
Abstract
The efflux transporter P-glycoprotein (P-gp) at the blood-brain barrier limits the cerebral uptake of various xenobiotics. To assess the sensitivity of [11C]metoclopramide to measure decreased cerebral P-gp function, we performed [11C]metoclopramide PET scans without (baseline) and with partial P-gp inhibition by tariquidar in wild-type, heterozygous Abcb1a/b(+/−) and homozygous Abcb1a/b(−/−) mice as models with controlled levels of cerebral P-gp expression. Brains were collected to quantify P-gp expression with immunohistochemistry. Brain uptake of [11C]metoclopramide was expressed as the area under the brain time-activity curve (AUCbrain) and compared with data previously obtained with (R)-[11C]verapamil and [11C]N-desmethyl-loperamide. Abcb1a/b(+/−) mice had intermediate P-gp expression compared to wild-type and Abcb1a/b(−/−) mice. In baseline scans, all three radiotracers were able to discriminate Abcb1a/b(−/−) from wild-type mice (2.5- to 4.6-fold increased AUCbrain, p ≤ 0.0001). However, only [11C]metoclopramide could discriminate Abcb1a/b(+/−) from wild-type mice (1.46-fold increased AUCbrain, p ≤ 0.001). After partial P-gp inhibition, differences in [11C]metoclopramide AUCbrain between Abcb1a/b(+/−) and wild-type mice (1.39-fold, p ≤ 0.001) remained comparable to baseline. There was a negative correlation between baseline [11C]metoclopramide AUCbrain and ex-vivo-measured P-gp immunofluorescence (r = −0.9875, p ≤ 0.0001). Our data suggest that [11C]metoclopramide is a sensitive radiotracer to measure moderate, but (patho-)physiologically relevant decreases in cerebral P-gp function without the need to co-administer a P-gp inhibitor.
Introduction
Considerable efforts have been dedicated to the development of radiotracers for positron emission tomography (PET) to measure the function of P-glycoprotein (P-gp, encoded in humans by the ABCB1 gene and in rodents by the Abcb1a and Abcb1b genes) at the blood-brain barrier (BBB).1 –3 P-gp is a membrane transporter belonging to the adenosine triphosphate-binding cassette (ABC) family and an important gatekeeper at the BBB that restricts the brain distribution of many structurally unrelated, clinically used drugs. 4 In addition, P-gp may modulate the neuropharmacokinetics of some central nervous system-active drugs5,6 and contribute to the removal of endogenous neurotoxic compounds from the brain, such as beta-amyloid, which plays an important role in the pathophysiology of Alzheimer’s disease. 7 P-gp has furthermore been implicated in the pathophysiology of drug-resistant epilepsy and Parkinson’s disease. 8
Selection of candidate compounds for PET tracer development has initially focused on molecules, which showed high efflux ratios in bidirectional transport assays in P-gp overexpressing cell lines and great differences in brain uptake between wild-type and Abcb1a/b knockout (Abcb1a/b(−/−)) mice, leading to the development of racemic [11C]verapamil, (R)-[11C]verapamil and [11C]N-desmethyl-loperamide as P-gp substrate radiotracers.9 –11 Candidate selection was based on the assumption that compounds which are efficiently transported by P-gp (i.e. “avid” P-gp substrates) would also display optimal sensitivity to detect moderate, but physiologically relevant changes in cerebral P-gp function as they are expected to occur in different disease conditions, during healthy ageing or in clinically relevant transporter-mediated drug-drug interactions. Contrary to these expectations, clinical PET studies in different conditions in which P-gp is either up- or downregulated revealed only very small, if any, changes in the brain distribution of racemic [11C]verapamil or (R)-[11C]verapamil.12 –22 To overcome the limited sensitivity of (R)-[11C]verapamil to detect moderate changes in P-gp function, we previously validated a partial P-gp inhibition protocol.21,23 –25 This protocol, which entails the intravenous (i.v.) administration of the Pgp inhibitor tariquidar prior to the PET scan at a dose that partially inhibits P-gp,21,23 –25 has also been used in combination with other P-gp substrate radiotracers (e.g. [18F]MPPF or [18F]MC225).26,27 However, this protocol is limited by the lack of availability of tariquidar for clinical use and the safety risk associated with the administration of a P-gp inhibitor in patients who receive concomitant medication. A further drawback of “avid” P-gp substrate radiotracers is their negligible baseline brain uptake which hinders the detection of an induction of P-gp function at the BBB. This led to the development of radiotracers which are less efficiently transported by P-gp and show substantial brain uptake when P-gp is fully functional (i.e. [11C]metoclopramide and [18F]MC225).28 –32 It is not known, however, whether such radiotracers display an adequate sensitivity to detect moderate, physiologically relevant changes in cerebral P-gp function.
In this work, we assessed the sensitivity of [11C]metoclopramide to measure cerebral P-gp function by studying wild-type, heterozygous (Abcb1a/b(+/−)) and homozygous (Abcb1a/b(−/−)) Abcb1a/b knockout mice as models with controlled levels of cerebral P-gp expression. Data were compared with data obtained with (R)-[11C]verapamil and [11C]N-desmethyl-loperamide in the same mouse models.23,33
Materials and methods
Chemicals and radiotracer synthesis
Unless otherwise stated, chemicals were purchased from Sigma-Aldrich Chemie GmbH (Schnelldorf, Germany) or Merck (Darmstadt, Germany). Metoclopramide ampoules (Paspertin®, 10 mg/2 mL) were obtained from a local pharmacy. The P-gp inhibitor tariquidar dimesylate was obtained from Haoyuan Chemexpress Co., Ltd (Shanghai, China) and freshly dissolved in 2.5% (w/v) aqueous dextrose solution (0.75 mg/mL) before each administration and injected i.v. into mice at a volume of 4 mL/kg body weight (corresponding to an administered dose of 0.75 mg/kg body weight). [11C]Metoclopramide was synthesised by 11C-methylation of O-desmethyl-metoclopramide (ABX advanced biochemical compounds, Radeberg, Germany) as previously described. 34 For i.v. injection into mice, [11C]metoclopramide was formulated in 0.9% (w/v) physiological saline and 10 µL Paspertin® solution (containing 0.05 mg of unlabelled metoclopramide) was added to each injected dose (to slow down the peripheral metabolism of the radiotracer).30,35 Radiochemical purity was >98%.
Animals
Female wild-type, Abcb1a/b(+/−) and Abcb1a/b(−/−) mice, all with a C57BL/6J genetic background, were generated at the KPM Radium Hospital (Oslo, Norway). Animals were housed in type III IVC cages under controlled environmental conditions (22 ± 3 °C, 40–70% humidity, 12-hour light/dark cycle) and had free access to standard laboratory rodent diet (LASQCdiet™ Rod16, Auto, LASvendi GmbH, Soest, Germany) and water. An acclimatisation period of at least one week was allowed before the animals were used in the experiments. The study was approved by the Austrian Federal Ministry of Education, Science and Research [2021-0.785.873] and the Intramural Committee for Animal Experimentation of the Medical University of Vienna. Study procedures were in accordance with the European Community’s Council Directive of September 22, 2010 (2010/63/EU) and the data reported in the study are in compliance with the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines.
