
Abstract
Post-stroke cognitive impairment and dementia (PSCID) affects many survivors of large vessel cerebral ischemia. The molecular pathways underlying PSCID are poorly defined but may overlap with neurodegenerative pathophysiology. Specifically, synaptic dysfunction after stroke may be directly mediated by alterations in the levels of amyloid beta (Aβ), the peptide that accumulates in the brains of Alzheimer’s disease (AD) patients. In this study, we use the transient middle cerebral artery occlusion (MCAo) model in young adult mice to evaluate if a large vessel stroke increases brain soluble Aβ levels. We show that soluble Aβ40 and Aβ42 levels are increased in the ipsilateral hippocampus in MCAo mice 7 days after the injury. We also analyze the level and activity of β-site amyloid precursor protein cleaving enzyme 1 (BACE1), an enzyme that generates Aβ in the brain, and observe that BACE1 activity is increased in the ipsilateral hippocampus of the MCAo mice. Finally, we highlight that treatment of MCAo mice with a BACE1 inhibitor during the recovery period rescues stroke-induced deficits in hippocampal synaptic plasticity. These findings support a molecular pathway linking ischemia to alterations in BACE1-mediated production of Aβ, and encourage future studies that evaluate whether targeting BACE1 activity improves the cognitive deficits seen with PSCID.
Introduction
Long-term morbidity in stroke survivors exerts significant costs for both individuals and society. Extensive effort has been put forth developing therapeutics that improve acute ischemic stroke outcomes, with important success achieved via advancements in thrombolytic therapy and endovascular thrombectomy.1,2 Despite these treatments, a large proportion of stroke survivors struggle with long-term neurologic consequences. An area of increasing concern is post-stroke cognitive impairment and dementia (PSCID).3–6 Current estimates suggest that cognitive deficits are detectable during the recovery phase in roughly one-third of stroke survivors. 4 PSCID affects survivors of all ages, with evidence that even 50% of young stroke patients (<50 years old) exhibit ongoing cognitive issues that persist up to 11 years after the original brain injury. 7 Though awareness of PSCID as a clinical entity is increasing, little in the way of therapeutic advances has been achieved due to a limited understanding of the relevant cellular and molecular pathways underlying neuronal dysfunction following a stroke. Furthermore, while extensive effort has been put forth in stroke research targeting neuroprotection, 8 less attention has been devoted to the development of neuro-restorative therapies that improve cognition after the ischemic injury has already evolved. Therefore, studying the mechanisms underlying PSCID should be a prime focus of stroke research to develop novel therapeutic targets that can be used to improve the lives of survivors.
Investigating the pathophysiology underlying PSCID and evaluating possible treatment strategies require preclinical models that consistently demonstrate impaired cognition along with cellular evidence of reduced synaptic function. Middle cerebral artery occlusion (MCAo) is a well-established model of ischemic stroke that causes both short- and long-term behavioral deficits in hippocampal-dependent learning and memory.9–11 Interestingly, prior studies demonstrate that the MCAo injury triggers prolonged, impaired hippocampal plasticity.10,12 These synaptic alterations occur despite the injury site being remote from the hippocampus or related structures, often with little evidence of hippocampal cell death. 12 Given that hippocampal cell loss is unlikely to be the primary mediator of the cognitive deficits, the search for novel molecular pathways altered by the MCAo injury are necessary to better understand the cause of impaired synaptic transmission.
Preliminary evidence exists that Alzheimer’s disease (AD)-related pathology is associated with impaired cognition after an ischemic stroke. In clinical studies, increased brain amyloid β (Aβ) deposition13,14 and alterations in serum soluble Aβ and tau levels15,16 have been described in cases of PSCID. Furthermore, animal models of AD-associated amyloidosis subjected to MCAo have increased Aβ deposition17,18 and progressive behavioral deficits up to 12 weeks following the initial stroke. 18 While evidence is limited that stroke directly initiates AD pathogenesis, these findings suggest that components of the neurodegenerative changes of AD may nevertheless play a role in the pathophysiologic mechanism of PSCID. Aβ-mediated synaptic dysfunction has been postulated to play a central role in the cognitive decline present in AD. 19 It is well-established that soluble oligomeric Aβ species attenuate long-term potentiation (LTP), a measurement of synaptic plasticity, when added to murine hippocampal slices.20,21 Several synaptic pathways likely mediate this effect, including inhibition of NMDA and AMPA receptors along with the blockade of neuronal glutamate uptake.20,22,23 While the effects of soluble forms of Aβ on hippocampal function have been extensively studied in mouse models of AD pathogenesis, a potential role of soluble Aβ in mediating the synaptic deficits after experimental ischemic stroke has yet to be evaluated.
In this study, we use the transient MCAo model in mice to evaluate how a large vessel stroke affects soluble Aβ levels in the brain and to determine if altering the production of Aβ during the recovery period improves hippocampal plasticity. We hypothesize that large vessel ischemic stroke results in sustained elevations of endogenous Aβ that causes impaired hippocampal synaptic plasticity in surviving mice. Using a transient MCAo experimental model in mice, we measure the levels of endogenous soluble Aβ in both the cortex and hippocampus after a stroke. We observe an increase in hippocampal levels of soluble Aβ in the ipsilateral hemisphere following the MCAo stroke with no changes in cortical Aβ levels. We also demonstrate that MCAo stroke increases the activity of the Aβ-producing protein beta-secretase 1 (also known as beta-site amyloid precursor protein enzyme 1 or BACE1) in the hippocampus, an enzyme that has been extensively targeted from a therapeutic standpoint in AD clinical research. 24 Finally, we highlight that in vivo pharmacologic inhibition of BACE1 activity during the recovery period after a stroke significantly improves hippocampal synaptic function. Our findings emphasize a novel role for BACE1-mediated production of Aβ in mediating the synaptic dysfunction that occurs after a stroke and suggest this pathway should be further studied in stroke survivors suffering from PSCID.
Material and methods
Experimental animals
All animal use procedures were approved by the University of Colorado Institutional Animal Care and Use Committee and met United States Public Health guidelines as dictated by the National Institutes of Health (NIH). Adult male C57Bl/6 wild type (Wt) mice (aged 8–12 weeks) acquired from the Charles River Laboratory were used in this study. Animals were housed on a 14:10-hr light-dark schedule and had ad libitum access to standard rodent chow and tap water. The ARRIVE 2.0 guidelines for animal experiments were adhered to in all experiments for this study. 25 Groups for this study included both MCAo and sham-treated animals that were analyzed 7 days after the procedure. Any animals that did not survive the full 7 days were not included in the analysis. All animals were randomly assigned to the experimental groups and blinding was used both during the MCAo surgeries and by the individuals who performed all of the experiments.
