
Abstract
Stroke pathology and its treatments conventionally focus on the brain. Probing inflammation, a critical secondary cell death mechanism in stroke, has been largely relegated to the brain. To this end, peripheral inflammation has emerged as an equally potent contributor to the onset and progression of stroke secondary cell death. Here, we review novel concepts on peripheral organs displaying robust inflammatory response to stroke. These inflammation-plagued organs include the spleen, cervical lymph nodes, thymus, bone marrow, gastrointestinal system, and adrenal glands, likely converging their inflammatory effects through B and T-cells. Recognizing the significant impact of this systemic inflammation, we also discuss innovative stroke therapeutics directed at sequestration of peripheral inflammation. This review paper challenges the paradigm of a brain-centered disease pathology and treatment and offers a peripheral approach to our stroke understanding.
Introduction
Stroke is an incredibly debilitating disorder, killing nearly 130,000 people each year and ranked as the 5th leading cause of death in the United States. 1 Furthermore, stroke is the leading cause of long-term disability, causing paralysis, motor dysfunction, and more. 2 Stroke is defined as a lack of blood flow to the brain leading to extensive infarction, either due to ischemia or hemorrhage. 3 For ischemic strokes, the mortality rate is near 25%, while 50% of patients die from hemorrhagic stroke. 4
Despite these grave statistics and the large incidence of stroke, few therapies are available, and most are inadequate at reversing stroke damage. 5 Current treatments include tissue plasminogen activator (tPA) and mechanical thrombectomy (MT) to combat ischemic clot, however, both must be given in a limited time window. Furthermore, tPA has an associated risk of hemorrhage and suppressed anti-inflammatory regulatory T-cell (T-reg) responses.6–8 Compounding the primary insult, secondary cell death exacerbates ischemic stroke. The ensuing cascade of subsequent cell death involves multiple mechanisms, notably inflammation, altogether playing a key role stroke progression, as well as offering novel therapeutic targets.
Importantly, exorbitant inflammation causes significant impairment of the neurovascular unit. 9 The common causes of stroke include atherosclerosis, hypertension, diabetes, or infection, all of which promote a systemic pro-inflammatory environment. 10 While central inflammation ensues after stroke, secondary cell death entails an equally robust systemic inflammation. Elevated C-reactive protein, leukocyte, and cytokine measurements after stroke exemplify systemic inflammation. 11 Targeting systemic inflammation may lead to tremendous breakthroughs in developing effective stroke treatments. This review will outline the major roles of inflammation in stroke, with a pertinent focus on central (i.e. brain), peripheral organ, and lymphocyte involvement in stroke pathogenesis (Figure 1). Furthermore, an overview of potential therapies targeting peripheral inflammation in stroke will be summarized.
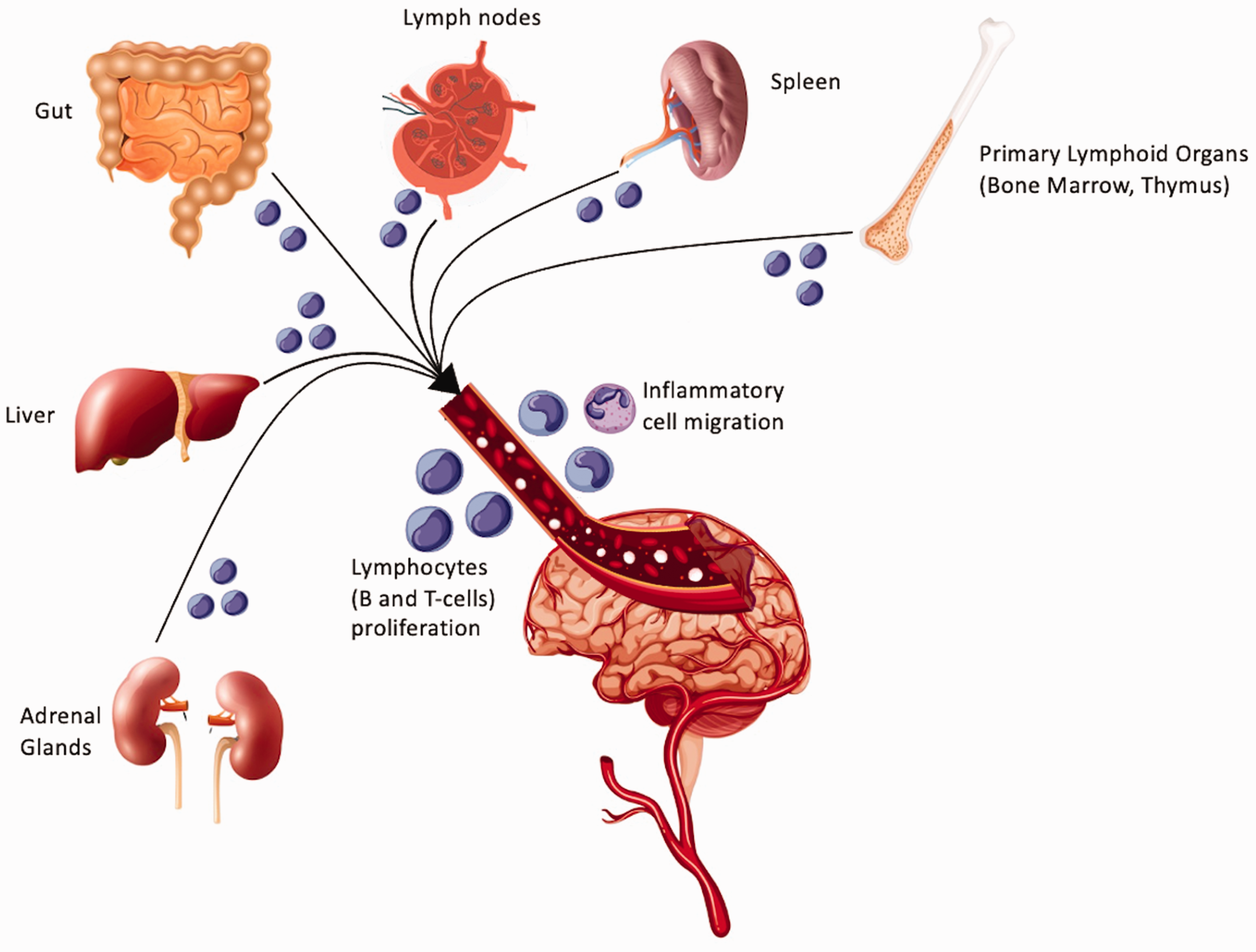
Peripheral inflammation in stroke. This diagram exemplifies the multiple components of the systemic inflammatory response. Influences from the adrenal glands, liver, gut, lymph nodes, spleen, bone marrow, thymus, and lymphocytes all contribute to peripheral inflammation and worsened stroke outcomes.
Neuronal death after stroke and subsequent inflammation
Cell death after stroke is primarily due to oxygen and nutrient deprivation, however, necrosis and consequent reperfusion contributes to even further cell death. 12 The proposed secondary cell death mechanisms for stroke include excitotoxicity, oxidative stress, free radical accumulation, mitochondrial dysfunction, impaired neurogenesis, angiogenesis, vasculogenesis, and inflammation. 5 Ischemic injury to multiple cell types within the neurovascular unit may succumb to cell death; thus, while many of the topics discussed here may pertain to neurons, such ischemic cascade of cell damage involves impairment to the entire neurovascular unit. After cerebral vessel blockage, neurons suffer from ATP depletion and ultimately release glutamate to the extracellular space. As a result of ATP depletion, membrane protein pumps vital to cell survival and functionality, the Na+/K+ ATPase and Na+/Ca2+ ATPases, fail. Sodium buildup causes cellular edema and rupture. Calcium becomes overly abundant in the cell as well, leading to mitochondrial swelling and mitophagy (programmed mitochondrial death). 13 Furthermore, large amounts of calcium flood the neurons and activate lipases, proteases, and other cell death pathways. 14 Without ample mitochondria to produce ATP and remove excess calcium, cytochrome C, an apoptotic activator, is released from the few remaining mitochondria and promotes apoptosis. 13 Neurons also release ATP and Fas ligand to activate apoptosis. Cell death leads to high levels of reactive oxidative species (ROS) and mitochondrial dysfunction, which causes further ATP depletion. 15 Products of cell death signal to extracerebral inflammatory cells, such as leukocytes, macrophages, and neutrophils, which release pro-inflammatory cytokines and exacerbate cell death.16,17 Macrophages have two differentiations, pro- or anti-inflammatory.1,2,18,19 Cell death products induce pro-inflammatory macrophage differentiation. 20 Inflammatory macrophages release tumor necrosis factor α (TNF-α), interleukin-1β (IL-1β), matrix metalloproteinase (MMP) 3 and 9, and ROS to exacerbate inflammation. 12 The abundance of pathways resulting from cerebral ischemia offer many potential therapeutic targets. Ultimately, however, a major consequence of ischemia is an excessive inflammatory environment. Whereas this ischemic stroke-induced inflammation appears to arise centrally, within the brain, equally compelling evidence that acutely and chronically widespread systemic inflammation accompanies ischemic stroke. Accordingly, cognizant of the tandem central and peripheral inflammatory events necessitates a dual-pronged approach in our understanding of the pathology and treatment of ischemic stroke.
