–12 mmHg). 42 Current clinical guidelines outline a stepwise approach to managing elevated ICP, first seeking to reduce pressure via conservative measures, and subsequently escalating the intervention level in accordance with response to treatment.56,59 When BBB integrity is compromised, as frequently reported in the clinical literature, pharmacological management with hyperosmolar agents can paradoxically worsen injury, causing rebound elevations in ICP and brain herniation60 –63, thus often necessitating surgical intervention via DC.12 –14 Indeed, the only randomized controlled clinical trial of ICP monitoring in patients undergoing neurocritical care was negative, likely owing to the suite of non-specific side effects of ICP-lowering therapies. 64 Critically, management of elevated ICP must not be at the expense of CPP. 57 If autoregulation is preserved, a CPP of ∼70 mmHg is tolerated and may reduce ICP whilst maintaining cerebral blood flow to ischemic tissue. However, if autoregulation fails, elevated CPP increases cerebral blood volume, further exacerbating ICP. 56
The reduction in ICP observed following NK1-R treatment in this study addresses several limitations to current ICP management. Firstly, treatment produced a substantial reduction in ICP, cerebral edema and midline shift when administered consecutively on days 1–3 post-stroke. Although treatment ceased at 3-days, ICP measured at 6-days post-stroke was within ovine physiological limits, 42 suggesting that NK1-R antagonist administration was effective in reducing BBB permeability alterations, as demonstrated by reduced contrast extravasation in the stroke hemisphere on T1 MP-RAGE images, thus preventing a significant evolution of edema and accompanying rise in ICP over the ensuing days. Treatment was also effective in reducing established elevated ICP, cerebral edema and midline shift 43 following delayed administration on day 5 post-stroke. Taken together, these findings suggest an extended therapeutic window for NK1-R antagonist treatment, and an enduring ICP-lowering effect as compared to mannitol which requires repeat bolus administration approximately every 6-hours. 65 Secondly, NK1-R antagonist administration maintained CPP at ∼80 mmHg in both treatment regimens, suggesting the mechanism of ICP reduction was not due to a reduction in the size of the vascular compartment, minimizing CPP related compromise to ischemic tissue. Finally, IV administration of the NK1-R antagonist circumvents the need for invasive surgery and thus potential for iatrogenic injury and postoperative complications. Taken together with previous studies demonstrating that treatment is as effective in reducing ICP as DC following permanent MCAo, 18 our results suggest NK1-R antagonist treatment is effective in reducing ICP in both permanent and transient MCA territory infarction.
The reduction in cerebral edema and midline shift observed in both early and late treatment groups suggests that the mechanism of action may differ depending upon the treatment time-point. Indeed, early treatment is most likely exerting effects via attenuation of neuroinflammation/BBB permeability changes and resultant development of vasogenic edema, in line with our previous observations in rat and ovine stroke models.17,18,27 Given neurogenic inflammation is well characterized to occur in the acute phase post-stroke,15,17 –19 it is unlikely that delayed treatment at 5-days meaningfully impacted on this injury pathway. Instead, at 5-days post-stroke when cerebral edema is established, the most likely mechanism of action was in the facilitation of bulk fluid movement from the brain parenchyma to the vasculature, which may explain the significant reduction in parenchymal water content observed in this treatment group. Indeed, aquaporin-4 channels on astrocytic end feet are known to facilitate excess fluid clearance in vasogenic edema, 66 although further confirmatory studies are required.
Despite a reduction in BBB permeability observed following NK1-R administration on MRI, no significant differences in albumin pixel intensity on IHC were observed between groups, with only an increase in the peri-infarct region relative to the contralateral hemisphere. Although surprising, these albumin findings are consistent with our current observation of no differences in caveolin-1 immunoreactivity between groups. Such findings are not in keeping with our previous studies 18 where we have demonstrated that NK1-R antagonist treatment decreased both albumin and caveolin-1 reactivity compared to vehicle following permanent MCAo. A decrease in claudin-5 immunoreactivity in the late NK1-R group was also observed, an inconsistent finding when compared with permanent MCAo, where we saw no change in claudin-5 between groups. Although it is possible that the differences in the albumin, caveolin-1 and claudin-5 results observed between our studies may be attributed to the different models used (permanent versus transient), it is most likely a consequence of the small sample size and greater lesion heterogeneity in the transient model and thus the results should be interpreted with this in mind. In terms of identifying the mechanism driving reduced edema in the NK1-R treated animals, the current IHC findings do not conclusively support superior involvement of either caveolin-1 or claudin-5. It is important to highlight a key limitation in that shams were not conducted, thereby limiting the comparisons to post-stroke observations only. Future analysis of the NK1-R antagonist in a larger cohort (including shams), should also explore the effects of treatment on integrity of other BBB constituents (immunoglobulin G, zonula occludens-1, matrix metalloproteinases), and neuroinflammation (microglia, astrocytes, pro-inflammatory cytokines) to further elucidate the mechanisms of action in the setting of transient ischemic stroke.