MCAo model
Focal reversible cerebral ischemia was induced using an intraluminal filament technique as previously described with some modifications. 10 Isoflurane anesthesia (3–4% for induction, 1.5–2% for maintenance) was provided, and focal cerebral ischemia was induced by placing the filament into the right middle cerebral artery. Body temperature was maintained throughout the procedure by keeping the mice on a warm heating pad. Adequate occlusion was confirmed with a laser Doppler probe located over the ipsilateral parietal cortex. Successful occlusion was considered a blood flow reduction to less than 25% of baseline. Following confirmation of reduced blood flow, the anesthetic was discontinued and the animals were returned to their individual cages. After 60 minutes, the filament was removed. Sham mice were exposed to anesthesia and arterial access was obtained without occlusion.
BACE1 inhibitor drug treatment
The BACE1 inhibitor verubecestat was purchased from Selleck chemicals (MK-8931). Prior to treating the mice, the compound was re-suspended in DMSO and then dissolved in 20% beta-cyclodextran to its final concentration. Three hours prior to tissue collection, either verubecestat (30 mg/kg) or a control (20% beta-cyclodextran) solution was administered to the MCAo or sham animals via oral gavage. Animals were then returned to their cages prior to being used for experiments.
Brain sample collection
For the ELISA, qPCR, and BACE1 activity experiments, animals were anesthetized by exposure to isoflurane (3.0%) followed by a cardiac perfusion of cold phosphate-buffered saline (PBS) for 3 minutes. Brains were removed from the skull and the hippocampus was dissected from the cortex. The bilateral hippocampus and cortex tissue were flash frozen and stored at −80°C. For immunohistochemistry experiments, animals were anesthetized by exposure to isoflurane followed by a trans-cardiac perfusion of cold PBS for 5 minutes followed by 4% paraformaldehyde in PBS for 5 minutes. The brain was removed from the skull and stored at 4°C in paraformaldehyde. After 24 hours, the brains were transferred to cryoprotection solution and stored at 4°C until sectioning.
Tissue ELISA
Extraction of soluble mouse Aβ was performed using a modification of a previously described protocol. 26 Frozen brains were hand-homogenized in 50 mM NaCl with protease inhibitor (cOmplete Protease Inhibitor Cocktail, Millipore Sigma) for 75 seconds, followed by the addition of 10% diethylamine in 50 mM NaCl. The tissue lysate was then sonicated at 20% amplitude for 1 minute (1 second on: 1 second off intervals) and incubated for 1 hour at 4°C. The samples were centrifuged at 4°C for 1 hour at 100,000 g in a high-speed centrifuge. The supernatant was removed and then neutralized by adding 10% (v/v) of 0.5 M Tris-HCl (pH 6.8). Samples were then vortexed and stored at −80°C. ELISA assays specific to either Aβ40 or Aβ42 (Fujifilm Wako Chemicals) were used in all described experiments. A prior study has verified the specificity of these assays for mouse Aβ. 27 Samples were diluted with the diluent from the kit, then loaded onto the plate to incubate overnight at 4°C. Plates were washed with the cold, diluted wash solution from the kit 5 times before the HRP-conjugated antibody solution was added. The plates were then incubated at 4°C for 1 hour in the solution before being washed again 5 times. The TMB solution was added and incubated at room temperature for 30 minutes before the plate was read using a BioTek Synergy 2 plate reader. All tissue ELISA values were normalized to total protein content in the sample as measured with a Pierce BCA Protein Assay Kit (ThermoFisher Scientific).
Quantitative PCR
RNA was isolated from frozen hippocampal tissue using an RNAqueous-4PCR Total RNA Isolation kit (AM1914) from ThermoFisher Scientific. The amount of RNA was subsequently quantified using a NanoDrop One machine (ThermoFisher) and water was used to dilute all samples to equal amounts. cDNA synthesis was performed using the iScript cDNA Synthesis kit (BioRad, 1708890). Real time quantitative PCR measurements were performed by mixing the cDNA with ThermoFisher TaqMan primers and SsoAdvanced Universal Probes Supermix (BioRad, 1725281). The primer used for BACE1 was Mm00478671_m1. For all mRNA measurements, values were normalized to 18S ribosomal RNA.
Immunohistochemical staining and image acquisition
Fixed brains in cryoprotection solution were sectioned on a freezing sliding microtome to obtain 50 µm coronal sections. The tissue sections were stored in cryostorage solution at 4°C prior to use. Sections used for immunohistochemistry were washed 3 times for 5 minutes each in PBS. Sections were then incubated for 1 hour at room temperature in 5% normal donkey serum (NDS) in 0.3% Triton in PBS (PBST), followed by an overnight incubation at 4°C in primary antibody (monoclonal rabbit BACE1 antibody, Cell Signaling Technology, 1:100 dilution) in 3% NDS in 0.3% PBST. The tissue was then washed 3 times for 5 minutes each in PBS. The sections were incubated in the dark at room temperature for 1 hour in Alexa Flour-conjugated secondary antibody (Jackson Immuno Research) which was diluted in 0.3% PBST and 3% NDS. Following a second period of three 5 minute washes, the sections were also stained with Hoescht (Tocris) at room temperature for 5 minutes, diluted in PBS and then washed 3 times for 5 minutes before being mounted on slides. Images were acquired on an Olympus Fluoview confocal microscope.
Image quantification
BACE1 staining was quantified using ImageJ software (NIH). BACE1 staining was outlined by drawing a region of interest (ROI) using ImageJ on 8-bit images around the CA3 region. ImageJ was then used to calculate the mean intensity of BACE1 staining. For the final analysis, each data point represents one mouse and consists of the average of 2 coronal sections per mouse.
BACE activity assay
The tissue was lysed in a 1% Triton buffer with a protease inhibitor. All tissue lysates were used immediately after homogenization and centrifugation to ensure no degradation of enzyme activity. Beta-secretase activity was measured using a beta-secretase activity fluorogenic assay kit (Millipore Sigma). The lysed tissue was loaded into the well plate with the diluent and the fluorescent substrate, and a reading was immediately taken at time 0 on a BioTek Synergy 2 plate reader. The plate was then read a second time after an incubation at 37°C for 2 hrs. All fluoroscopic readings were normalized to total protein level from the tissue sample as measured with a Pierce BCA Protein Assay Kit (ThermoFisher Scientific).