Local neuroinflammatory responses to ischemic stroke
After ischemic insult, damage associated molecular proteins (DAMPs) activate local immune and inflammatory cells.15,18 An example of these activated cells include neutrophils. Neutrophils have high levels of proteolytic proteins and ROS, contributing to cell death.26,32 Another example are microglia which can differentiate into pro-inflammatory (M1) or anti-inflammatory (M2) phenotypes, however, the direct polarization into two subtypes is debated, as some researchers suggest a more fluid intersection between inflammatory and anti-inflammatory microglia. 21 M2 microglia can be further differentiated into M2a, M2b, or M2c microglia. While subtype M2a plays a role in neurogenesis, M2b and M2c are more involved in cleaning up the dead neurons. 22 In the case of central nervous system (CNS) injury, M1 microglia predominate. 18 These inflammatory microglia release TNF-α and IL-1β, which further promote M1 differentiation. Microglia also release ROS and reactive nitrogen species. 18 In some cases, M2 microglia will dampen the M1 response, 23 however, in severe ischemia, M1 microglia perpetuate an ongoing inflammatory cycle, leading to excitotoxicity, oxidative stress, mitochondrial dysfunction, blood brain barrier disruption, and even further inflammation.1,2 The inflammasome generated by these cells, and subsequent increases in IL-1 and TNF-α, induces an amplified inflammatory cascade via cytokine and chemokine production by endothelial cells and astrocytes.18,19 Yet another glial cell, astrocytes, are activated as well. Astrocytes have a plethora of receptors which can modulate the central inflammatory response. 24 In response to IL-1α, TNF-α, and complement component subunit 1q (C1q) released by microglia, astrocytes act to enhance neuronal injury. Additionally, these glial cells can release IL-15 to increase CD8+ T-cells and natural killer (NK) cell migration to the injury.25,26 In some cases, astrocytes can release IL-33 or C-C Motif Chemokine Ligand-1 (CCMK-1) to reduce inflammation via the migration of T-reg cells. 24 In mice without IL-33, this protective signaling is absent and T-reg cells do not mitigate stroke damage. 27 Cytokines, chemokines, and ROS released by these local immune modulators play a significant role in stroke pathogenesis.
The weakened blood-brain barrier diminishes the Central nervous systems’ immune privilege
The blood brain barrier (BBB) barrier typically prevents extraneous cells and signals, such as immune cells and pro-inflammatory signals, from entering the CNS. 28 The neurovascular unit (NVU) of the CNS includes the BBB and is mainly composed of neurons, glial cells, endothelial cells (EC), vascular smooth muscle cells (VSMC), pericytes, and a basement membrane including collagen IV. The NVU allows for oxygen and glucose exchange while prohibiting toxic accumulations of cytokines and inflammatory cells in the brain. 29 With the newfound permeability and NVU damage following ischemic insult, however, DAMPs, cytokines, and chemokines are exuded into the systemic circulation to recruit peripheral macrophages, neutrophils, and lymphocytes30,31 (Figure 2). Cellular adhesion molecules, such as intercellular adhesion molecule-1 (ICAM-1), are upregulated, allowing for neutrophils and microglia to flood the injury, followed closely by T lymphocytes.32,33 While stroke induces massive changes within the CNS, many factors such as cytokines, chemokines, the sympathetic nervous system, and DAMPs also induce peripheral organs to activate pro-inflammatory responses, ultimately instigating a systemic inflammatory response. 12
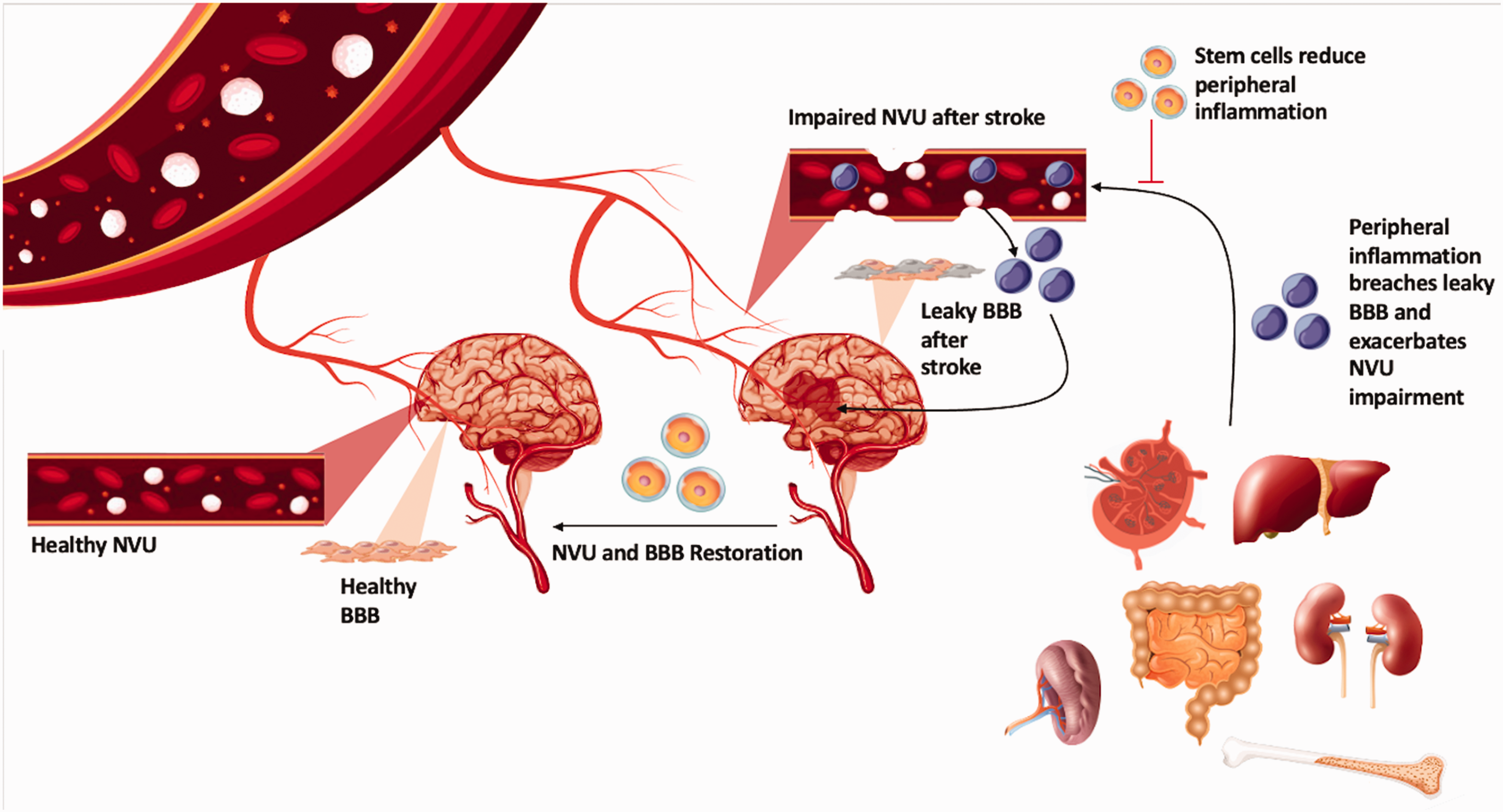
Neurovascular Unit and Blood-Brain Barrier permeability allows for peripheral inflammatory contributions to neuroinflammation. This image exemplifies the peripheral organs’ adrenal glands, liver, gut, lymph nodes, spleen, bone marrow, and thymus to contribute to neuroinflammation due to impaired permeability of the Neurovascular Unit (NVU) and Blood-Brain Barrier (BBB).