Experimental design
An overview of animal groups examined in this study is given in Table 1. Mice were grouped together in cages per genotype and the investigators knew the group allocation during the experiments. No randomisation was applied in the group allocation. Groups of wild-type, Abcb1a/b(+/−) and Abcb1a/b(−/−) mice underwent a baseline PET scan with [11C]metoclopramide. Two additional groups of wild-type and Abcb1a/b(+/−) mice underwent a [11C]metoclopramide PET scan after partial inhibition of P-gp achieved by i.v. injection of tariquidar over 1 min at a dose of 0.75 mg/kg body weight at 30 min before the start of the PET scan. This dose of tariquidar was selected based on previous work in which the dose-response relationship of tariquidar to increase the brain uptake of [11C]metoclopramide was assessed in rats. 36
Overview of animal groups and numbers examined with [11C]metoclopramide.
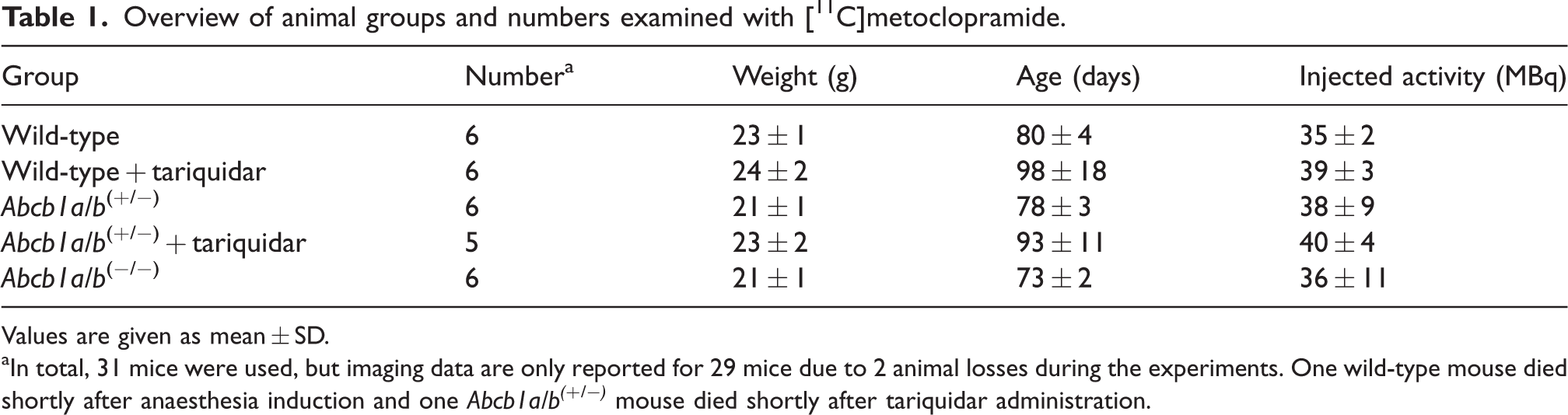
Values are given as mean ± SD.
In total, 31 mice were used, but imaging data are only reported for 29 mice due to 2 animal losses during the experiments. One wild-type mouse died shortly after anaesthesia induction and one Abcb1a/b(+/−) mouse died shortly after tariquidar administration.
PET imaging
Imaging experiments were performed under isoflurane/medical air anaesthesia. Animals were warmed throughout the experiment and body temperature and respiratory rate were constantly monitored (SA Instruments Inc., Stony Brook, NY, USA). Anaesthetised mice were positioned on a heated double imaging chamber (m2m imaging Corp., Cleveland, OH, USA) and the lateral tail vein was cannulated. A microPET Focus220 scanner (Siemens Medical Solutions, Knoxville, TN, USA) was used for PET imaging. [11C]Metoclopramide (38 ± 7 MBq in a volume of 100 µL, spiked with 0.05 mg of unlabelled metoclopramide, molar activity: 0.25 ± 0.05 GBq/µmol) was i.v. administered over 1 min and a 60-min dynamic PET scan was initiated with the start of radiotracer injection. List-mode data were acquired with a timing window of 6 ns and an energy window of 250–750 keV. A 10-min transmission scan was performed before each PET scan for attenuation correction using a rotating 57Co-point source. At the end of the PET scan, a blood sample was collected from the retro-bulbar plexus and animals were killed by cervical dislocation while still under deep anaesthesia. Radioactivity in weighted blood aliquots was measured in a gamma counter (Hidex Automatic Gamma Counter, Hidex Oy, Turku, Finland). From three animals per mouse model the brain was perfused with physiological saline and whole brains were removed and snap frozen in liquid nitrogen-cooled isopentane and stored at −80°C for immunohistochemical analysis of P-gp.
PET data analysis
The person analysing the data knew the group allocation. The PET data were sorted into 23 time frames with a duration increasing from 5 s to 10 min. PET images were reconstructed using Fourier re-binning of the 3-dimensional sinograms followed by a 2-dimensional filtered back-projection with a ramp filter giving a voxel size of 0.4 × 0.4 × 0.796 mm³. The standard data correction protocol, including normalisation, attenuation- and decay correction, was applied to the data. Using the medical image data examiner software AMIDE, 37 the brain was manually outlined as a region of interest to derive time-activity curves (TACs) expressed in units of standardised uptake value (SUV) ((radioactivity per g/injected radioactivity) × body weight). The area under the brain TAC (AUCbrain, SUV × min) was calculated from 0 to 30 min after radiotracer injection using Prism 8.0 (GraphPad Software, La Jolla, CA, USA). The time period from 0 to 30 min was chosen for AUC calculation to enable comparison of our data with a previous study in rats, in which AUCbrain was also calculated from 0 to 30 min. 36 [11C]Metoclopramide data were compared with results previously obtained with (R)-[11C]verapamil and [11C]N-desmethyl-loperamide in the same mouse models obtained from the same source.23,33 For the PET scans with (R)-[11C]verapamil and [11C]N-desmethyl-loperamide, no carrier was added to the radiotracer (molar activity at the time of injection: 20-60 GBq/µmol).23,33
Immunohistochemistry
The frozen brain tissue was cut in 14 µm thick slices and mounted on slides. The slides were incubated for 15 min at room temperature (rt) in 4% (w/v) aqueous paraformaldehyde (PFA). In order to block the PFA, the slides were incubated for 5 min at rt in phosphate-buffered saline (PBS) containing 50 mM of ammonium chloride. Then the cuts were permeabilised with frozen methanol/acetone followed by PBS solution containing 0.1% (v/v) Triton X-100. Several washes with PBS were carried out between each of these steps. The non-specific sites were saturated by incubating the slides for 1 h at rt in a PBS solution containing 5% (v/v) Bovine Albumin Serum and 0.5% (v/v) Tween 80. Each slide was incubated for 1 h in the presence of anti-P-gp primary antibody (1:100, P-glycoprotein Recombinant Rabbit Monoclonal Antibody [#MA5-35257], Invitrogen). After several washes with PBS to remove excess antibody, the slides were left for 30 min at rt in the presence of a solution containing the secondary antibody (1:1000, AlexaFluor 488 goat anti-rabbit, Invitrogen). Finally, all the slides were rinsed again, then mounted with mounting medium containing 4’,6-diamidino-2-phenylindole (DAPI) for labelling the nuclei (ProLongTM Diamond Antifade Mountant with DAPI, Invitrogen). The brain tissue slides were analysed using a Zeiss AxioCam fluorescence microscope (Carl Zeiss, Germany). For each brain of wild-type (n = 3), Abcb1a/b(+/−) (n = 3) and Abcb1a/b(−/−) (n = 2) mice four rectangular areas (0.323 × 0.323 μm) were selected. In each of the four areas, eight brain microvessels were selected and fluorescence intensity was quantified using ImageJ software.