Hippocampal slice preparation
Hippocampal slices were prepared at 7 days after recovery from MCAo in adult mice following treatment with either verubecestat or cyclodextran as described above. Animals were anesthetized with 3% isoflurane in an oxygen enriched chamber. Mice were trans-cardiac perfused with ice-cold (2–5°C), oxygenated (95% O2/5% CO2) artificial cerebral spinal fluid (aCSF) for 2 min prior to decapitation. The brains were then extracted and placed in the same aCSF. The aCSF was composed of the following (in mmol/L): 126 mM NaCl, 2.5 mM KCl, 25 mM NaHCO3, 1.3 mM NaH2PO4, 2.5 mM CaCl2, 1.2 mM MgCl2 and 12 mM glucose. 28 Horizontal hippocampal slices (300 μm thick) were cut with a Vibratome 1200 (Leica), transferred to a holding chamber containing aCSF, and allowed to equilibrate for at least 1 hr at room temperature prior to recording.
Electrophysiology
Synaptic evoked field potentials were recorded from hippocampal slices that were placed on a temperature controlled (31 ± 0.5°C) interface chamber perfused with aCSF at a rate of 1.5 ml/min. Field excitatory post-synaptic potentials (fEPSP) were produced by stimulating the Schaffer collaterals and recording in the stratum radiatum of the CA1 region. The fEPSP’s were adjusted to 50% of the maximum slope and test pulses were evoked every 20 seconds. Paired-pulse responses were recorded using a 50-ms interpulse interval (20 Hz) and expressed as a ratio of the slopes of the second pulse over the first pulse. Maximum slope of fEPSP was plotted against stimulus intensity (0–80 uA). Only slices in which the I/O curve fell within this range were used for this analysis. A 20-min stable baseline was established before delivering a theta burst stimulation (TBS) consisting of a train of four pulses delivered at 100 Hz in 30-ms bursts repeated 10 times with 200-ms interburst intervals, similar to as described previously. 28 Following TBS, the fEPSP was recorded for 60 min. Analog fEPSP’s were amplified (1000×) and filtered through a pre-amplifier (Model LP511 AC, Grass Instruments) at 1.0 kHz, digitized at 10 kHz and stored on a computer for later off-line analysis (Clampfit 10.4, Axon Instruments). The derivative (dV/dT) of the initial fEPSP slope was measured. The averaged 10-min slope from 50–60 min after TBS was divided by the average of the 10-min baseline prior to TBS to determine the amount of potentiation. 28 For time course graphs, normalized fEPSP slope values were averaged and plotted as the percent of baseline. For statistical analysis, each data point represents an individual hippocampal slice and no more than 2 slices were from an individual animal. A minimum of 3 to 4 animals (2–3 sections per animal) were used for each electrophysiology experimental group to assure biologic variability.
Statistical analysis
All statistical analyses were performed using GraphPad Prism v9.4 and presented as arithmetic mean ± standard deviation (SD). Group size for the Aβ ELISA studies was determined using a power analysis that would detect a 40% change between two groups based upon average Aβ levels from prior studies (standard deviation of 25 pg/mg for Aβ40 and 5 pg/mg for Aβ42, alpha error of 5% and beta error of 80%). Based upon these results 6 mice per group are required. For the LTP experiments, detection of a 40% difference with a standard deviation of 20 requires a group of 6 per treatment paradigm (alpha < 0.05 with 80% power). For the ELISA studies, qPCR results, BACE1 level measurements, and BACE1 activity analysis, the data was investigated using a 2 way ANOVA followed by a Tukey’s multiple comparisons test to assess for significance between treatment groups and brain hemisphere. For the electrophysiology studies, variability of LTP was greatly different between experimental groups, therefore Brown-Forsyth and Welch one-way ANOVA followed by Dunnett’s test for multiple comparisons was performed. Normality tests were performed with verification that all data conformed to normality. For all studies significance was assigned for a p value of less than 0.05. A summary of the statistics for all relevant ANOVA comparisons is provided in Supplemental Table 1.
Results
Transient MCAo stroke increases hippocampal, but not cortical, soluble Aβ levels
An ischemic stroke induced via the MCAo model has previously been shown to increase brain Aβ plaque formation in mouse models of AD-associated amyloidogenesis.17,18 To determine whether an acute ischemic stroke can alter soluble Aβ, we exposed Wt mice to a transient MCAo stroke and analyzed the levels of both Aβ40 and 42 in surviving animals. A transient stroke model was chosen to mimic vasculature reperfusion that commonly occurs either spontaneously or with reperfusion-based therapies in human stroke patients. We decided to analyze Aβ levels 7 days after the MCAo injury to allow time for the injury to evolve and minimize the possibility any observed effects were from the acute neurotoxicity phase after stroke onset. Aβ was extracted from the brain tissue using a diethylamine-based protocol that has been previously shown to isolate steady-state levels of soluble Aβ. 26 In the ipsilateral cortex, no significant differences were observed in the amount of Aβ40 (50.09 ± 7.87 pg/mg versus 62.61 ± 21.72 pg/mg, p = .23) or Aβ42 levels (9.43 ± 2.08 pg/mg versus 12.00 ± 3.17 pg/mg, p = 0.13) when comparing sham to MCAo-treated animals 7 days after the injury (Figure 1(a)). Similarly, the contralateral cortex also did not exhibit any differences in Aβ40 (52.93 ± 6.31 pg/mg versus 43.35 ± 7.09 pg/mg, p = 0.42) or Aβ42 (8.42 ± 0.84 versus 8.14 ± 1.60 pg/mg, p = 0.97) in sham compared to MCAo mice. However, MCAo stroke did increase hippocampal Aβ levels in the injured hemisphere (Figure 1(b)). Soluble Aβ40 levels were elevated by 71.1% (118.7 ± 24.77 versus 203.2 ± 103.8 pg/mg, p = 0.04) and Aβ42 levels were increased by 91.7% (23.21 ± 3.92 pg/mg versus 44.49 ± 20.00 pg/mg, p = 0.03) in MCAo mice in the ipsilateral hippocampus. No statistically significant differences were observed in Aβ40 levels (160.4 ± 42.78 versus 139.6 ± 20.60 for MCAo versus sham animals, p = 0.63) or Aβ42 levels (34.38 ± 10.57 versus 28.77 ± 6.13 pg/mg, p = 0.63) in the hippocampal tissue from the contralateral hemisphere. These results demonstrate that MCAo stroke leads to a brain region and hemisphere-specific increase in soluble Aβ levels 7 days after the injury.