Also within the NVU, intravascularly, the coagulation cascade and complement system are activated after vessel occlusion.14,34 The coagulation cascade can directly instigate inflammation. 34 C3a and C5a, anaphylatoxins and products of the complement system, increase ROS production, secretion of proinflammatory cytokines, and immune cell degranulation. 15 Within the CNS, microglia are thought to amplify the complement system activation and astrocytes have several receptors for C1q, C3a, C5a, and complement receptor 3. 35 The C3a receptor plays a notable role in stroke, as mice with a knockout C3a receptor or C3a receptor antagonist show reduced ischemic injury and improved functionality. 36 Deficiencies in mannose binding lectin, another activator of the complement system, also demonstrate improved clinical outcomes after stroke. 37 Via the complement system and DAMPs, a plethora of local pro-inflammatory pathways are activated. The subsequent release of ROS and inflammatory cytokines, such as IL-6, IL-1, and TNF-α, may be immunoprotective at first, 38 but overactivity causes cell death and further damage to the BBB and NVU. 39
While ischemic stroke targets the brain primarily, parallel homeostatic imbalances to the peripheral inflammation-plagued organs, like the spleen, cervical lymph nodes, thymus, bone marrow, gastrointestinal system, and adrenal glands, trigger systemic inflammation, likely via B and T cell mobilization, in response to stroke acting via dependent, as well as independent of direct DAMP signaling pathways. Ultimately, the weakened BBB allows for activation of peripheral inflammatory responses along with detrimental immune and inflammatory cell migration to the ischemic area.
Peripheral inflammation and its contribution to neuroinflammation
Evidence for peripheral inflammation after stroke is supported by a wide number of studies examining pro-inflammatory biomarkers in the periphery of patients with stroke. In humans, prolonged peripheral inflammation correlates to worse clinical outcomes. 12 At an organ level, spleen and thymic shrinkage is noted after stroke, parallel to an increase in systematic lymphocyte concentrations. 40 Cerebral antigens activate lymph nodes, which ultimately further contribute to this lymphocyte surge.41,42 The bone marrow also exudes a plethora of pro-inflammatory cells which exacerbate peripheral inflammation. 43 The gut, via the recently established gut-brain axis, impacts peripheral inflammation by altered permeability and dysbiosis after stroke. 44 The adrenal glands also play a role via the HPA axis and subsequent release of cortisol and catecholamines.45,46 Thus, it is vital to examine peripheral inflammation for stroke therapy development. Multiple organs, including the spleen, cervical lymph nodes, thymus, bone marrow, gastrointestinal system, and adrenal glands, likely elicit B and T cell immune responses in conferring stroke-induced peripheral inflammation (Table 1).
Inflammation-plagued peripheral organs.
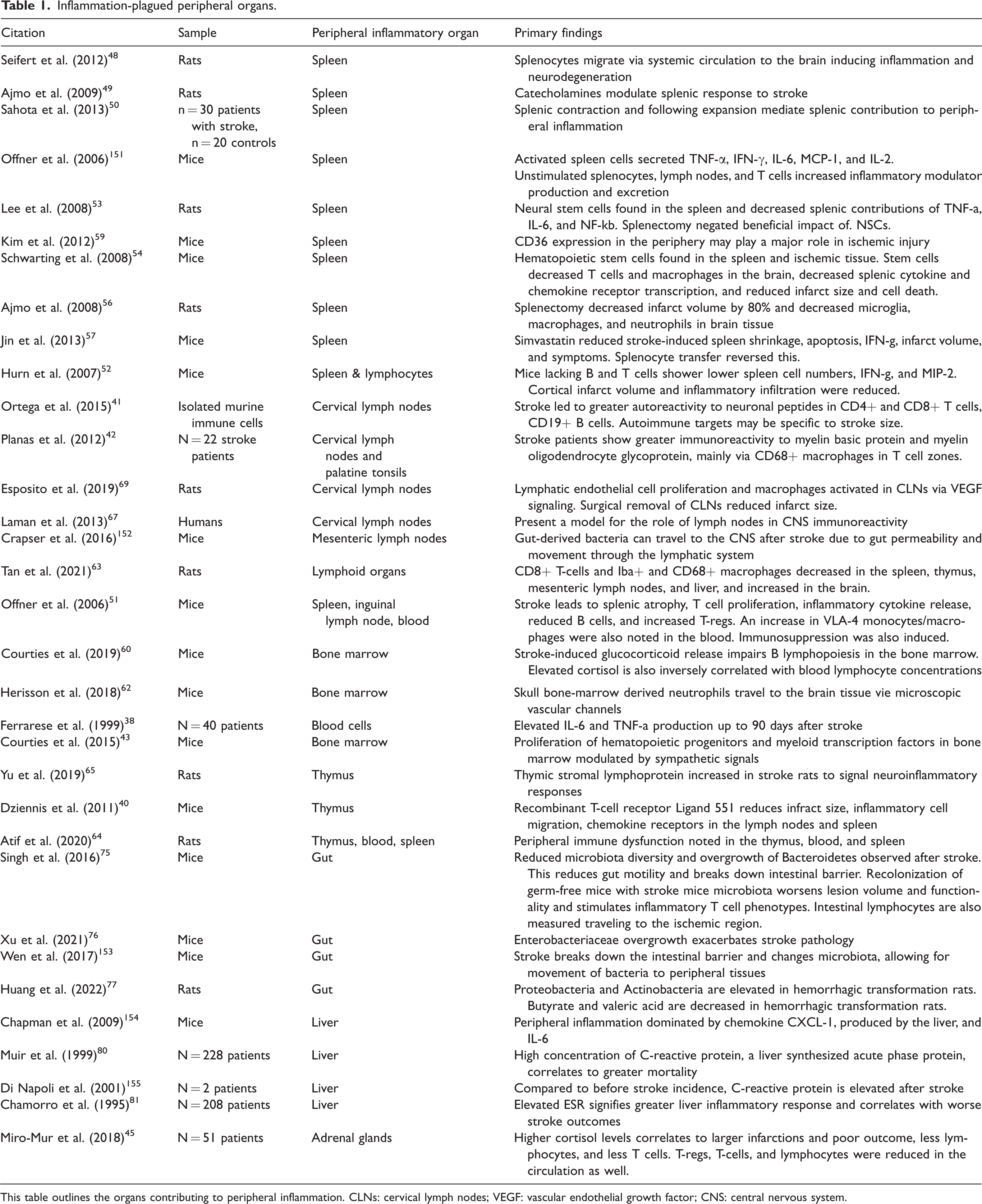
This table outlines the organs contributing to peripheral inflammation. CLNs: cervical lymph nodes; VEGF: vascular endothelial growth factor; CNS: central nervous system.
Spleen
The spleen is a vital organ to consider in peripheral inflammation given its storage function for platelets, macrophages, and immune cells.1,2,47 In a rat model of middle cerebral artery occlusion (MCAO), the spleen is significantly reduced in size compared to controls. This is due to splenic release of lymphocytes, monocytes, NK cells, and neutrophils. 48 Also, after stroke, the spleen size is reduced due to sympathetic nervous system signaling from the CNS inducing splenic contraction. MCAO in rats increases catecholamine levels but blocking alpha or beta receptors mitigates spleen shrinkage. 49 This phenomenon is also observed clinically. In 30 patients with suspected stroke, spleen size was measured daily and notably decreased. 50 The measurement of splenic chemokines also demonstrates the role of the spleen in stroke pathology. In mouse models of MCAO, elevated levels of TNF-α, IFN-γ, IL-6, monocyte chemoattractant protein-1, and IL-2 are released from spleen cells. Splenocytes also showed increased macrophage inflammatory protein (MIP)-2, C-C Motif Chemokine Receptor (CCR)-2, CCR7, CCR8, MIP-2, interferon gamma-induced protein-10, CCR1 and CCR2, all inflammatory cytokines and chemokines. 51 Furthermore, mRNA of IL-1β, TNF-α, IFN-γ, and IL6 are increased within the spleen after stroke.52–55 The direct observation of splenic changes post-ischemic injury exemplifies the spleen’s significant role in stroke pathology. The spleen’s role is further elucidated by changes in peripheral inflammatory responses when the spleen is compromised.