Statistical analysis
Statistical analysis was performed using Prism 8.0 software. The GINGER tool of the Institute of Clinical Biometrics at the Medical University of Vienna, available at https://clinicalbiometrics.shinyapps.io/GINGER/, was used for sample size calculations. Based on previous data on the variability of the outcome parameter AUCbrain in mice and assuming an effect size ( = difference in mean value between groups divided by standard deviation within groups) of 2.5, the minimum required sample size was 5 in order to detect differences between 5 groups with a two-sided significance level of 0.05 and a power of 80%. After assessment of normal distribution of the data using the Shapiro-Wilk normality test, differences among groups were assessed by one-way ANOVA followed by a Tukey’s multiple comparison test (to compare all groups) or a Dunnett’s multiple comparison test (to compare all groups with a control group). The Pearson correlation coefficient (r) was calculated to assess correlations. The level of statistical significance was set to a p-value of less than 0.05. Values are given as mean ± standard deviation (SD).
Results
We performed [11C]metoclopramide PET scans in groups of wild-type, Abcb1a/b(+/−) and Abcb1a/b(−/−) mice and compared these data with data obtained with (R)-[11C]verapamil and [11C]N-desmethyl-loperamide in the same mouse models.
At the end of the PET scan, brains were collected from all three mouse models and analysed for P-gp with immunohistochemistry, which confirmed normal, intermediate and no P-gp expression levels in the brains of wild-type, Abcb1a/b(+/−) and Abcb1a/b(−/−) mice, respectively (Figure 1(a)). Quantitative analysis of images revealed a 31% reduction (p ≤ 0.001) in cerebral P-gp staining in Abcb1a/b(+/−) mice as compared with wild-type mice (Figure 1(b)). PET summation images for all three radiotracers are shown in Figure 2 and the corresponding brain TACs in Figure 3. As an outcome parameter for the brain uptake of [11C]metoclopramide, AUCbrain was calculated from 0 to 30 min after radiotracer injection (Figure 4, Table S1). In wild-type mice, AUCbrain was highest for [11C]metoclopramide (11.5 ± 1.2 SUV × min), intermediate for (R)-[11C]verapamil (8.5 ± 0.9 SUV × min) and lowest for [11C]N-desmethyl-loperamide (6.1 ± 0.3 SUV × min). All three radiotracers had significantly higher AUCbrain values in Abcb1a/b(−/−) mice relative to wild-type mice and this difference between the two mouse models was highest for (R)-[11C]verapamil (4.6-fold, p ≤ 0.0001) and comparable for [11C]metoclopramide (2.5-fold, p ≤ 0.0001) and [11C]N-desmethyl-loperamide (2.45-fold, p ≤ 0.0001) (Figure 4). Among the three radiotracers, only [11C]metoclopramide showed significantly higher AUCbrain values in Abcb1a/b(+/−) mice relative to wild-type mice (1.46-fold, p ≤ 0.001, Figure 4(a)). For (R)-[11C]verapamil and [11C]N-desmethyl-loperamide, AUCbrain values in Abcb1a/b(+/−) mice were not significantly different from wild-type mice (Figure 4(b) and (c)). There was a statistically significant negative correlation between AUCbrain of [11C]metoclopramide and ex vivo-measured P-gp fluorescence intensity (r = −0.9875, p ≤ 0.0001) (Figure 5).
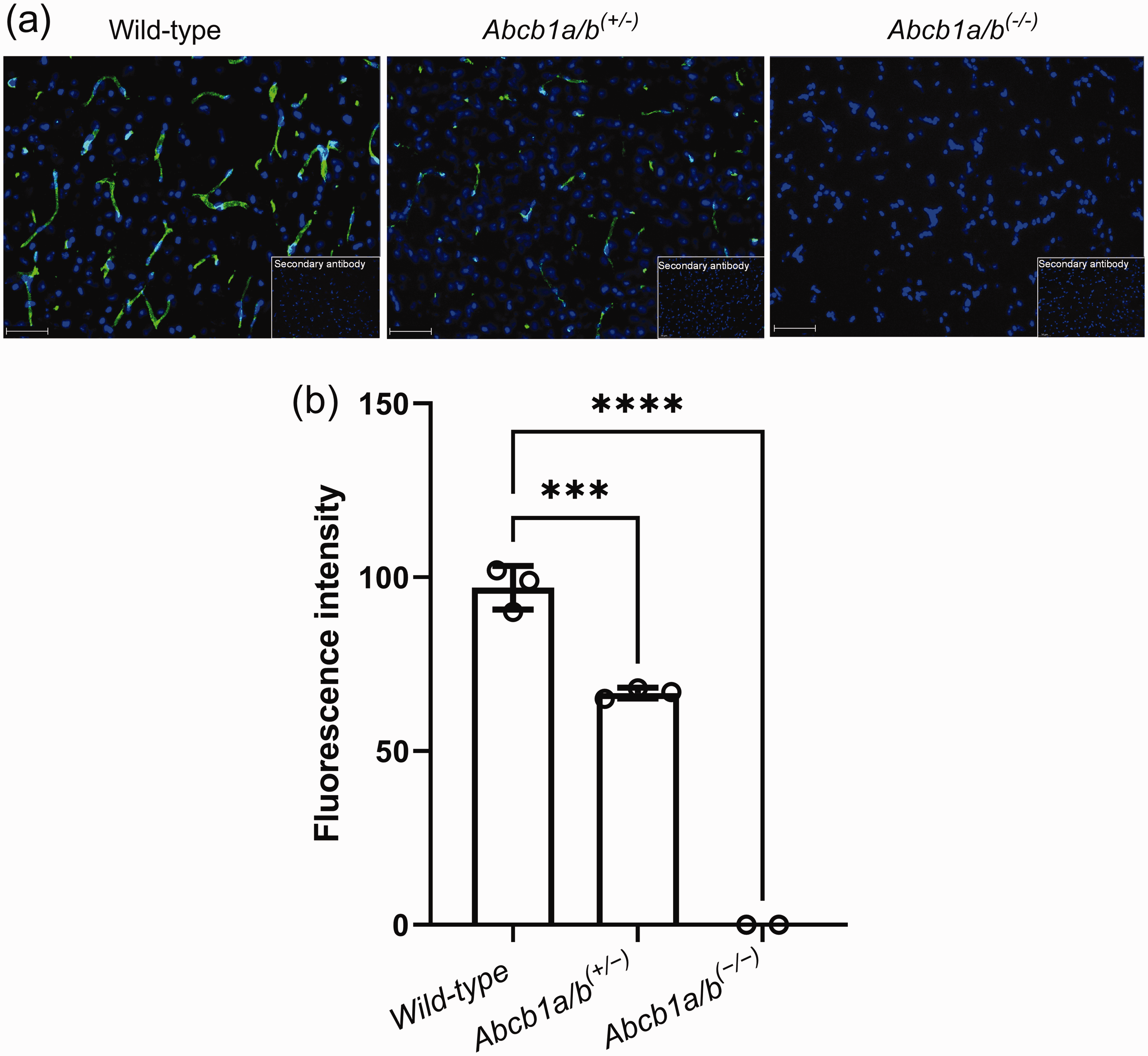
Immunohistochemistry data for the employed mouse models. Immunohistochemical staining of P-gp (green) in brain microvessels and nuclei counterstained with DAPI (blue) in frozen brain sections of wild-type, Abcb1a/b(+/−) and Abcb1a/b(−/−) mice viewed by fluorescence microscopy (scale bar = 50 µm) (a). Inserts show negative controls (no primary antibody). Quantitative evaluation of stained P-gp in brain microvessels of wild-type (n = 3), Abcb1a/b(+/−) (n = 3) and Abcb1a/b(−/−) mice (n = 2) (b). Data are shown as mean ± SD (except for Abcb1a/b(−/−) mice). ***, p ≤ 0.001; ****, p ≤ 0.0001 (one-way ANOVA followed by a Dunnett’s multiple comparison test against the wild-type group).
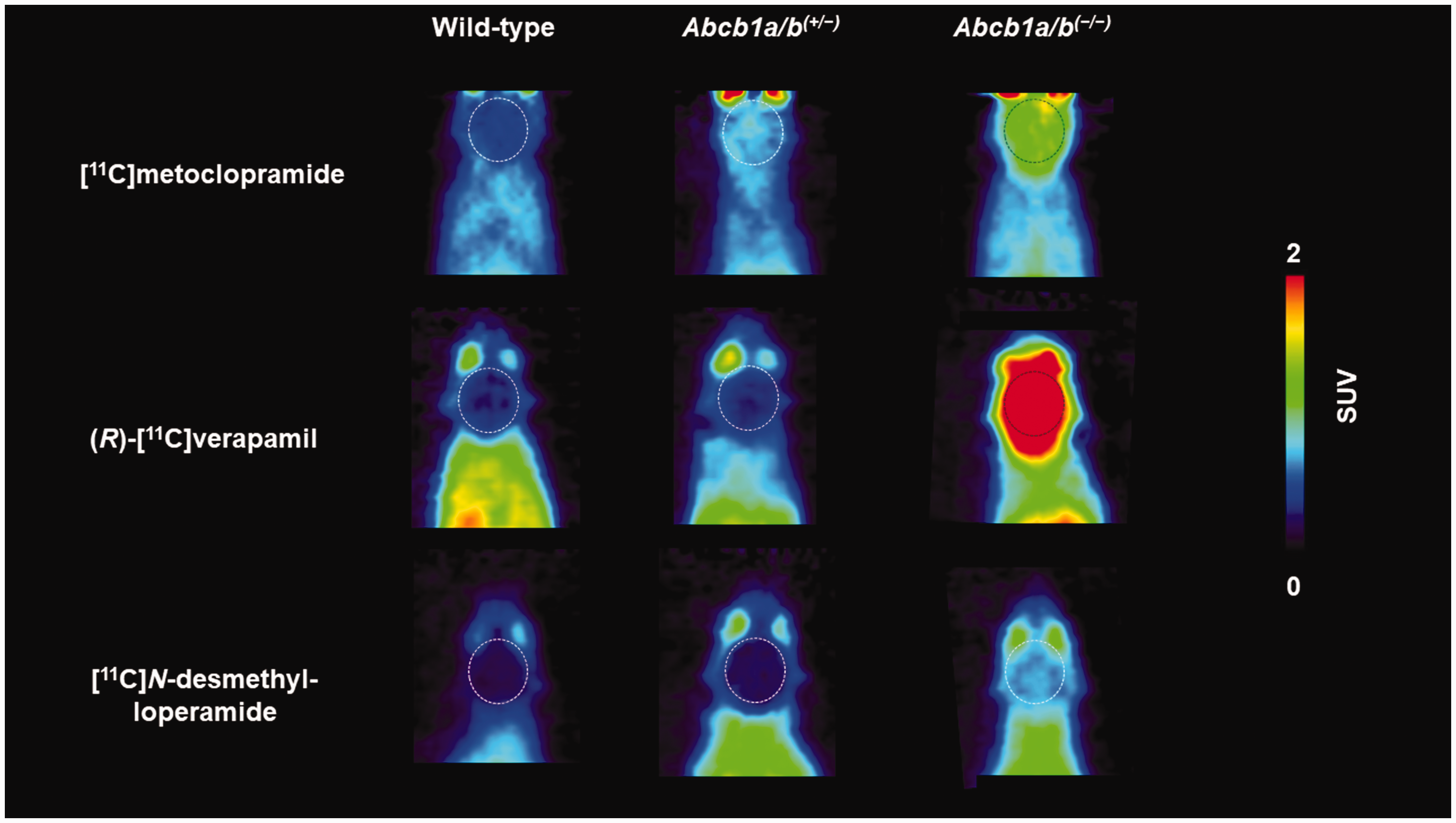
Representative coronal PET summation images (0–30 min) obtained after i.v. injection of [11C]metoclopramide, (R)-[11C]verapamil or [11C]N-desmethyl-loperamide in wild-type, Abcb1a/b(+/−) and Abcb1a/b(−/−) mice. Raw data for (R)-[11C]verapamil and [11C]N-desmethyl-loperamide are taken from previously published work.23,33 Whole brain region is highlighted with a broken line. All images are scaled to the same intensity (0–2 standardised uptake value, SUV).