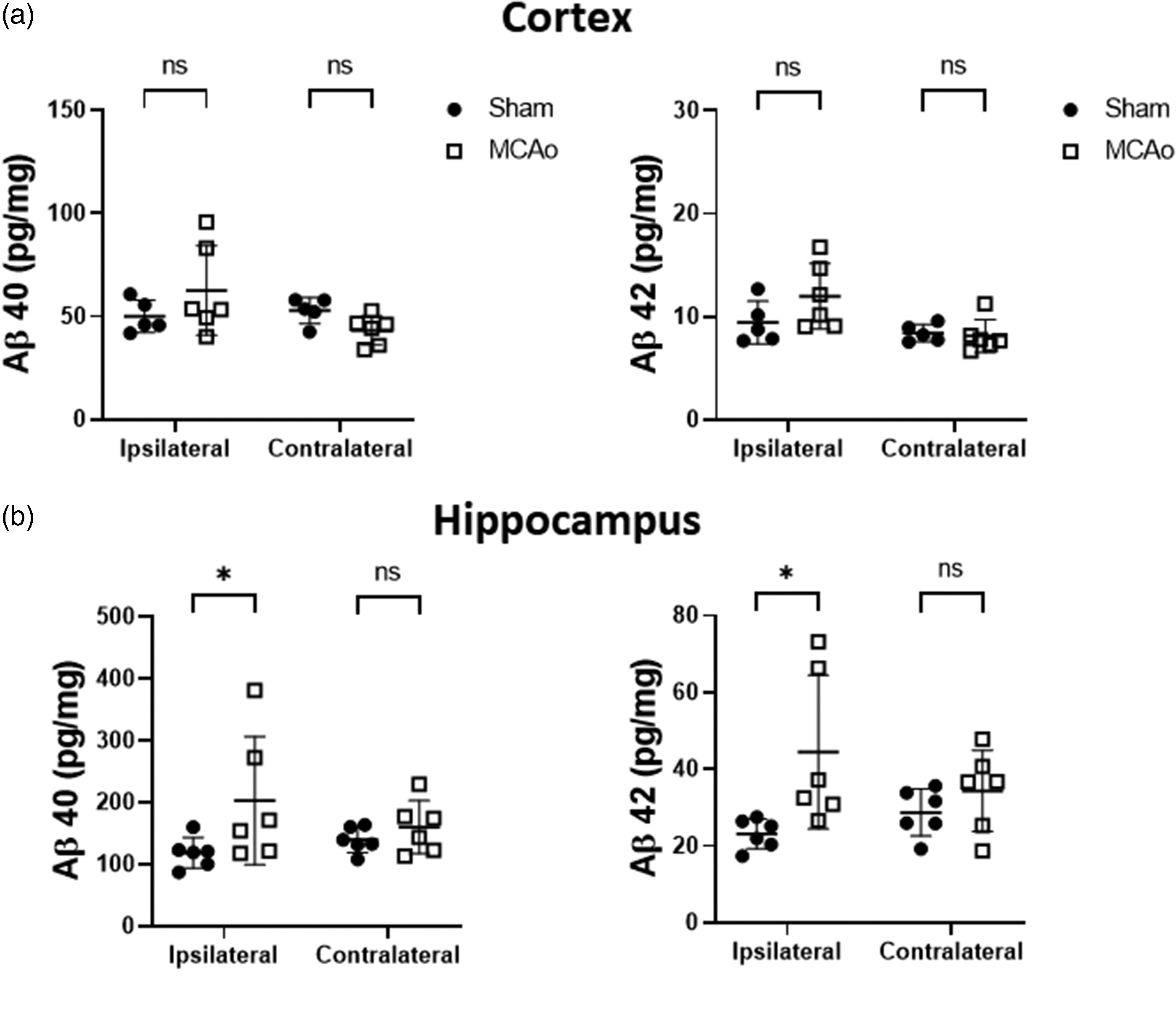
MCAo stroke increases hippocampal, but not cortical, soluble Aβ levels. The amount of tissue Aβ40 and Aβ42 were measured 7 days following either a sham or MCAo procedure in both the (a) cortex and (b) hippocampus. All Aβ levels are shown as total Aβ normalized to total tissue protein (n = 5–6 animals per group, data shown as mean ± SD, *p < 0.05 compared with sham controls).
MCAo stroke increases hippocampal BACE1 activity but not protein levels
BACE1-mediated cleavage of the amyloid precursor protein (APP) is the primary pathway for Aβ production in the brain. 24 BACE1 upregulation has been observed following ischemia in different stroke models.18,29 We therefore examined whether BACE1 levels were increased in the hippocampus following a MCAo stroke, possibly explaining the observed increase in Aβ levels. Quantitative PCR analysis of BACE1 mRNA levels did not demonstrate a statistically significant difference in levels in either the ipsilateral or contralateral hippocampus (Figure 2(a)). Immunohistochemical analysis of BACE1 levels in the CA3 hippocampal region did not demonstrate a difference in protein expression when comparing MCAo and sham animals (Figure 2(b) and (c)).