Interestingly, removal of the spleen has a significant role on ischemic damage, thus, this organ is a notable target for stroke therapies. In rats with MCAO, splenectomies result in >80% reduced infarction volume as well as decreased microglia, macrophages, and neutrophil infiltration. 56 After splenectomy, adding splenocytes reverses this therapeutic effect. 57 Radiation induced reduction of spleen functionality also shows reduced infarct volumes and reduced microglia, T-cell infiltration, and apoptotic neurons. 58 These results strengthen the evidence supporting the spleen’s major role in peripheral inflammation following stroke. While splenectomies may be extreme for clinical treatment of stroke, a more directed subset of splenocytes may be optimal targets for therapies. For instance, increased CD36 expression in mouse models of stroke is observed. 59 CD36 plays a major role in the migration of monocytes and macrophages to the ischemic region. Thus, directly targeting CD36 splenocytes may be a more conservative option for mitigating spleen-induced peripheral inflammation. More discussion on reducing splenic inflammatory effects will be discussed in the therapeutics section.
Primary lymphoid organs
In addition to the spleen, other immune organs likely contribute to peripheral inflammatory responses. Primary lymphoid organs are responsible for the initial maturation of inflammatory mediators. Within the bone marrow, B-cells are matured, and myeloid cells are synthesized. Similarly, the thymus allows for T-cell maturation. Thus, it is understandable that these organs contribute significantly to peripheral inflammation after stroke. Blood cell activation is involved in peripheral cytokine amplification, as lipopolysaccharide (LPS) applied to cell cultures of stroke patients exhibits enhanced IL-6 and TNF-α production (compared to non-stroke controls). 38 After MCAO in mice, tyrosine hydroxylase and norepinephrine levels are elevated in the bone marrow within 1 day, leading to primarily myeloid hematopoietic stem cell proliferation. 43 Bone marrow B lymphopoiesis may be related to glucocorticoid release following HPA axis activation in stroke, as discussed below in the section on adrenal glands. Deleting the glucocorticoid receptor reduces B-cell proliferation after stroke in mice models. Additionally, elevated cortisol correlates to amplified lymphocytes in the blood. 60 In addition to lymphocytes, bone marrow derived mononuclear cells wildly shape the inflammatory response to stroke depending on various differentiation possibilities. 61 The mechanism linking immune invasion and bone marrow may involve microscopic vascular channels within the bone marrow, as shown by murine models of stroke and myeloid cells invasion pathways. 62 The role of bone marrow in stroke pathogenesis is further exemplified by bone marrow mononuclear stem cell therapeutics, which will be discussed later.
Within the thymus, changes are also notable following stroke. Following an inflammatory eruption after stroke in mice, splenic and thymic atrophy are observed. 40 In rat models of ischemic stroke, CD8+ T cells and macrophages were decreased in a plethora of peripheral organs, including the spleen, thymus, mesenteric lymph node, and liver, but increased in the brain. These findings suggest major peripheral contributions of immune cells to ischemic brain regions. 63 Additionally, flow cytometry demonstrates a thymic role in balancing inflammatory cell populations. 64 Thymic stromal lymphopoietin (TSLP), an epithelial cell-derived cytokine found in the thymus, induces a Th2 response. After MCAO in rats, TSLPR is increased in neurons and glia, thus increasing Th2 activation as well. 65 The thymus and bone marrow are the primary sites of lymphocyte maturation; thus, these organs play a substantial role in any reaction involving lymphocyte proliferation. By studying these organs’ contributions to stroke pathology, early intervention of peripheral inflammation may be achievable. The increase in myeloid cells and enhanced Th2 response are likely significant contributors to the peripheral inflammation seen after stroke and notable therapeutic targets.
Cervical lymph nodes
Another significant site of T and B cell priming includes the lymph nodes. Prior to the discovery of a perivenular lymphatic system, termed the “glymphatic system”, the brain was not believed to be connected to the lymph nodes. 66 Now, however, a substantial lymphatic network is known to connect CNS antigens to adaptive immune system responses instigated within lymph nodes. 26 Two predominant lymphatic pathways exist from the CSF to the nasal mucosa and deep cervical lymph nodes, or, alternatively, from the brain parenchyma via capillaries and arteries to cervical lymph nodes (CLNs).67,68 Following central inflammation in stroke and resultant BBB damage, cerebral antigens are found activating B and T-cells in CLNs.41,42 After focal cerebral ischemia in rats, lymphatic endothelial cells proliferation and macrophage activation is significant within CLNs. CLN involvement is believed to be regulated by vascular endothelial growth factor (VEGF) signals, as blocking VEGF receptor 3 (VEGFR3) can reduce this activation and mitigate infarct damage. Cell cultures of macrophages also demonstrate activation after co-culturing with VEGF-stimulated lymphatic endothelial cells. The removal of CLNs, like blocking VEGF signaling pathways, also reduces stroke damage in mice. 69 In addition to the CLNs, other local lymphatic organs contain brain-derived antigens after stroke. MHC-II presenting cells within T-cell areas can be localized in the palatine tonsils after stroke. 42 As mentioned previously, T-cell and macrophage shifts have been noted within mesenteric lymph nodes. 63 Moreover, increases in TNF-α, IL-6, IL-2, and IFN-γ secretions were noted in inguinal lymph nodes after mice underwent MCAO. 51 Even more widespread, there have been a few case reports suggesting transient ischemic attacks are complications of Kikuchi–Fujimoto disease (KFD), or histiocytic necrotizing lymphadenitis, characterized by local lymphadenopathy.70,71 Given these findings and the more recent understanding of the brain’s lymphatic connections, novel treatments may be explored to depress the CLNs apparently detrimental impacts on stroke damage.
Gastrointestinal system
While seemingly less intertwined with the afore-mentioned immune organs, the gut contains more than 70% of the body’s immune system and the largest macrophage population in the body. 44 Immune responses are relayed via immune components of the gut, Peyer’s patches, lamina propria immune cells, intraepithelial lymphocytes, and mesenteric lymph nodes. 15 The gut microbiota can influence these immune responses and ultimately drive gut-mediated peripheral inflammation. Recently, a significant interaction between the CNS and GI system has been discovered, termed the gut-brain axis. 72 The gut receives parasympathetic and sympathetic signals from the CNS to control gut motility, secretions, permeability, microbiota, and immune cell activity. 73 In turn, the gut can influence the brain via vagal signals, endotoxins from microbiota, and metabolites released from microbiota, including neurotransmitters, short chain fatty acids, indoles, and bile acids. 73 After stroke, however, these signals are disrupted due to aberrant sympathetic nervous system activation, ultimately impacting the gut microbiome. 74 After stroke, changes in the concentration of Firmicutes, Bacteroidetes and Actinobacteria, prevalent bacteria within the gut are seen, as well as decreased microbiota diversity. 75 Additionally, post-stroke increases in Enterobacteriaceae have significant detrimental effects on clinical outcomes in mice. 76 Germ-free mice recolonized with dysbiotic, post-stroke microbiota showed greater lesion volume and poorer outcomes compared to mice given a normal microbiota. After dysbiotic recolonization, inflammatory T-cell populations were also elevated within the gut and brain. 75 Gut dysbiosis after stroke also promotes hemorrhagic transformation, a severe complication of ischemic stroke. 77 Through microbiome transformation, it is apparent that there is a dual communication network between the gut and the brain significantly impacting stroke outcomes. With antibiotic treatments or fecal transplants, these peripheral impacts may be substantially controlled. Other components of gut functionality can impact stroke outcomes as well.