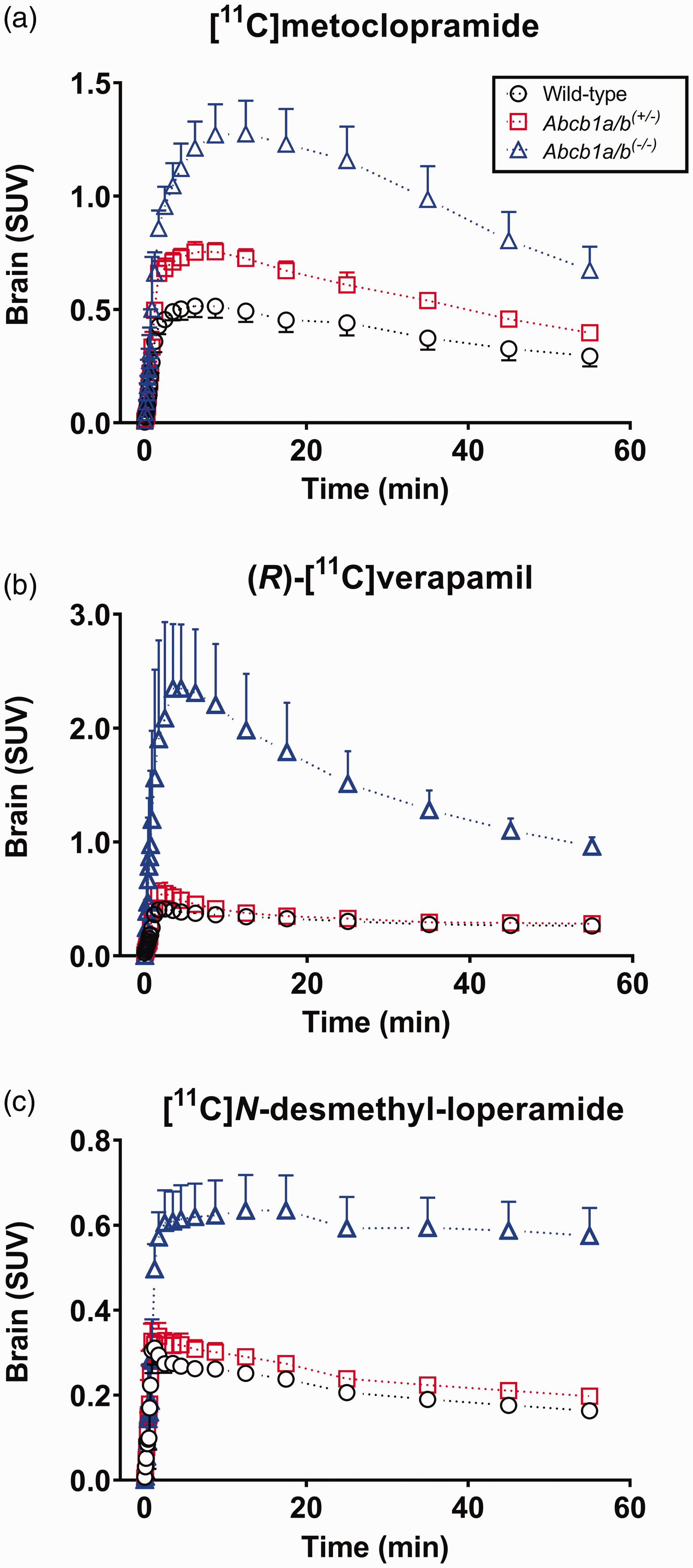
Mean (SUV ± SD) whole-brain time-activity curves obtained after i.v. injection of [11C]metoclopramide (a, n = 6 per group), (R)-[11C]verapamil (b, n = 3–5 per group) or [11C]N-desmethyl-loperamide (c, n = 4–5 per group) into wild-type, Abcb1a/b(+/−) and Abcb1a/b(−/−) mice. Data for (R)-[11C]verapamil and [11C]N-desmethyl-loperamide are taken from previously published work.23,33.
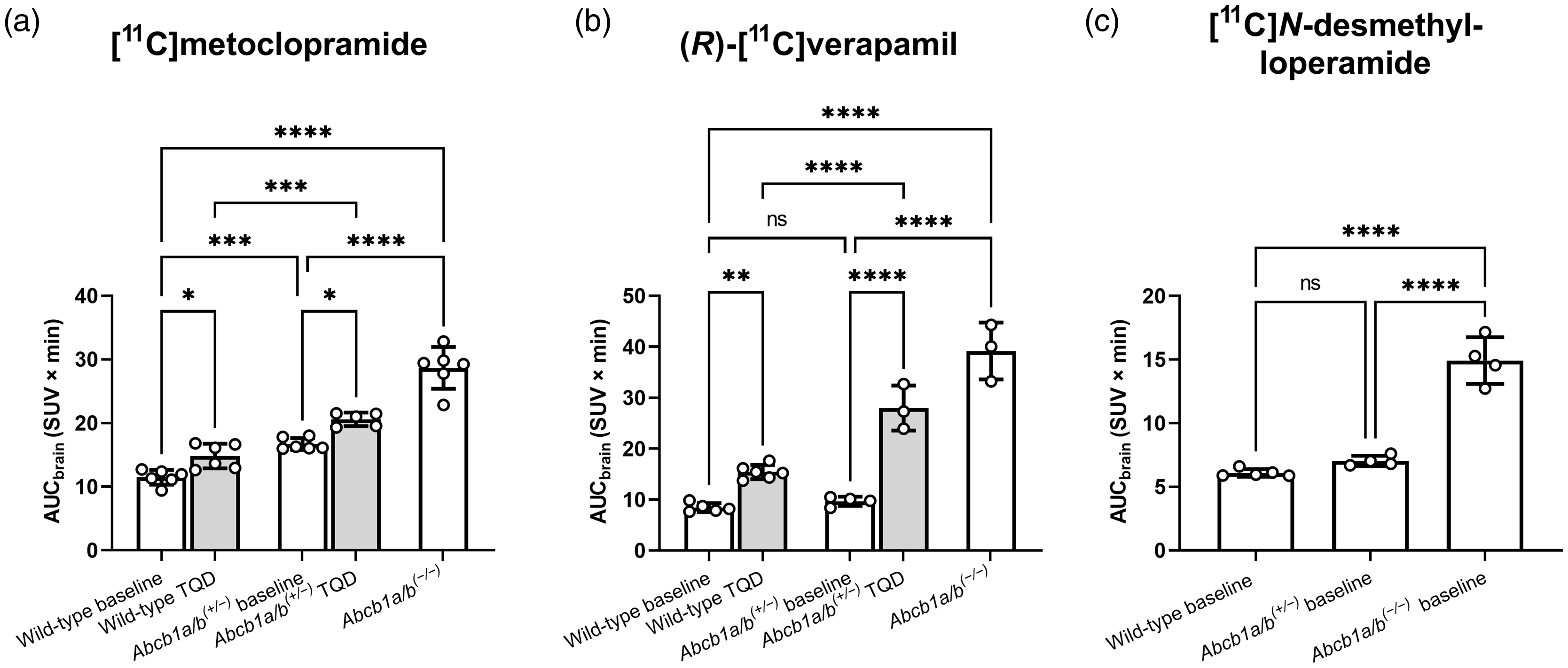
Area under the whole-brain time activity-curve (AUCbrain, SUV × min) calculated from 0 to 30 min after i.v. injection of [11C]metoclopramide (a, n = 5–6 per group) or (R)-[11C]verapamil (b, n = 3–6 per group) into wild-type and Abcb1a/b(+/−) mice, without (baseline) and with partial P-gp inhibition with tariquidar (TQD) given either at a dose of 0.75 mg/kg at 30 min ([11C]metoclopramide) or at dose of 4 mg/kg at 120 min ((R)-[11C]verapamil) before the start of the PET scan, and in Abcb1a/b(−/−) mice. In (c), baseline AUCbrain values are shown for [11C]N-desmethyl-loperamide in the three mouse models (n = 4–5 per group). Data for (R)-[11C]verapamil and [11C]N-desmethyl-loperamide are taken from previously published work.23,33 Data are given as mean ± SD. ns, not significant; *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001 (one-way ANOVA followed by a Tukey’s multiple comparison test).