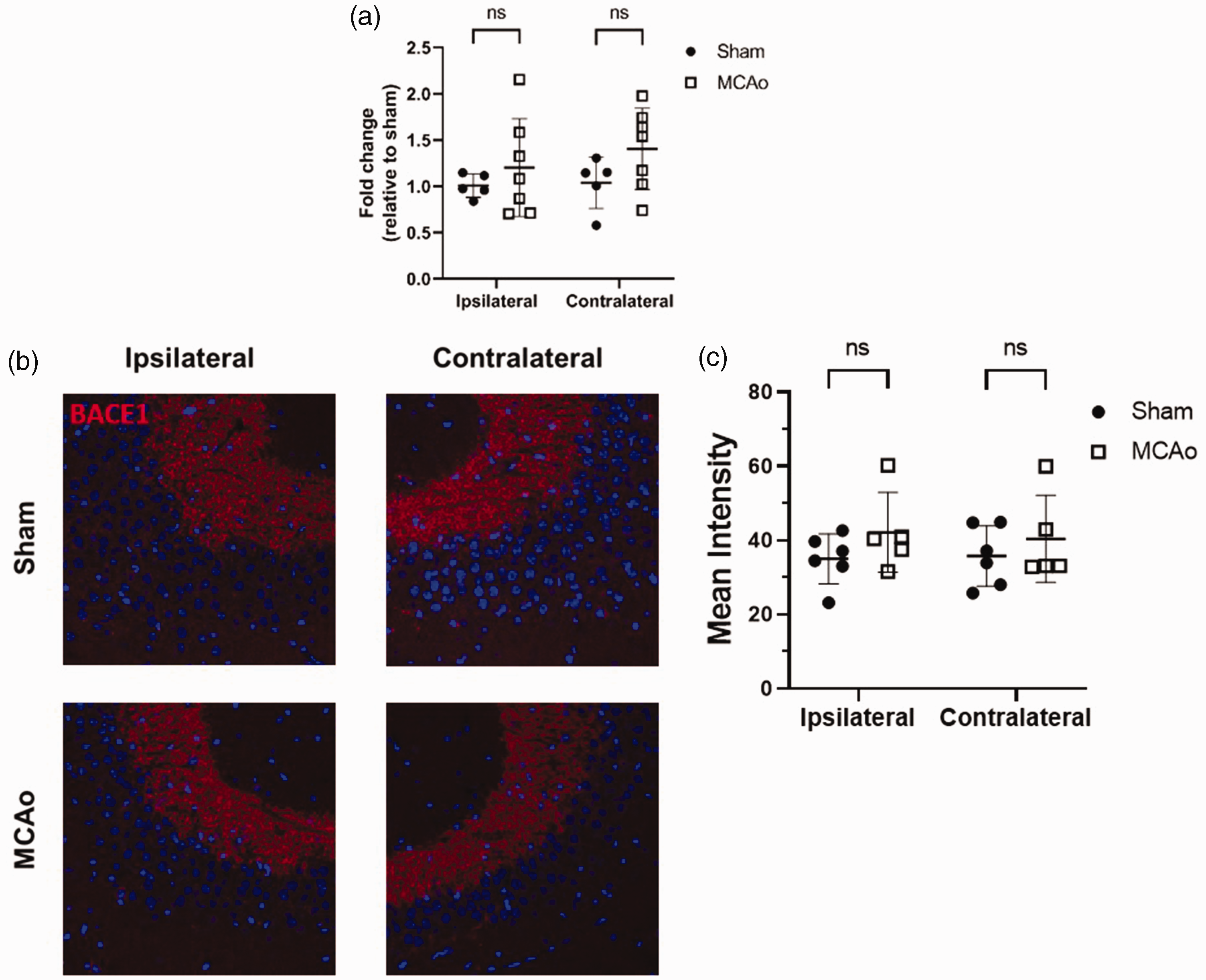
BACE1 mRNA and protein levels are not changed following an MCAO stroke. (a) Quantitative PCR levels of BACE1 mRNA in the hippocampus from both sham and MCAo mice (n = 5–7 animals per group, data presented as mean ± SD) and (b) Representative images displaying BACE1 (shown in red) immunohistochemistry of the CA3 hippocampal region from sham and MCAo brain. Note that nuclei-staining is shown in blue. (c) Quantification of the mean intensity of BACE1 staining in the hippocampus (n = 5–6 animals per group, each point represents the average of two slice from each animal, data presented as mean ± SD).
Despite the fact that BACE1 levels did not change in our MCAo cohort, it remains possible that the ischemia associated with the MCAo stroke affects BACE1 activity. A prior study has highlighted that an acute increase in BACE1 activity can be seen in the cortex of animals 24 hrs after the injury when using a transient ischemic mouse model. 29 We therefore measured the amount of BACE1 activity in the hippocampus using a fluorogenic-based BACE1 activity assay. Fresh tissue lysate from both the ipsilateral and contralateral hippocampal hemispheres of sham and MCAo mice 7 days after the procedure were analyzed. Interestingly, we observed a significant increase in BACE1 activity in the ipsilateral hippocampus (46.2% increase, p = 0.0053), but not the contralateral hippocampus, 7 days after the MCAo stroke injury (Figure 3). Taken together, these results suggest that increased BACE1 activity leads to an elevation in Aβ levels following a transient MCAo stroke.
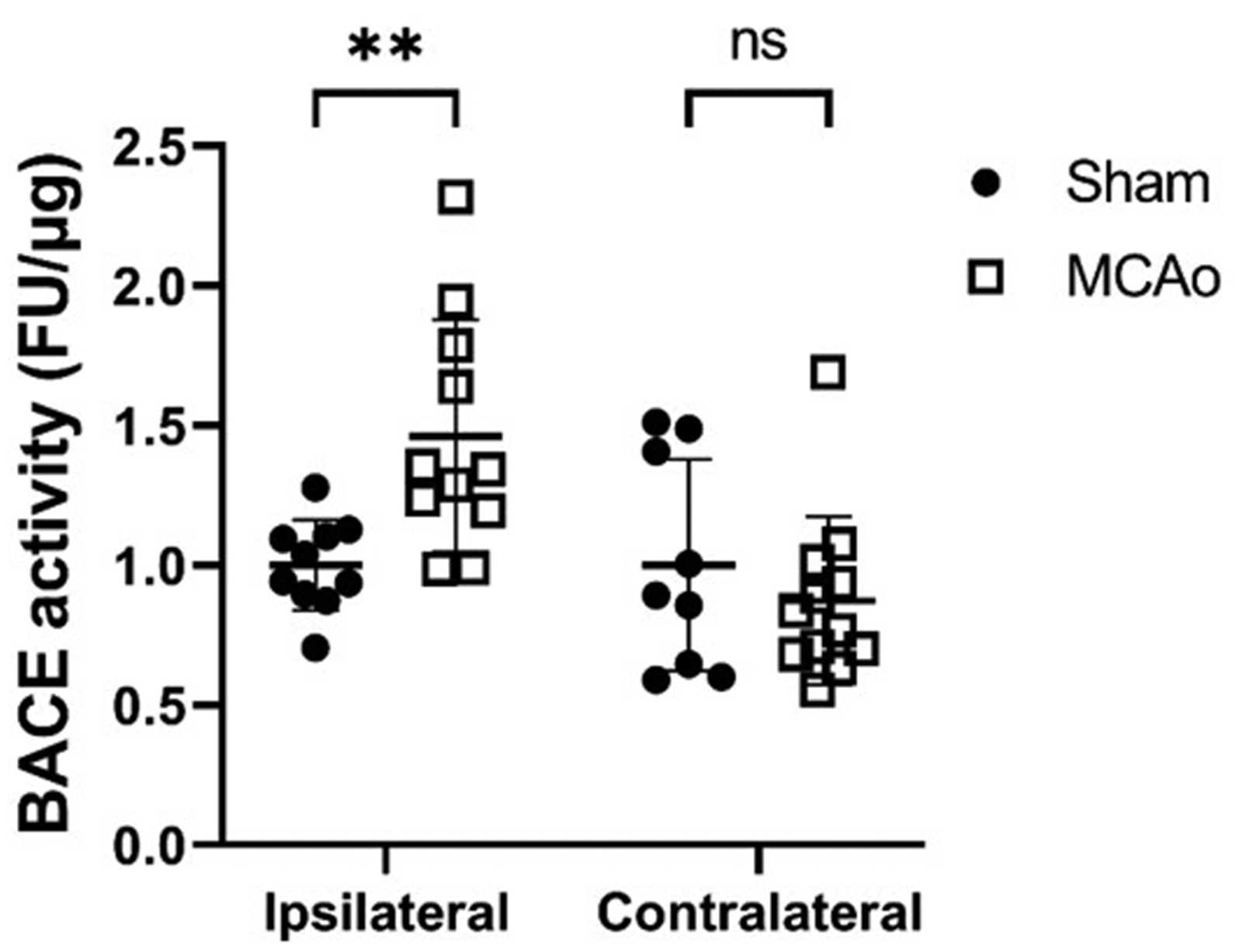
Hippocampal BACE1 activity is increased in the setting of a MCAo stroke. BACE1 activity was measured in hippocampal tissue lysates from both sham and MCAo animals and total relative activity is shown in comparison to sham animals. Note that for this experiment two groups of sham and MCAo mice were analyzed and the data was subsequently pooled. There was a significant change in ipsilateral BACE1 activity levels for each of the two separate experiments. (n = 9–12 animals per group, data presented as mean ± SD, *p < 0.05 compared with sham controls).