Alongside microbiota alterations, disrupted endothelial cell integrity, mucus secretions, and inflammatory cell concentrations are noted after stroke. 73 These changes in gut homeostasis contribute to gut dysmotility, dysbiosis, leaky gut, gut hemorrhage, or sepsis. These complications are also correlated with poor stroke outcomes, proportional to stroke severity.44,75 Despite being seemingly separated from the CNS, the gut plays a major role in stroke pathology. In fact, intestinal immune cells have been recorded traveling throughout the meninges to the ischemic region itself. 78 Whether it is via microbiota composition or general functionality, the gut is a vital target for peripheral inflammation therapeutics.
Also, within the digestive system, the liver is impacted after stroke. As noted, the complement system is activated and perpetuates inflammation in the ischemic region. Complement proteins are synthesized within the liver. After experimental stroke, neutrophil-selective CXC chemokine, keratinocyte chemoattractant, is elevated. This chemokine is produced by the liver. 10 Additionally, IL-6 and IL-1, two cytokines elevated after stroke, induce liver production of acute phase proteins, modulators of inflammation. 79 Concentrations of C-reactive protein, an acute phase protein, are elevated in the plasma after acute stroke and can convey survival outcomes. 80 Erythrocyte sedimentation rate is also prolonged after stroke, signifying increased concentrations of acute phase proteins, predicting poor short-term outcomes, and suggesting impending stroke recurrence.79,81 Understanding the liver’s role in stroke induced inflammation is incredibly important given this organ’s large role in inflammatory protein production. Targeted treatments blocking this additional inflammatory protein production could significantly reduce peripheral inflammatory cascades.
Adrenal glands
The adrenal glands are heavily connected to the CNS and can also alter stroke pathology due to the hypothalamic-pituitary-adrenal (HPA) axis. 12 After stroke, levels of IL-1 are elevated due to release from local inflammatory mediators. 15 IL-1 triggers corticotropin releasing hormone from the hypothalamus. This induces the HPA axis, ultimately increasing cortisol production and release from the adrenal glands. Within 1 day after stroke, cortisol increases rapidly, and higher levels correlate to poor clinical outcomes and exacerbated infarct volumes. Additionally, larger infarction sizes are related to lymphocytopenia lower concentrations of T-cells and T-regs. 45 Cortisol elevations after stroke are also associated with greater mortality and morbidity. 82 Adrenal glands are also stimulated to release catecholamines from the medullary region. 12 Working with adrenal steroids, catecholamines induce immune cell release from splenic reserves and allow these cells to be brought to the ischemic region of the CNS. 83 This level of signaling further ties the CNS to the spleen, while also involving the autonomic nervous system. Catecholamines can further impact many of the peripheral inflammatory organs, inducing a pro-inflammatory response. 46 Each of these organ systems are heavily intertwined, ultimately exacerbating stroke pathology and contributing to poor clinical outcomes due to peripheral inflammation.
B and T-cells
B and T cells may represent the convergence of many of the peripheral organs in mounting the systemic inflammation after stroke. While a wide array of immune cells are involved in stroke peripheral and neural inflammation, B and T cells are the common convergence amongst the above peripheral organs. For instance, one of the lymph nodes’ primary roles includes activating the adaptive immune system, such as lymphocytes. In fact, within 3 days, concentrations of CNS antigens proportional to infarct size are noted within CNLs, propagating T and B-cell maturation and action. 42 After ischemic stroke, B cells, regulatory anti-inflammatory B cells (B-regs), and CD8+ T-regs are increased, while CD4+ T-regs are transiently decreased. 84 Lymphocyte differentiation is incredibly important in determining stroke outcomes, as some lymphocytes, such as CD4 + Th1, Th-17, and CD8+ T-cells are pro-inflammatory, but regulatory lymphocytes and CD4 + Th2 T-cells are anti-inflammatory. 30 The Th1 CD4+ phenotype may be the most inflammatory, as patients with predominantly Th1 responses to stroke show poorer clinical outcomes.85,86 DAMPs released from dying neurons are prominent inducers of T-cell immune responses. Notably, high-mobility group box 1 (HMBG1) and heat shock protein (Hsp) 70, 32, and 27 bind Toll-Like Receptors (TLR) on macrophages and induce the NF-kB signaling pathway, a major pro-inflammatory pathway.87–91 Hsp70 induces CD4+ T-cells to shift into their pro-inflammatory, Th1 phenotypes. 92 DAMPs activate receptors for advanced glycation end products (RAGE) to shift T-cell proliferation from T-reg, which are anti-inflammatory, to Th-17, which are pro-inflammatory. 93 Within 24 hours, CD4+ T-cells proliferate within the brain 94 and CD8+ T-cells are seen as soon as 3 hours after ischemic insult.95,96 The role of T-cells in stroke is further fortified by the presence of IL-21, an interleukin released by CD4+ T-cells, in cadaver stroke tissue. 97 Furthermore, antagonism of IL-21 in mice reduces stroke damage. 97 Interestingly, BBB damage and subsequent neuronal antigen release, such as myelin basic protein, NMDA receptors, and microtubule-associated protein, can also trigger inflammatory T-cell responses in the form of autoimmune reactions. 68 On the other hand, in mice with severe combined immunodeficiency (low B and T-cell levels), infarct volumes are significantly reduced, suggesting an overall pro-inflammatory contribution of T or B-cells after stroke. 52 Removing CD4+ T-cells directly results in improved neurogenesis and functional recovery, while removing T-reg cells establishes the opposite result. 98 This plethora of studies strongly supports the role of T-cells in inflammation and offers many potential targets for anti-inflammatory treatments. In addition to T-cells, B-cells also play a vital role in adaptive immunity and peripheral inflammatory responses.
In contrast to T-cell responses, regulatory B-cells modulate an anti-inflammatory response.99–101 In B-cell-deficient mice, stroke damage functional deficits, inflammatory cell proliferation, and mortality risk are increased. 100 Adoptive transfer of B-cells demonstrates reduced infarct volume after transient MCAO in mice. B-cells migrate to the CNS after stroke, ultimately improving motor and cognitive functionality and neurogenesis. Blocking B-cell proliferation with Rituximab, a CD20 monoclonal antibody, inhibits these beneficial effects. 99 Non-regulatory B-cells, however, may enhance infarct formation after interaction with the thymus independent type 2 antigens and subsequent IgA production. 102 IgA plasma cell infiltration after stroke impairs cognition in mice and patients with vascular dementia and stroke. 103 Despite these varying results insinuating B-cells’ role in peripheral inflammation, depletion of B cells using anti-CD20 antibody do not impact the size of infarcts nor functionality in mice. 104 Thus, the overall role of B cells in stroke recovery or pathogenesis is questionable. Ultimately, shifting the adaptive immune response towards anti-inflammatory, regulatory B and T-cells is an ideal therapy, while mitigating long-term activation of pro-inflammatory lymphocyte differentiation.
Aside from the standard B cells, CD4+, CD8+, and Treg cells, recent studies have elucidated rarer subsets of T and B cells involved in ischemic stroke (Table 2). One example includes CD8+CD122 T cells. Systemic CD8+CD122+ T cells require no antigen presentation and release IL-10 or directly regulate inflammation by cell contact. 105 In fact, they may be even better at this immunosuppression than the typically studied Tregs. 106 In mouse models of ischemic stroke, these cells are predominant directly after the ischemic insult and release epidermal growth factor–like transforming growth factor (ETGF) and IL-10, suggesting an immunomodulatory role in stroke. Importantly, these cells migrate using CXCR3. 107 Interestingly, when mouse models of ischemic stroke are treated with IL-10 producing B cells prior to injury, CD8+CD122+ Tregs are noted in the spleen and ischemic region. In addition to the CXCR3 signaling, there may be B and T-cell crosstalk optimizing anti-inflammatory lymphocyte proliferation. 108 Thus, further research on CD8+CD122+ differentiation and CXCR3 migration may unveil promising cell based therapies for stroke. On the contrary, another T cell subset, CD3+CD4−CD8− T cells (or double negative T cells (DNTs)), seen in preclinical and clinical settings may promote neuroinflammation following stroke. Via the Fas ligand/PTPN2/TNF-a pathway, the migration of these cells to the ischemic region results in amplified neuroinflammation in mice. Proinflammatory microglia are also more prevalent, suggesting these DNTs prompt further inflammatory immune cell migration to the CNS. Blocking the Fas ligand/PTPN2/TNF-a reduces inflammatory damage and cell death. 109 Blocking the activity of these DNTs in stroke patients using TNFa inhibitors, such as adalimumab or infliximab, may be beneficial as future stroke therapies. Among the rarer B cell subtypes observed after stroke, CD11bhigh B cells may worsen neuroinflammation after stroke. This B cell population increases in prevalence with age, as does the risk of stroke. In mice models and ex vivo, CD11bhigh B cells enhanced microglial phagocytosis and TNF-a production. While phagocytosis may be beneficial in clearing necrotic tissue, TNF-a is a notorious pro-inflammatory cytokine. 110 Thus, further research on CD11bhigh B cells is needed prior to therapeutic development for stroke, especially given the overlap between CD11bhigh B cells, age, and stroke prognoses. As demonstrated, there are several therapies focused on the regulation of T and B-cell activity, and, given these cells apparent role in stroke pathology, these therapies should be tested in patients with stroke.