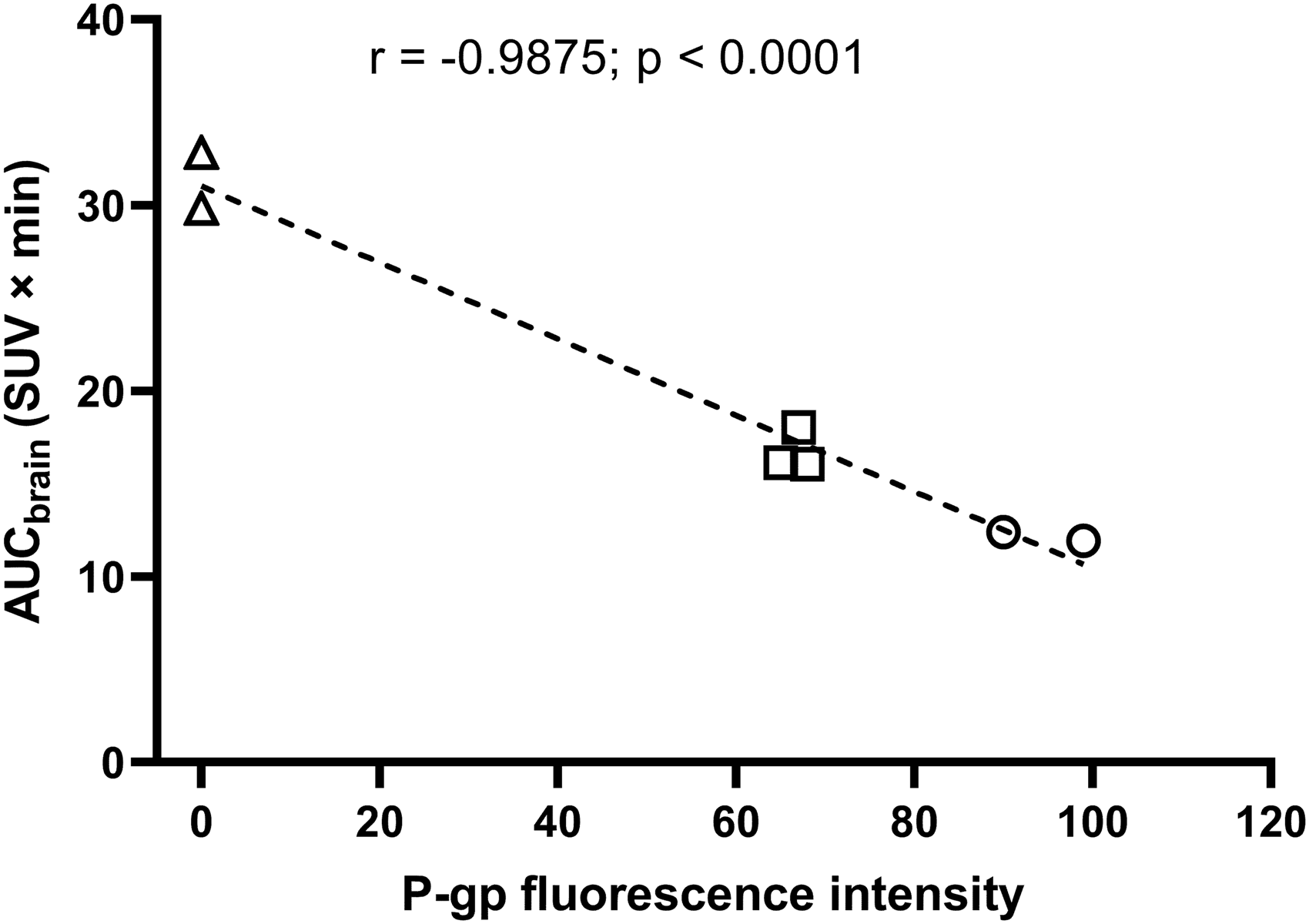
Correlation between PET-measured AUCbrain of [11C]metoclopramide with P-gp fluorescence intensity in brain microvessels measured with immunohistochemistry in wild-type (n = 2, circles), Abcb1a/b(+/−) (n = 3, squares) and Abcb1a/b(−/−) mice (n = 2, triangles). One wild-type mouse was not included in the correlation because it received tariquidar pre-treatment before the PET scan. r = Pearson correlation coefficient.