BACE1 inhibition improves hippocampal synaptic function after MCAo stroke
Hippocampal synaptic plasticity is decreased in mice following a MCAo stroke.10,12 Furthermore, soluble Aβ has been demonstrated to exert significant synaptotoxicity, 20 and increasing endogenous mouse Aβ levels has been shown to reduce synaptic density and plasticity in the hippocampus. 30 To determine the effects of decreasing Aβ levels via BACE1 inhibition on hippocampal synaptic plasticity, we treated both sham and MCAo mice on day 7 with a known BACE1 inhibitor (verubecestat, 30 mg/kg) and then measured hippocampal LTP 3 hours following administration from fresh brain slices. This verubecestat dose has been shown to decrease cerebral spinal fluid Aβ levels by 90% and brain tissue levels by 66% three hours after administration. 31 To verify that verubecestat would affect Aβ levels in our ischemic mice, we first measured Aβ levels in the hippocampus 3 hours after treatment via an ELISA. As expected, we saw a significant decrease in Aβ42 levels in the verubecestat-treated group (Figure 4(a), 42.9% decrease, p = 0.0072). For the LTP experiments, extracellular field recordings were performed from ipsilateral hippocampal slices collected 3 hours after drug administration. Representative field excitatory post-synaptic potential (fEPSP) tracings are shown in Figure 4(b) from sham and MCAo mice treated with either cyclodextran (vehicle) or verubecestat (30 mg/kg). Figures 4(c) and (d) shows that sham-operated, vehicle-treated mice exhibit robust LTP following a brief TBS (40 pulse, 100 Hz), increasing the fEPSP slope to 166 ± 25.5% (n = 6). In contrast, stroke operated, vehicle treated mice demonstrate impaired LTP (124 ± 9.6% (n = 7) 60 minutes post-TBS. Following treatment with verubecestat, we saw an increase in LTP in sham mice compared to the vehicle group though this was not statistically significant when analyzed with an ANOVA (225 ± 55.4%; n = 6). Of note, subgroup analysis did show significance when comparing these two groups via an unpaired T-test (p = 0.04). Importantly, MCAo mice treated with verubecestat demonstrated a significant increase in LTP compared to the control MCAO group (219 ± 45.3%; n = 6; p < 0.05) 60 min post TBS, highlighting improvement in synaptic plasticity following BACE1 inhibition.
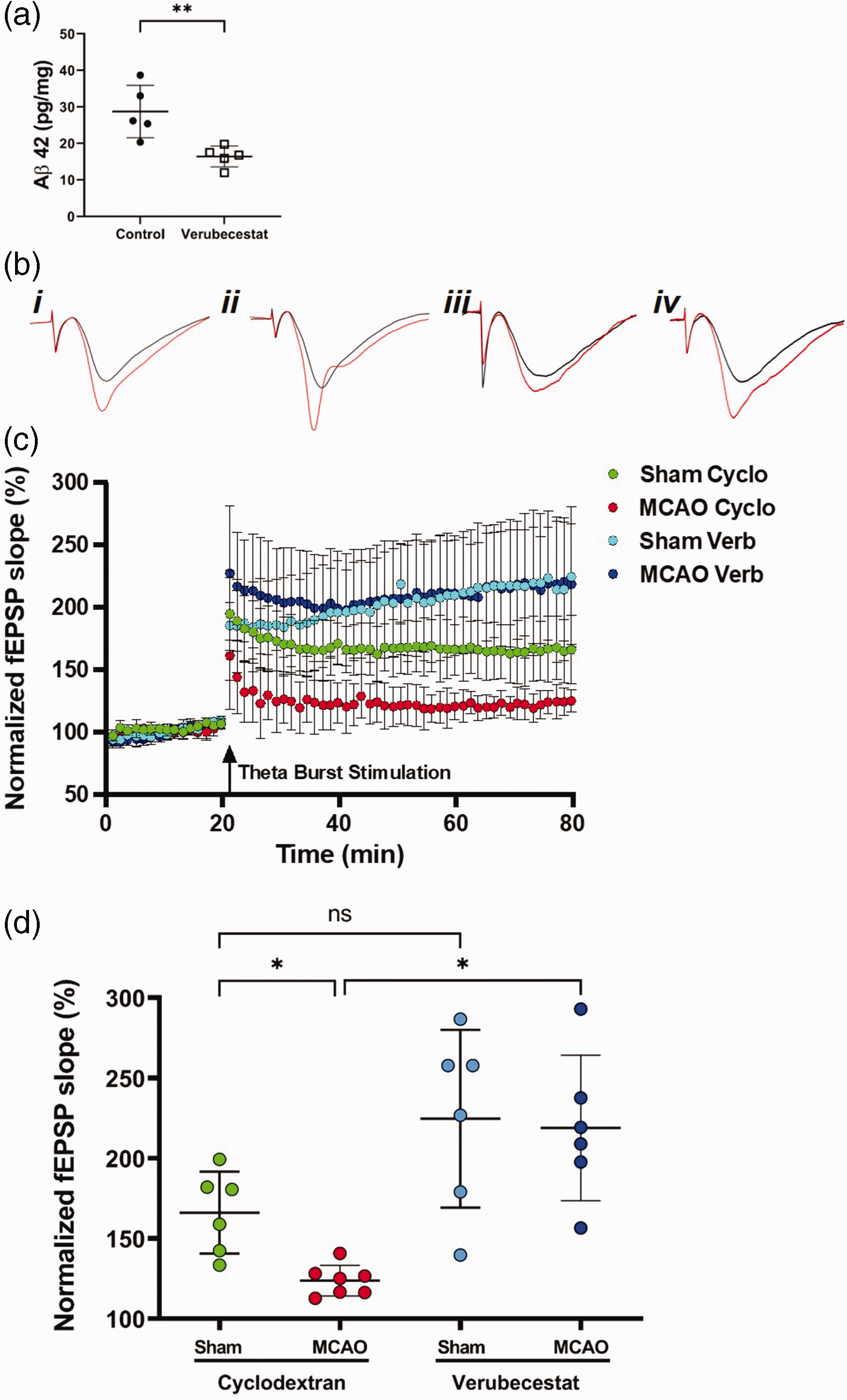
BACE1 inhibition restores synaptic plasticity 7 days following MCA stroke. (a) Effect of BACE1 inhibition on Aβ42 levels 3 hours post-treatment (n = 5, data represented as mean ± SD *p < 0.005 compared with sham controls). (b) Representative fEPSP traces from i) sham with vehicle (cyclodextran), ii) sham with verubecestat (30 mg/kg, iii) MCAo with vehicle and iv) MCAo with verubecestat. Black line indicates pre-TBS trace and red line post-TBS trace. (c) Time course of fEPSP slope (mean ± SD) from sham (pink-filled circles) and MCAo (red-filled circles) treated with vehicle (cyclodextran), and sham mice (light blue-filled circles) and MCAo (dark blue-filled circles) treated with verubecestat. Arrow indicates timing of theta-burst stimulation (40 pulses) and (d) Quantification of change in fEPSP slope after 60 min following TBS normalized to baseline, set to 100%. *p < 0.05. Data are presented as mean ± SD.