Rarer subsets of lymphocytes alter stroke prognosis.
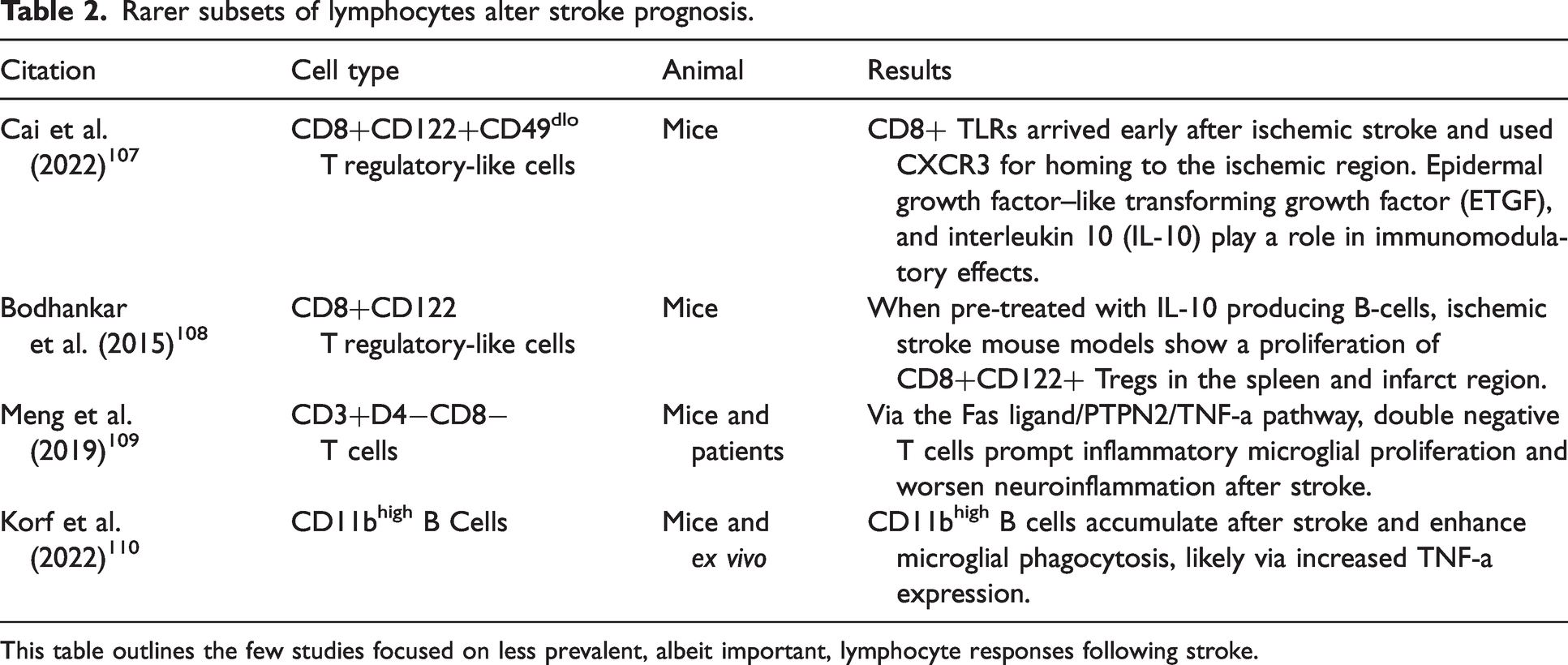
This table outlines the few studies focused on less prevalent, albeit important, lymphocyte responses following stroke.
Immunodepression
The peripheral inflammation following stroke is incredibly detrimental, but even further complications occur following the exacerbated inflammatory response. In an effort to dampen the detrimental impacts of excessive inflammation, stroke patients show severe immunodepression. 15 Reduced white blood cell counts (leukocytopenia) are notable in stroke patients. 111 Furthermore, T-cell proliferation and related cytokines such as TNF, IL-2, IL-12, IFN-γ are decreased after stroke.112,113 This exposes patients to high risk of infection. In 30% of patients, urinary tract infections and pneumonia are diagnosed after stroke. 12 Pneumonia is the most common consequence of subsequent immunodepression and is a life-threatening complication. 114 Post-stroke infection severity is related to stroke severity due to paralleled leukocytopenia. 112 In a study on murine models of stroke, more severe strokes required 1000-fold less bacteria to instigate pneumonia. 115 This consequent immunodepression after extreme peripheral inflammation is vital to consider when developing stroke therapeutics targeting peripheral inflammation. Given the high risk of infection in immunodepressed patients after stroke, a careful balance must be obtained in mitigating inflammation, while not putting patients at a more severe risk of post-stroke infection.
Potent inflammation-directed therapeutics
Cognizant that inflammation is a major cell death pathway in stroke, anti-inflammation-based therapeutics targeted to the afore-mentioned organs and the B and T-cell activation stand as potent strategies for reducing stroke damage. 15 Thus far, several organs have been described as potential targets for mitigating peripheral inflammation in stroke. A plethora of potential targets exists for pharmaceutical and cell-based therapies to improve clinical outcomes for stroke patients. To maintain focus of this paper on stem cell therapy, the topic of pharmaceuticals and immunomodulation or immune cell therapies is provided in the Supplemental Material, while stem cell therapy is presented here.
Stem cell therapy
In addition to pharmaceutical approaches, several clinical and preclinical trials have investigated the use of cell therapy in stroke treatment,5,116 however, the use of stem cells to directly target peripheral inflammation is less abundantly reported in the literature. Stem cells are hypothesized to act via a by-stander effect to induce an anti-inflammatory influence. 117 These cells can induce neurogenesis, angiogenesis, oligodendrogenesis, vasculogenesis, and synaptogenesis by secreting anti-inflammatory cytokines and signals promoting repair. 118 Stem cell treatments in murine models of MCAO show reduced levels of pro-inflammatory cytokines TNF-a, IL-1b, and IFN-g gene transcription and increased transcription of the anti-inflammatory cytokine, IL-10.119,120 A vital role of stem cells in modulating stroke damage involves their role in restoring NVU impermeability. For instance, mesenchymal stem cells (MSCs) can secrete several growth factors to enhance angiogenesis, gliogenesis, and neurogenesis, reduce neuroinflammation, and differentiate into endothelial cells, glial cells, and neurons, all of which are vital to the NVU. Similarly, endothelial progenitor cells are demonstrated to promote angiogenesis, aiding in neural tissue recovery and NVU reestablishment after stroke (Reviewed in Moon et al. 121 ). Importantly, the effects of these stem cells may be more related to their signaling molecules, rather than simply replacing damaged cells. 122 Thus, further research is warranted on the use of stem cells to restore BBB stability and the NVU. Serum-derived exosomes can also lessen endothelial cell apoptosis and autophagic BBB destruction, further protecting the CNS from peripheral inflammation. 123 This shift in immune and inflammatory transcription profiles may play a major role in modulating peripheral inflammation. Additionally, it is theorized that MSCs work predominately in the periphery, while NSCs stimulate CNS immunomodulation. 124 Thus, further analysis of stem cell therapies in peripheral organs and B and T-cell activity may open many therapeutic opportunities (Table 3).
Stem cell sequestration of peripheral inflammation.