To test whether [11C]metoclopramide shows like (R)-[11C]verapamil improved sensitivity to measure cerebral P-gp function under conditions of partial P-gp inhibition, we additionally performed [11C]metoclopramide PET scans in groups of wild-type and Abcb1a/b(+/−) mice which were pre-treated with a low dose of the P-gp inhibitor tariquidar (0.75 mg/kg). [11C]Metoclopramide and (R)-[11C]verapamil brain TACs in wild-type and Abcb1a/b(+/−) mice without and with partial P-gp inhibition with tariquidar are shown in Figure S1 and the corresponding AUCbrain values are shown in Figure 4 and Table S1. For [11C]metoclopramide, AUCbrain was significantly and to a similar extent increased in wild-type mice (1.29-fold, p ≤ 0.05) and Abcb1a/b(+/−) mice (1.23-fold, p ≤ 0.05) after partial P-gp inhibition relative to the respective untreated groups (Figure 4(a)). For (R)-[11C]verapamil, AUCbrain increase after partial P-gp inhibition was higher in Abcb1a/b(+/−) mice (2.89-fold, p ≤ 0.0001) than in wild-type mice (1.82-fold, p ≤ 0.01) (Figure 4(b)). For [11C]metoclopramide, differences in AUCbrain between Abcb1a/b(+/−) mice and wild-type mice were similar under baseline conditions (1.46-fold higher in Abcb1a/b(+/−) mice, p ≤ 0.001) and after partial P-gp inhibition (1.39-fold higher in Abcb1a/b(+/−) mice, p ≤ 0.001) (Figure 4(a)). For (R)-[11C]verapamil, significant differences in AUCbrain between Abcb1a/b(+/−) mice and wild-type mice were only obtained after partial P-gp inhibition (1.81-fold higher in Abcb1a/b(+/−) mice, p ≤ 0.0001), but not under baseline conditions (Figure 4(b)).
At the end of the [11C]metoclopramide PET scans, venous blood samples were collected from all investigated groups and measured in a gamma counter, revealing no significant differences in radioactivity content among all groups (Figure S2).
Discussion
In this study, we could demonstrate that [11C]metoclopramide has a better sensitivity than (R)-[11C]verapamil and [11C]N-desmethyl-loperamide to detect an approximately 30% reduction in cerebral P-gp expression as it occurs in Abcb1a/b(+/−) mice relative to wild-type mice. Moreover, we could show that [11C]metoclopramide does not require the co-administration of a P-gp inhibitor to increase its sensitivity for the measurement of P-gp function, as is the case for (R)-[11C]verapamil.21,23 –25 These findings further pave the way for the clinical use of [11C]metoclopramide as an improved PET probe to measure cerebral P-gp function in health and disease.
Metoclopramide is an antiemetic drug, which exerts its pharmacological effect through inhibition of dopamine D2 and serotonin 5-HT3 receptors in the brain (i.e. the chemoreceptor trigger zone within the area postrema of the medulla oblongata). 38 [11C]Metoclopramide was shown to be transported by P-gp at the rat, non-human primate and human BBB.28,30,31 It is not a substrate of breast cancer resistance protein (BCRP, ABCG2), another important efflux transporter at the BBB. 30 Co-injection of unlabelled metoclopramide revealed a lack of specific binding of [11C]metoclopramide in the human brain (e.g. to dopamine D2 receptors). 39
To assess the sensitivity of [11C]metoclopramide to measure cerebral P-gp function, we used in our study wild-type, Abcb1a/b (+/−)and Abcb1a/b(−/−) mice as models with controlled levels of cerebral P-gp expression. We could show with immunohistochemical analysis that Abcb1a/b(+/−) mice have an approximately 30% reduction in cerebral P-gp expression relative to wild-type mice (Figure 1), which is in good agreement with earlier findings in another mouse strain 40 as well with our own results in the same mouse model. 23 Previous studies which measured cerebral P-gp function with [11C]metoclopramide, (R)-[11C]verapamil or [11C]N-desmethyl-loperamide used compartmental modelling approaches to describe the brain uptake of these PET probes, i.e. in terms of the total volume of distribution (VT) or the influx rate constant from plasma into the brain (K1).28,41,42 These modelling approaches require the measurement of a metabolite-corrected arterial plasma input function, which is not possible to perform in mice due to their small blood volume. We therefore used AUCbrain as a simplified and model-independent parameter to compare the brain uptake of the three PET probes in mice. Previous studies have shown a good correlation between AUCbrain values and modelling-derived brain VT or K1 values for [11C]metoclopramide and [11C]N-desmethyl-loperamide31,43 and in a study by Breuil et al. 36 AUCbrain has been used as a parameter to compare the sensitivity of the three PET probes to assess cerebral P-gp inhibition in rats. The use of AUCbrain to describe the brain uptake of [11C]metoclopramide in our study is further supported by our observation that blood radioactivity concentrations measured at the end of the PET scan did not differ among the investigated mouse groups suggesting that differences in brain uptake were not caused by differences in the peripheral disposition of the radiotracer (Figure S2). This is consistent with a previous study reporting no changes in the plasma kinetics of metoclopramide in P-gp-deficient mice compared with wild-type mice. 44
The “avid” P-gp substrate (R)-[11C]verapamil is very efficiently transported by P-gp at the BBB leading to an excellent imaging contrast between conditions of full P-gp function and complete absence of P-gp function (i.e. wild-type and Abcb1a/b(−/−) mice) (Figure 2). However, a complete absence of P-gp function is very unlikely to occur under pathophysiological conditions and it is therefore of great importance that effective PET tracers for P-gp are also capable of detecting a moderate decrease in P-gp function. In clinical PET studies, the brain uptake of (R)-[11C]verapamil or racemic [11C]verapamil was only modestly or not at all increased under conditions of decreased cerebral P-gp expression (i.e. in elderly people, patients with Alzheimer’s and Parkinson’s disease, people with genetic polymorphisms).12 –18,22 In line with this, the baseline brain uptake of (R)-[11C]verapamil was not significantly different between Abcb1a/b(+/−) mice and wild-type mice despite the approximately 30% lower P-gp expression in Abcb1a/b(+/−) mice (Figure 4(b)). Partial inhibition of P-gp with tariquidar increases the sensitivity of (R)-[11C]verapamil to detect an age- or disease-induced decrease in P-gp expression.23,24 This can be seen in our data in which significant differences in brain uptake of (R)-[11C]verapamil between Abcb1a/b(+/−) mice and wild-type mice were only observed when P-gp was partially inhibited (Figure 4(b)). However, a pre-requisite for the applicability of the partial P-gp inhibition protocol to detect differences in P-gp function between two study groups (e.g. patients and control subjects) is that identical plasma levels of tariquidar can be achieved in both groups, which may often not be possible. Moreover, the applicability of this protocol is hampered by safety issues and the limited availability of tariquidar for clinical use.