We then examined the paired-pulse ratio (PPR), a measure of pre-synaptic transmitter release probability.32,33 Paired-pulse responses were recorded from CA1 pyramidal cells in the stratum radiatum using a 50-ms (20 Hz) interpulse interval applied to the Schaffer collateral pathway (Supplemental Figure 1a). We did not observe differences in the PPR in any of the groups (1.41 ± 0.05% (n = 6), 1.46 ± 0.07% (n = 6), 1.36 ± 0.05% (n = 7), 1.41 ± 0.07% (n = 6) in sham-vehicle, sham-verubecestat, MCAo-vehicle and MCAo-verubecestat, respectively.) This suggests that impairment or enhancement of LTP seen at this time point is not due to changes in the pre-synaptic probability of release. Input/output functions of fEPSP’s versus stimulation amplitude were established at the beginning of each recording prior to TBS to examine CA3 axonal intrinsic excitability and synaptic transmission. The slope of each recording was measured individually and compared to control slices (Supplemental Figure 1b). Interestingly, we observed a significant reduction in the slope of MCAo animals compared to sham control with no effect of drug treatment. In summary, these results demonstrate that a single dose of the BACE1 inhibitor verubecestat given 7 days after an MCAo stroke rescues LTP deficits in the injured hippocampus.
Discussion
Significant attention has been devoted to understanding the relationship between small-vessel vascular dementia and AD. 34 However, the importance of studying the interaction between the pathophysiology of cognitive dysfunction following a large vessel stroke with the pathology of AD is only recently gaining attention in the field of stroke research. 3 Significant work is needed to elucidate the mechanistic underpinnings of this relationship at a molecular level. In this study, we examine how soluble Aβ levels change following a transient MCAo ischemic stroke in both the hippocampus and cortex. We specifically observe that 7 days after the ischemic injury, hippocampal Aβ40 and Aβ42 levels were increased in the ipsilateral hippocampus. No statistically significant changes in Aβ levels were observed in the contralateral hippocampus or either cortical hemisphere. We also demonstrate that the activity of BACE1 is upregulated in the ipsilateral hippocampus 7 days after a stroke, suggesting that increased Aβ production may in part be causing the rise in soluble Aβ. To our knowledge, this is the first study demonstrating that a transient ischemic stroke increases endogenous levels of soluble Aβ in the hippocampus. Our results also highlight that large vessel ischemia may lead to the activation of pathways involved in Aβ generation.
An elevation in hippocampal Aβ following a stroke has several important implications for understanding and treating PSCID. First, elevations in soluble Aβ may directly lead to impaired synaptic function in the recovery period after a stroke. Soluble forms of Aβ have been shown to impair LTP when applied to hippocampal slices in vitro and when increased in animal models.21,35,36 Furthermore, Krohn and colleagues demonstrate using a mouse model with genetic deletions of proteins involved in Aβ clearance that chronic elevations in mouse soluble Aβ decrease hippocampal LTP and reduce hippocampal synaptic density. 30 Second, chronic increases of soluble Aβ levels in the brain after a stroke may lead to deposition of Aβ into plaques, ultimately leading to neuronal toxicity and cell death. Prior studies using the MCAo model have analyzed Aβ pathology in the murine brain and have shown changes in Aβ plaque deposition. Garcia-Alloza et al. demonstrated that amyloid plaques were increased 7 days after a stroke in a mouse model of AD-associated amyloidosis. 17 Nguyen and colleagues highlighted that MCAo stroke in wild type mice promotes the deposition of Aβ in white matter tracts, but only in animals that were 18 months old. 18 Early increases in soluble Aβ after a stroke may be a catalyst for the development of amyloid accumulation in the brain, but this hypothesis needs further verification. Finally, targeting soluble Aβ levels in stroke survivors represents a potential approach for improving cognition in individuals that suffer from PSCID, though significant further work is needed confirming our findings before trials can be conducted in human patients.