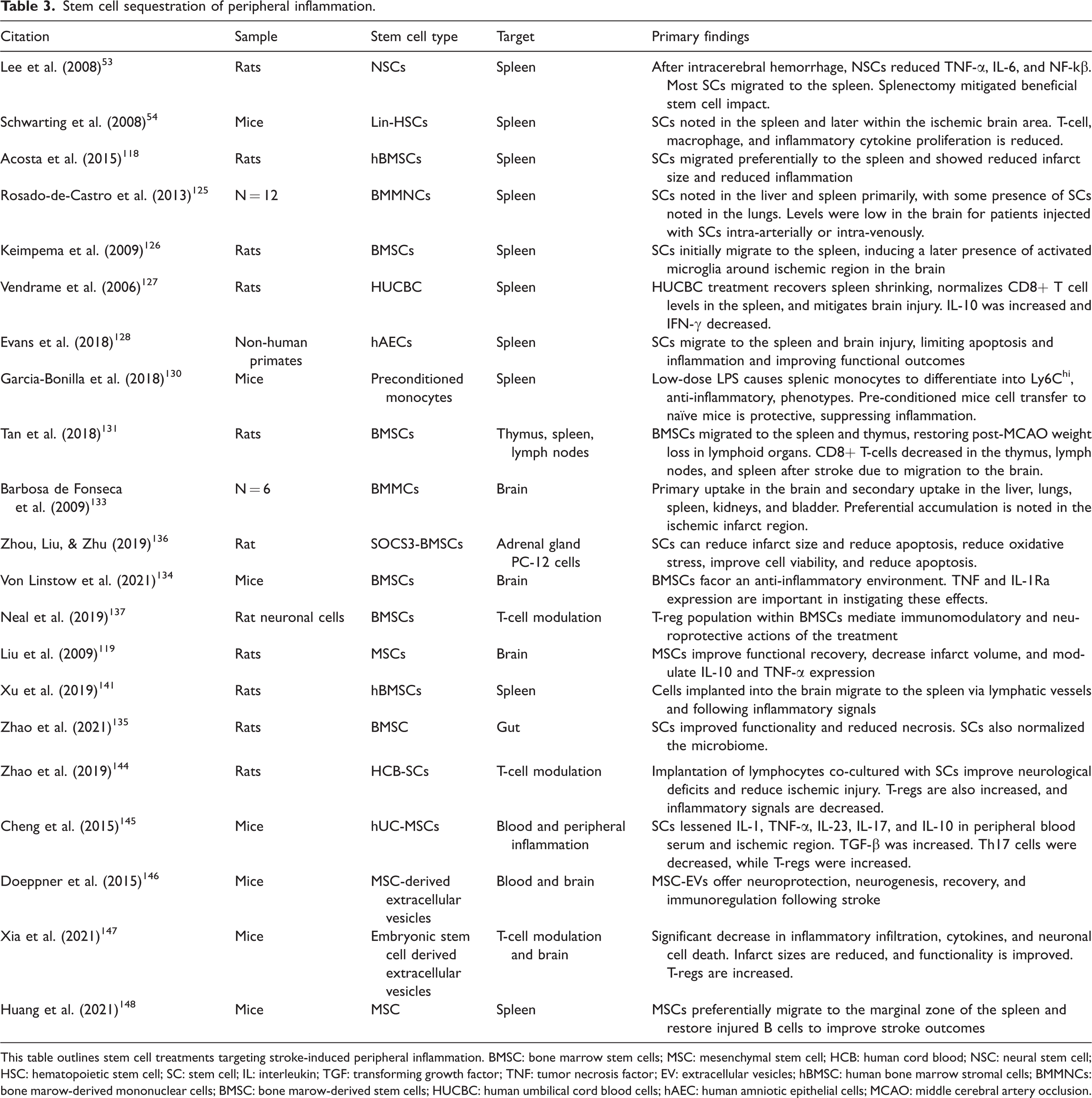
This table outlines stem cell treatments targeting stroke-induced peripheral inflammation. BMSC: bone marrow stem cells; MSC: mesenchymal stem cell; HCB: human cord blood; NSC: neural stem cell; HSC: hematopoietic stem cell; SC: stem cell; IL: interleukin; TGF: transforming growth factor; TNF: tumor necrosis factor; EV: extracellular vesicles; hBMSC: human bone marrow stromal cells; BMMNCs: bone marow-derived mononuclear cells; BMSC: bone marow-derived stem cells; HUCBC: human umbilical cord blood cells; hAEC: human amniotic epithelial cells; MCAO: middle cerebral artery occlusion.
Spleen
Considering the studies demonstrating the therapeutic effect of splenectomy on stroke outcomes, it is likely that replacement of splenocytes with stem cells will have equally beneficial results, without associated asplenic complications. Intravenous neural stem cell administration shows neuroprotection and reduced inflammatory infiltrates, primarily through their migration to the spleen. 53 In ischemic stroke models, lineage-negative bone marrow-derived hematopoietic stem cells and precursor cells reduced injury in mice reduces neuronal death, infarct size, and immune cell invasion. Splenic contribution to inflammation was also seemingly reduced, given the lower concentrations of cytokines and chemokine receptor transcription in splenic cells. 54 It is likely that stem cells play a large role in spleen-mediated peripheral inflammation, as intravenous bone marrow stem cells (BMSCs) are noted to preferentially migrate to the spleen compared to the ischemic brain tissue.118,125 Even with this surprising migratory partiality, significant therapeutic effects are eminent after stem cell administration. BMSCs may exert their effects through microglial activation. 126 Rats who undergo MCAO with following human umbilical cord blood cells (hUC-MSCs) show restored splenic weight, normalized CD8+ T-cell counts, and improved ischemic damage. 127 Human amnion epithelial cells, or placental stem cells, also migrate preferentially to the spleen and brain, where they reduce apoptosis, inflammation, ischemic damage, and immunosuppression, and improve functionality. 128 MultiStem, composed of multipotent adherent bone marrow cells, is currently being studied in a clinical trial for stroke patients. In addition to measuring clinical outcomes, spleen size will also be measured to examine the impact of MultiStem on spleen-induced peripheral inflammation. 129 To further strengthen these cell-based therapies, pre-conditioning may be used. Ex vivo lipopolysaccharide exposure can induce monocytes to adopt a Ly6Chi phenotype, a notable neuroprotective differentiation. When these monocytes are injected into mice with MCAO, stroke damage is reduced. Using cell-tracking, these ideally differentiated monocytes are found traveling form the spleen to the brain and meninges. 130 Ultimately, the spleen seems to be a major target for peripheral inflammation in stroke, and stem cells recognize this pattern as demonstrated through preferential migration. Promising results in preclinical studies on splenic peripheral inflammation suggests clinical advancements and treatments are, hopefully, in the near future.
Primary lymphoid organs
In addition to the spleen, stem cells commonly migrate to the thymus. Furthermore, the bone marrow offers a plethora of stem cell generative capabilities. To mitigate inflammatory B and T-cell proliferation, stem cell therapies targeting primary lymphoid organs’ peripheral inflammatory effects are a beneficial target of stroke therapies. Stem cells directly targeting the thymus is understudied, however, some studies exemplify therapeutic effects of stem cells on the thymus following stroke. BMSCs lessen neurological deficits and weight loss in rat models of stroke. Weight loss occurred primarily in the spleen and thymus, and BMSCs were able to reverse changes in both organs. BMSCs also ameliorated lymphoid organ inflammatory responses. 131 While not in stroke, autologous hematopoietic stem cell transplantation (HSCT) in patients with multiple sclerosis doubled CD4+ T-cells and reduced memory T-cells. Ultimately, these findings establish the role of stem cells in regenerating the thymic T-cell profile, while also reducing inflammation. 132 Similar approaches may be beneficial in modulating thymic inflammatory responses to stroke.