As compared to verapamil and loperamide, metoclopramide is less efficiently transported by P-gp as reflected by a lower efflux ratio in a bidirectional transport assay in a human P-gp overexpressing cell line (efflux ratio, metoclopramide: 1.4, verapamil: 2.1, loperamide: 3.8). 45 In addition, the passive membrane permeability (Papp) of metoclopramide is higher than that of verapamil and loperamide (Papp, metoclopramide: 17.7 ± 2.61 × 10−6 cm/s, verapamil: 12.6 ± 1.83 × 10−6 cm/s, loperamide: 2.7 ± 1.07 × 10−6 cm/s). 45 These properties lead to higher baseline brain uptake of [11C]metoclopramide than for (R)-[11C]verapamil and [11C]N-desmethyl-loperamide (Figure 2) and a smaller difference in brain uptake between Abcb1a/b(−/−) mice and wild-type mice for [11C]metoclopramide (2.5-fold) than for (R)-[11C]verapamil (4.6-fold) (Figure 4). For [11C]N-desmethyl-loperamide, the difference in brain uptake between Abcb1a/b(−/−) mice and wild-type mice is comparable to that of [11C]metoclopramide despite the fact that [11C]N-desmethyl-loperamide is more efficiently transported by P-gp than [11C]metoclopramide (Figure 4). This is caused by the brain uptake of non-P-gp transported radiolabelled metabolites of [11C]N-desmethyl-loperamide in mice, which blunt the differences in brain uptake between the two mouse models. 33 When correcting brain-to-plasma ratios of total radioactivity for the contribution of radiolabelled metabolites, the difference in brain uptake between conditions of complete P-gp inhibition and baseline in wild-type mice is 18.3-fold for [11C]N-desmethyl-loperamide and 14.1-fold for (R)-[11C]verapamil. 33 As opposed to [11C]N-desmethyl-loperamide and (R)-[11C]verapamil, [11C]metoclopramide lacks brain-penetrant radiolabelled metabolites in rats and mice.30,35 It may be hypothesised that due to its distinct properties as compared with (R)-[11C]verapamil and [11C]N-desmethyl-loperamide, [11C]metoclopramide may possess a better sensitivity to measure moderate changes in cerebral P-gp function. 46 Breuil et al. 36 have recently addressed this question by comparing the increase in brain uptake of all three PET probes in response to the administration of a low dose of tariquidar (1 mg/kg) in rats. This study revealed a comparable increase in the brain uptake of [11C]metoclopramide (2.1-fold) and (R)-[11C]verapamil (2.4-fold) in response to tariquidar (1 mg/kg) administration, while the brain uptake of [11C]N-desmethyl-loperamide was unaffected. 36 However, since P-gp has multiple substrate/inhibitor binding sites 47 the dose-response relationship to pharmacological P-gp inhibition will depend on the employed substrate-inhibitor pair. As such, the relationship between reduced P-gp expression and partial P-gp inhibition is not clear. Therefore, it is not known whether good sensitivity of a PET tracer to measure P-gp inhibition with one selected inhibitor may translate to a good sensitivity to measure (patho)physiological changes in P-gp expression/function. Moreover, it cannot be predicted whether all “weak” P-gp substrates will possess enhanced sensitivity to detect moderate changes in P-gp function at the BBB.
Given these caveats about the interpretation of previous methods, we therefore propose to use Abcb1a/b(+/−) mice, which are naturally generated in the breeding of Abcb1a/b(−/−) mice, as a better suited model to assess the sensitivity of newly developed PET tracers to measure decreased P-gp function at the BBB. Our present findings, which reveal a better sensitivity for [11C]metoclopramide than for (R)-[11C]verapamil and [11C]N-desmethyl-loperamide, are supported by results from studies in humans, which demonstrated that [11C]metoclopramide was able to detect an age-related decrease in cerebral P-gp function leading to greater changes in its brain distribution as compared with (R)-[11C]verapamil.24,39 However, based on these previous human data 24,39 no firm conclusions could be made whether [11C]metoclopramide is more sensitive than (R)-[11C]verapamil as the studies were not done in the same cohorts and it is known whether the elderly subjects examined with [11C]metoclopramide had the same level of decrease in P-gp expression/function as the subjects examined with (R)-[11C]verapamil. In contrast, our present study includes a systematic comparison of these radiotracers in transgenic mouse models with well-defined decreases in cerebral P-gp expression. Moreover, our earlier work has also shown that [11C]metoclopramide can measure pharmacological P-gp induction in mice. 35
Taken together, our results establish [11C]metoclopramide as a sensitive PET probe to measure moderate decreases in cerebral P-gp function as they may occur in certain diseases, during healthy ageing or due to drug-drug interactions or genetic factors, without the need to co-administer a P-gp inhibitor. We furthermore propose to use heterozygous rather than homozygous transporter knockout mice as an appropriate model to test the sensitivity of newly developed PET tracers to measure efflux transporter function at the BBB.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X231202336 - Supplemental material for [11C]metoclopramide is a sensitive radiotracer to measure moderate decreases in P-glycoprotein function at the blood-brain barrier
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X231202336 for [11C]metoclopramide is a sensitive radiotracer to measure moderate decreases in P-glycoprotein function at the blood-brain barrier by Severin Mairinger, Sarah Leterrier, Thomas Filip, Mathilde Löbsch, Jens Pahnke, Irene Hernández-Lozano, Johann Stanek, Nicolas Tournier, Markus Zeitlinger, Marcus Hacker, Oliver Langer and Thomas Wanek in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by the Austrian Research Promotion Agency (FFG) [882717 PETABC, to Oliver Langer], the Austrian Science Fund (FWF) [I4470-B EPIFLUX and KLI 694-B30, to Martin Bauer and Oliver Langer], Norges forskningsrådet/Norway [327571 PETABC, to Jens Pahnke] and the French National Agency for Research [ANR-19-CE17-0027 EPIFLUX, to Nicolas Tournier]. PETABC is an EU Joint Programme - Neurodegenerative Disease Research (JPND) project. PETABC is supported through the following funding organisations under the aegis of JPND (![]() ): NFR #327571 – Norway, FFG #882717 – Austria, BMBF #01ED2106 – Germany, MSMT #8F21002 – Czech Republic, VIAA #ES RTD/2020/26 – Latvia, ANR #20-JPW2-0002-04 – France and SRC #2020-02905 – Sweden. The projects receive funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement #643417 (JPco-fuND).
): NFR #327571 – Norway, FFG #882717 – Austria, BMBF #01ED2106 – Germany, MSMT #8F21002 – Czech Republic, VIAA #ES RTD/2020/26 – Latvia, ANR #20-JPW2-0002-04 – France and SRC #2020-02905 – Sweden. The projects receive funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement #643417 (JPco-fuND).
Acknowledgements
The authors would like to acknowledge the efforts of the management teams at the Medical University of Vienna and at the AIT Austrian Institute of Technology GmbH to enable the successful transfer of the former AIT Preclinical Molecular Imaging Group to the Medical University of Vienna.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
S.M., J.P., N.T., M.Z., M.H, T.W. and O.L. contributed to the concept and design of the study. S.M., T.F., M.L., J.S. and T.W. performed animal studies. S.L. performed immunohistochemistry. J.P. was responsible for animal generation. T.W., S.M., S.L., O.L. and I.H-L. analysed the data. O.L., S.M., T.W. and I.H-L. wrote the first draft of the manuscript and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Supplementary material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.