Although NK1-R antagonist treatment efficacy has previously been demonstrated in ovine permanent stroke, 18 this was the first study to perform PK assessment. Analysis in a large animal species is possible given their significant blood volume, comparable to humans (sheep: ∼4 L; humans: ∼5 L), allowing for repeated blood collections. Ruminants have an intermediary metabolic clearance, and despite anatomical variation of their gastrointestinal system, metabolism is largely analogous between poly- and mono-gastric species. 67 The findings reported herein demonstrated highly variable PK parameters. Although the estrous cycle was not determined in this small cohort of animals, no significant differences in CL were observed between sexes. With regards to plasma concentrations over time, there was no evidence of a secondary adsorption or distribution peak seen up to 48-hours following administration (data not shown). Given PK analyses were performed in animals receiving the delayed treatment regimen alone, this may provide explanation as to why outcomes were most improved in delayed compared with early treatment regimens. In animals receiving early treatment on days 1–3 post-stroke, although repeat dosing was performed, given the average t1/2 was 4.7-hours, it is unlikely that concentrations of NK1-R antagonist would have been substantial by day 6 when ICP recording and MRI were performed. Nevertheless, if treatment attenuated neurogenic inflammation and reduced BBB dysfunction in the first 1-3-days, efficacious outcomes are still plausible when assessed at 6 days, as shown by the response to ICP and edema reported herein. While investigation of PK within both regimens is desirable, it was beyond the scope of the current study. It is also possible that peak concentrations were missed due to the 4 hourly blood collection (i.e. maximum peak could have been <4-hours). Future compartmental analysis of PK in a larger cohort of animals could also determine NK1-R antagonist concentrations in the brain versus the plasma, which may inform efficacy at the site of action.
Limitations to our study include the change in MRI hardware and lack of sequential acquisitions across post-stroke time-points. In clinical studies, scans obtained within 24-hours of stroke are important to determine ischaemic injury evolution on subsequent scans (for example see 2 ). Although the brains of larger animal species enable utilization of clinical neuroimaging equipment,68,69 as MRI was only obtained at a single time point, it was difficult to reliably delineate the infarct from the surrounding edema, particularly due to the potential for T2 shine through on DWI images acquired at 6-days post-stroke. Given infarct volume is a major predictor of the extent and clinical severity of cerebral edema, 70 sequential imaging is essential in future studies to comprehensively evaluate the impact of treatment on injury evolution. Continuous ICP monitoring was also not possible as the system used is not telemetric and thus could not be used in freely moving animals, necessitating anesthesia for the monitoring period. Opening the skull and underlying dura to install the intraparenchymal probes also resulted in a small amount (<1 mL) of cerebrospinal fluid (CSF) leakage. Studies investigating CSF dynamics in sheep report a formation rate of approximately 5.73 ± 0.51 mL/hour at an average resting pressure of 8.75 ± 1.0 cmH2O, with CSF formation constant over a wide range of pressures. 71 Although continuous CSF leakage was not observed in this study, by performing ICP recording on a single day we sought to reduce the influence of repeat dural opening and probe reinsertion on pressure dynamics. Of note, ICP monitoring in the neurointensive care unit is frequently performed in bed-bound patients who are often sedated and/or mechanically ventilated. Given ICP is heavily contingent on body position 72 monitoring animals under anesthesia ensured head positioning was standardized between animals and allowed control of physiological parameters such as respiration. Although efforts were made to maintain animals within normal ovine limits, pCO2 was significantly higher in vehicles compared with NK1-R antagonist treated animals. Given the inverse relationship between pCO2 and cerebral blood flow, this may have influenced ICP recordings, although no differences in CPP were seen between groups.
In conclusion, this study demonstrated that NK1-R antagonist treatment is effective in reducing ICP when given either before or after the establishment of elevated ICP and is associated with a reduction in cerebral edema, midline shift and BBB dysfunction. Taken together, the findings from this study provide evidence for the clinical evaluation of the NK1-R antagonist as a novel therapy for the treatment of cerebral edema and elevated ICP following stroke with reperfusion.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X241241907 - Supplemental material for NK1 tachykinin receptor antagonist treatment reduces cerebral edema and intracranial pressure in an ovine model of ischemic stroke
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X241241907 for NK1 tachykinin receptor antagonist treatment reduces cerebral edema and intracranial pressure in an ovine model of ischemic stroke by Annabel J Sorby-Adams, Oana C Marian, Isabella M Bilecki, Levi E Elms, Nawaf Yassi, Rebecca J Hood, Janet K Coller, Shannon M Stuckey, W Taylor Kimberly, Tracy D Farr, Anna V Leonard, Emma Thornton, Robert Vink and Renée J Turner in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Data availability
The data that support the findings of this study are available from the senior author, [RJT], upon reasonable request.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Health and Medical Research Council (NHMRC) Australia (Project grant GNT1082556) and by the Neurosurgical Research Foundation (Adelaide, Australia) (RJT). ASA was supported by the Peter Couche Foundation PhD top-up scholarship.
Acknowledgements
We thank Associate Professor Stephanie Reuter Lange (University of South Australia) for her invaluable assistance with the modelling of the PK parameters. We would also like to acknowledge the facilities and technical assistance of the National Imaging Facility, a National Collaborative Research Infrastructure Strategy capability, at SAHMRI Preclinical Imaging Research Laboratories.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The NK1-R antagonist drug (EU-C-001) was kindly donated by PresSura Neuro, however, they had no input in the experimental design, conduct of the study, nor data analysis.
Authors’ contributions
ASA, ET, AVL and RJT conceived and designed the experiments. ASA, OCM, IMB, LEM and RJT conducted the experiments. ASA, JKC, RJH, SMS, TDF, WTK and NY analysed the data. ASA, JKC, AVL and RJT wrote the manuscript. ASA, OCM, IMB, LEM, JKC, NY, AVL, RJH, ET, TDF, WTK, SMS, RV and RJT edited the manuscript.
Supplementary material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.