Bone marrow contributions to peripheral inflammation can be mitigated by bone marrow derived stem cells.28,54,116,118,119,125,126,129,133,134 Bone marrow-mononuclear cells (BMMCs) travel to the liver, lungs, spleen, kidneys, and bladder in patients with stroke and can mitigate stroke damage and neuronal death by modulating peripheral inflammation.118,125,133 In addition to these migratory paths, BMSCs can also interact with other peripheral inflammation organ systems, such as the gut microbiome and adrenal glands in murine models.135,136 BMSCs with elevated IL-1 receptor antagonist expression notably reduce TNF levels and impact inflammation after stroke in mice. 134 Furthermore, BMSCs interact with T-regs, as noted above, to decrease inflammatory reactions to stroke in rats. 137 Other variations of BMSCs, hematopoietic stem cells and MSCs, are also shown to modulate immune cell infiltration and cytokine signaling in the brain in murine models.54,119 Thus, while few studies have targeted bone marrow induced peripheral inflammation directly, the use of these stem cells clearly demonstrates therapeutic potential in targeting bone marrow responses to stroke.
Lymph nodes
Despite an equally vital role in lymphocyte proliferation, direct stem cell modulation of lymph node function is not reported in stroke; however, stem cells have been used for lymphedema. Like stroke, lymphedema is defined by chronic inflammatory mechanisms from cancer, trauma, and parasite infection. Thus, analysis of stem cell treatment in this illness may offer therapeutic benefit for stroke inflammation. Adipose-derived stem cells show varied results at reducing lymphedema in animals and humans. 138 Multipotent adult progenitor cells also restore lymphatic system functionality in lymphedema, offering another possible approach. 139 A novel approach to changing lymph node functionality involves the generation and implantation of synthetic lympho-organoids using lymph node stromal progenitors and scaffolds. These organoids can recover lymphatic drainage and perfusion, while also responding to antigens. 140 Thus, this therapy may be useful in reestablishing a normalized immune response following stroke. While initiatives should be made to establish stem cell treatment within lymph nodes, this may be more difficult due to the plethora of lymph nodes impacted by stroke. In fact, lymph nodes may be indirectly modulated by stem cell treatment via the stem cells’ pathways to their final destinations. Human bone marrow MSCs implanted into the brain migrate to the spleen in rat models of stroke. Importantly, the pathway of these stem cells is through lymphatic vessels. 141 Further studies involving the lymphatic systems role in stroke are needed to determine the optimal role of stem cells in lymph nodes to reduce peripheral stroke inflammation.
GI system
Few stem cell studies have examined the use of stem cells in modulating gut dysbiosis following stroke. In rats, treatment of MCAO with BMMCs demonstrates improved motor, learning, and memory abilities. Additionally, BMMCs reduced neuronal damage and death and modulated dysbiosis. 135 Other neuroinflammatory diseases, such as Parkinson’s disease treated with stem cells exemplify preferential stem cell migration to the gut and mitigated neural and intestinal inflammation. 142 Aside from the intestines, few clinical trials show BMMC migration to the liver following stroke.125,133 This stem cell migration may minimize the pathological increase in acute phase proteins, ultimately lessening liver-induced inflammation. The few studies reporting stem cell use to mitigate gut and liver contributions to peripheral inflammation establish a basis for future work in this realm. Additionally, the reported studies on stem cells lessening peripheral inflammation in the gut in other neuroinflammatory illnesses suggests gut-targeted stem cells may be incredibly beneficial for ischemic stroke.
Adrenal glands
A major contributor to described immunoreactivity following stroke involves activation of the sympathetic nervous system, 46 however, the use of stem cells to ameliorate the adrenal glands contributions to peripheral inflammation after stroke is understudied. Only one study was found implementing suppressor of cytokine signaling 3 (SOCS3)-modified BMSCs in adrenal glands for ischemic stroke damage. SOCS3 regulates inflammatory signaling from the adrenal glands, and overexpression has been showed to reduce infarct size in rats. 143 SOCS3-BMSCs reduce oxidative stress in adrenal cells, reduce infarct size, provide neuroprotection by reducing apoptosis, and strengthen adrenal cell viability. 136 Further support for stem cell studies targeting adrenal glands comes from studies demonstrating the therapeutic effect of blocking carvedilol, a pan adrenergic receptor blocker. By blocking adrenal gland catecholamines, spleen size was maintained, and stroke severity was decreased. 49 Stem cells may be able to similarly modulate this therapeutic response with less severe side effects associated with blocking adrenergic signaling. Thus, future research should aim to modulate adrenal contributions to corticotropic and sympathetic nervous system signaling after stroke using stem cells.
B and T cells
Similar to the pharmacological approaches in sequestering peripheral inflammation, ultimately, each described organ system plays a role in lymphocyte infiltration and phenotype following stroke. Thus, targeting these cells directly may offer the most downstream and widespread therapeutic value. In cell cultures, BMSCs induce T-reg differentiation in rat neuronal cells after oxygen glucose deprivation. Co-culturing stroke models of neurons with BMSCs and T-regs resulted in significant neuroprotection. Thus, BMSC treatment for stroke exhibits its effects through modulating the T-reg population. 137 These results transfer to pre-clinical studies as well. Treatment of rat models stroke with lymphocytes co-cultured with human cord blood-derived multipotent stem cells (HCB-SCs) improves functionality, ischemic damage, and increases T-reg proliferation. Inflammatory factors, such as the NLRP3 inflammasome, caspase-1, IL-1b, and NF-kB are also decreased following stem cell treatment. 144 The use of co-culturing to induce T-reg proliferation is a unique use of stem cells to modulate inflammatory lymphocyte profiles following stroke. Another stem cell option are hUC-MSC. These cells change T-cell differentiation after stroke, decreasing Th-17 cells and increasing T-regs within the periphery. 145 Extracellular vesicles derived from stem cells can also modulate this lymphocyte profile by balancing peripheral concentrations of B-cells, NK cells, and T-cells. Ultimately, these cells induce T-reg differentiation and improve motor and coordination function, angiogenesis, neurogenesis, inflammatory infiltration, neuronal death, and infarct volume.146,147 Regarding B-cells, MSCs are found to migrate to the marginal zone of the spleen, a splenic zone predominantly made of B-cells. After MSC treatment, stroke damage is reduced, and B-cell normalization is observed. 148 The major role of lymphocytes in stroke pathology suggests these cells are large proponents for targeted cell-based therapies. Lymphocytes are also a novel target given their widespread locations and clear maturation sites, such as the aforementioned immune organs.
Stem cells have demonstrated a profound ability to mitigate ischemic stroke damage centrally, however, further research should employ stem cells to mitigate peripheral inflammation. Furthermore, there are some concerns surrounding stem cell safety, as embryonic, fetal, or iPS-derived stem cells have some tumorigenic potential. Adult tissue-derived stem cells, such as BMSCs, however, have very little tumorigenic risk. 149 Confirming safety, optimal injection routes, and ideal concentrations for therapeutic benefit are needed prior to larger clinical trials employing stem cell therapies. 5
Conclusion
Stroke-induced peripheral inflammation propels detrimental effects that exacerbate central inflammation and worsen the clinical outcomes. The currently available stroke treatments, including tPA and MT, are imperfect with narrow therapeutic window and risk for hemorrhagic transformation, severely limiting the number of applicable patients. 150 By targeting systemic inflammation via peripheral organ-acting drugs and stem cells may expand these therapeutic windows, 142 allowing increased enrolment of ischemic stroke patients. In-depth investigations into the peripheral inflammation as key mediator of stroke pathology may offer innovative insights into new therapies designed to mitigate such inflammatory contributors. To this end, recognizing that the inflamed stroke brain receives significant input from inflammation-plagued peripheral organs warrants serious consideration of established as well as emerging brain-peripheral organ axes to fully sequester stroke inflammation-associated secondary cell death.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X221149509 - Supplemental material for The central role of peripheral inflammation in ischemic stroke
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X221149509 for The central role of peripheral inflammation in ischemic stroke by Molly Monsour, Cesar V Borlongan in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: C.V.B. was funded by the National Institutes of Health (NIH) R01NS090962, NIH R01NS102395, and NIH R21NS109575. Additionally, C.V.B. was funded and received royalties and stock options from Astellas, Asterias, Sanbio, Athersys, KMPHC, and International Stem Cell Corporation.
Acknowledgements
We acknowledge the artists Brgfx, macrovector, storyset, svstudioart, and pikisuperstar in designing vectors used for the final figure in this paper (downloaded from freepik.com).
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: C.V.B. has received consultant compensation from Chiesi Farmaceutici. C.V.B. also declares patents and patent applications related to stem cell therapy.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.