The increase in Aβ levels seen after the transient MCAo stroke was notably only present in the ipsilateral cortex, with no significant differences apparent in either the cortex or contralateral hippocampus. While injury severity likely plays a role in the differences between hippocampal hemispheres, the mechanisms underlying the regional effects between the cortex and hippocampus are likely more complex. For example, the type and extent of injury is different between the cortex and hippocampus in this model. While neuronal cell death exists in the cortex following a transient MCAo stroke, results from our group and others suggest that the hippocampus is largely devoid of neuronal loss.10,12,37 Impairments in hippocampal LTP following the MCAo stroke injury are therefore likely due to changes at a synaptic level, which may function to alter Aβ production. MCAo stroke has also been shown to impair connectivity between the cortex and hippocampus, potentially leading to global dysregulation of hippocampal Aβ levels in the setting of ischemia independent of direct injury. 38 Finally, aberrant hippocampal neurogenesis has been shown to exist in the adult rodent brain after a stroke and may contribute to an increase in Aβ levels in nascent neurons. 39 Further work will be needed to understand the region-specific effects on Aβ after an ischemic stroke.
The experiments in this study used a transient MCAo model that includes both an ischemic and reperfusion element. While debate continues in stroke research regarding the role of both permanent and ischemia-reperfusion models, 40 we chose the latter in this study for several reasons. First, the use of recanalization therapies including either intravenous lytic therapy or mechanical thrombectomy continue to increase as knowledge of their clinical benefit grows.41–43 Therefore, the number of stroke patients with a transient occlusion will continue to increase in the coming years. Second, spontaneous reperfusion occurs in a significant number of stroke patients, 44 and understanding the brain’s response to reperfusion injury remains relevant to develop therapeutics that improve long-term stroke outcomes. Finally, from a practical standpoint reperfusion increases the likelihood that treatment with verubecestat would allow the drug to reach the injured brain area in a timely manner. Nevertheless, we recognize the mechanisms underlying both brain injury and recovery between a permanent and transient stroke model likely differ, and it will therefore be critical for future studies to also characterize how a permanent ischemia model affects the BACE1-Aβ pathway.
Prior work from our group and others demonstrated that hippocampal synaptic function as assessed by LTP is reduced at both 7 and 30 days after a transient MCAo stroke.10,12 In the current study, we were able to rescue this deficit at 7 days by treating mice with the BACE1 inhibitor verubecestat. Our results present the first evidence that BACE1 inhibition enhances synaptic plasticity after a stroke. It is important to emphasize that targeting BACE1 in both Wt mice and AD models has produced mixed results in regards to synaptic regulation. 45 BACE1 inhibition has been shown to improve LTP deficits in Thy1-APP-TG rats, 46 and genetic deletion of the Bace1 gene restored LTP and increased dendritic spine density in mouse models of AD-related amyloidosis.47,48 However, studies using Wt mice have shown that both genetic and pharmacologic decreases in BACE1 levels can impair both synaptic plasticity and cognitive function.49,50 Several reasons likely exist for these discrepancies, including differences in the duration of BACE1 inhibition and extent of synaptic integrity prior to decreasing BACE1. The magnitude of Aβ reduction after BACE1 inhibition may also be relevant, as evidence exists that low levels of Aβ are necessary for neurotransmission and memory formation. 50 Unfortunately, cognitive deficits have been observed in clinical trials with AD patients in the setting of chronic BACE1 inhibition, 51 suggesting a need to modify the approach or dosing regimen with BACE inhibitors if they are to be tested in different patient populations in the future. In the current study, we assessed the effect of a short-term decrease in BACE1 by treating MCAo and sham animals with a single oral injection of verubecestat. In our experimental paradigm, we observed an increase in LTP with verubecestat treatment alone in the sham mice when the treatment subgroups were analyzed individually. The exact reason for this difference may be secondary to the acute lowering of endogenous Aβ levels, the decrease of a BACE1 substrate other than Aβ, or an off-target effect of the drug. To our knowledge, LTP has not been assessed in wild type mice after a single dose of verubecestat, and further studies are warranted to better understand the mechanism mediating improved cognition with acute lowering of BACE1.
Other mechanisms regulating Aβ levels in the brain may also be altered after a stroke and prove to be useful targets for decreasing Aβ levels in stroke survivors. For instance, perturbation of pathways regulating metabolism of soluble Aβ from the brain may be relevant. Upregulation of apolipoprotein E (apoE), a protein that binds Aβ and mediates Aβ clearance from the brain, occurs in multiple forms of brain injury. Chronic elevations of apoE may also occur after an ischemic stroke, leading to an impairment in Aβ clearance. Chronic neuroinflammation after a stroke has been proposed as a potential driver of PSCID, and could modulate Aβ levels through dysregulated microglia or astrocytic Aβ degradation. 52 Finally, disruption of both the blood-brain barrier and glymphatic system are present after an ischemic stroke,53,54 both of which play important roles in Aβ clearance. While beyond the scope of this study, future studies evaluating the contribution of these mechanisms to modulation of Aβ levels following an ischemic stroke are warranted.
In summary, our study demonstrates for the first time that MCAo stroke leads to an increase in endogenous, soluble hippocampal Aβ levels and BACE1 activity after the injury. We also show that inhibiting BACE1-mediated generation of Aβ production improves hippocampal function following a stroke. Taken together, our data provide a potential molecular pathway linking ischemia to altered neurodegeneration after a large vessel stroke and suggest that targeting reduction of Aβ levels may improve synaptic and cognitive deficits seen with PCSID. In the future, studies will be needed using the MCAo model to address the effect of long-term BACE1 inhibition following a stroke on synaptic plasticity and cognition in animal stroke models.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X231159597 - Supplemental material for Targeting BACE1-mediated production of amyloid beta improves hippocampal synaptic function in an experimental model of ischemic stroke
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X231159597 for Targeting BACE1-mediated production of amyloid beta improves hippocampal synaptic function in an experimental model of ischemic stroke by Jacob M Basak, Macy Falk, Danae N Mitchell, Kelley A Coakley, Nidia Quillinan, James E Orfila and Paco S Herson in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a mentored research training grant from the Foundation of Anesthesia Education and Research (JMB), NIH R01 NS092645 (PSH), and the Richard Traystman Endowed Fellow Developing Clinician Scientist Award from the University of Colorado School of Medicine (JMB).
Acknowledgements
We would like to thank Ben Wasserman for help with performing and optimizing the MCAo experiments for this study. We would also like to thank all members of the Neuronal Injury and Plasticity Program at the University of Colorado School of Medicine for their insight and discussion around the experiments in this study.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
JMB and PSH conceived the study and designed all of the performed experiments. JMB and MF carried out the ELISA, immunohistochemistry, and BACE1 enzyme activity assay studies. DNM and KC performed the MCAo surgeries. JEO designed and carried out the BACE1 inhibitor injection studies and performed the electrophysiology experiments. JMB, JEO, NQ, and PSH analyzed all data for the study. JMB, MF, JEO, and PSH wrote the manuscript. All authors were involved in editing and reviewing the manuscript.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.