
Abstract
Cerebral ischemia triggers inflammatory changes, and early complications and unfavorable outcomes of endovascular thrombectomy for brain occlusion promote the recruitment of various cell types to the ischemic area. Although anti-inflammatory M2-type macrophages are thought to exert protective effects against cerebral ischemia, little has been clarified regarding the significance of post-ischemic phase-dependent modulation of M2-type macrophages. To test our hypothesis that post-ischemic phase-dependent modulation of macrophages represents a potential therapy against ischemic brain damage, the effects on rats of an M2-type macrophage-specific activator, Gc-protein macrophage-activating factor (GcMAF), were compared with vehicle-treated control rats in the acute (day 0–6) or subacute (day 7–13) phase after ischemia induction. Acute-phase GcMAF treatment augmented both anti-inflammatory CD163+ M2-type- and pro-inflammatory CD16+ M1-type macrophages, resulting in no beneficial effects. Conversely, subacute-phase GcMAF injection increased only CD163+ M2-type macrophages accompanied by elevated mRNA levels of arginase-1 and interleukin-4. M2-type macrophages co-localized with CD36+ phagocytic cells led to clearance of the infarct area, which were abrogated by clodronate-liposomes. Expression of survival-related molecules on day 28 at the infarct border was augmented by GcMAF. These data provide new and important insights into the significance of M2-type macrophage-specific activation as post-ischemic phase-dependent therapy.
Introduction
Cerebral ischemia triggers inflammatory changes and early complications. Ischemia-reperfusion injury has recently been reported as a major cause of early complications and unfavorable outcomes after endovascular thrombectomy therapy in patients with acute ischemic stroke. 1 As a result, modulation of microglia/macrophage polarization and subsequent inflammatory response may represent a potential adjunctive therapy to recanalization.
In the perivascular space, ischemia and/or reperfusion activate perivascular macrophages and mast cells. 2 Mast cell degranulation releases vasoactive mediators such as proteases and tumor necrosis factor (TNF), then activated macrophages release pro-inflammatory cytokines. These pro-inflammatory mediators contribute to the endothelial expression of adhesion molecules and to blood-brain barrier damage that promotes the infiltration of neutrophils, lymphocytes, and monocytes. 3 Microglia/macrophages or monocyte-derived macrophages have been implicated in stroke-induced inflammation and injury. 4
Macrophages are mainly classified into M1 and M2 phenotypes.5–7 M1-type macrophages, characterized by increased secretion of pro-inflammatory mediators such as TNF-α, interleukin (IL)-1β, and IL-6, are present in cerebral ischemia as well as in myocardial infarction. M1-type macrophages may exacerbate neuronal death through the release of harmful mediators and nitric oxide 8 and impair axon regrowth. On the other hand, M2-type macrophages (anti-inflammatory macrophages) are thought to be mediated by cytokine IL-4 and arginase-1 (Arg-1), improving long-term neurological outcomes after stroke.9–12 Neurons in the ischemic penumbra generate IL-4, which could subsequently convert surrounding microglia/macrophages to the M2 phenotype. 10 Arg-1 is also associated with the reduction of inflammatory mediators, thereby promoting the survival of cortical neurons under ischemic/hypoxic conditions.12–14 Arg-1 is an enzyme that hydrolyzes the amino acid L-arginine to form ornithine and urea. Deletion of Arg-1 worsens ischemic injury, suggesting a protective role of Arg-1.13,15 In addition, M2-type macrophages accelerate axon growth/sprouting in cultured neurons, suggesting a critical role in tissue recovery and re-innervation.15,16 Post-stroke imbalances between M1- and M2-type macrophages may thus be associated with either expansion of neuronal ischemic damage or protective effects. However, the roles of phenotype-specific macrophage activation in cerebral ischemia and the timeline in which these effects manifest are not well understood. In particular, the therapeutic role of M2-type macrophage-specific modulation in a time-dependent manner against post-ischemic brain damage has not been studied.
Group-specific component protein-derived Macrophage-Activating Factor (GcMAF) is a mammalian protein with incredible potency for directly activating macrophages. 17 Human group-specific component protein (Gc protein) is a multifunctional serum protein with three common allelic variants: Gc1F, Gc1S and Gc2. Gc1 contains an O-linked trisaccharide [sialic acid-galactose-N-acetylgalactosamine (GalNAc)] on the threonine420 (Thr420) residue and can be converted to a potent GcMAF by selective removal of sialic acid and galactose, leaving GalNAc at Thr420. In contrast, Gc2 is not glycosylated. During inflammatory responses, Gc protein is converted to GcMAF, inducing macrophage phagocytic activity as an immunostimulatory agent. 18 GcMAF also counteracts the neuronal damage induced by oxaliplatin. 19 In our preliminary study using mice subjected to traumatic brain injury, we confirmed that GcMAF delivered in the subacute phase attenuated brain damage through modulation of M2-type macrophages.
Given this background, we hypothesized that after cerebral infarction, modulation of M2-type macrophages in a phase-dependent manner under post-ischemia conditions may provide a potential therapy against ischemic brain damage under optimal circumstances. To test our hypothesis, we used GcMAF to examine the phenotypic expression profile of macrophages and changes in the secretion of cytokines over time, and the role of the activation of M2-type macrophages by GcMAF delivered in the acute and subacute phases of ischemia.
Material and methods
Study approval
This study was approved by the ethics committee of Tokushima University Graduate School of Biomedical Sciences in compliance with the animal care guidelines of Tokushima University, Tokushima, Japan. All animal experiments were conducted in accordance with the National Institutes of Health Guidelines for the Care and Use and the ARRIVE guidelines of Laboratory Animals. 20 Seven-week-old male Wistar rats obtained from Charles River Laboratories Japan, Inc. (Yokohama, Japan) were housed in a temperature- and humidity-controlled room (approximately 23°C and 50%, respectively) under a 12-h light cycle (08:00–20:00) and were allowed free access to food and water. Before all surgical procedures, the animals were anesthetized by 2–4% isoflurane inhalation. Body temperature was monitored and maintained at 37°C using a warming plate (NISSIN, Tokyo, Japan). Investigators involved in all surgical procedures, drug treatments, and end-point assessments were blinded to the group to which each animal had been assigned.
Focal cerebral ischemia induction
Rats were randomly allocated to a sham group or a group subjected to middle cerebral artery occlusion (MCAO), which was induced with a 4-0 monofilament suture, as described previously.21–24 Blood flow to the region surrounding the MCA was measured with a laser Doppler flow probe (Unique Medical, Osaka, Japan) to confirm MCA occlusion, with the probe placed on the temporal bone surface at a site 1 mm posterior to the bregma and 3 mm inferior to the temporal line.21–24 A reduction in regional cerebral blood flow (rCBF) to less than 30% of the baseline indicated successful MCAO. The suture was withdrawn after 120 min to allow reperfusion. Ipsilateral blood flow was restored to approximately 80–100% of the baseline value. The extent of rCBF decrease before and the rCBF increase after reperfusion were identical in all rats with successful MCAO. Rats consistently displayed a circling behavior, decreased resistance to lateral push, forelimb flexion, and shoulder adduction. Rats with incomplete MCAO (approximately 10%) were excluded from further experiments.
Measurement of infarct volume, assessment of phagocytosis and neurological deficit
Sliced brain tissues were immersed in a 2% 2,3,5-triphenyltetrazolium chloride (TTC) solution in phosphate-buffered saline (PBS). The extent of ischemic infarction was traced and the integrated volume was calculated using a BZ-X710 microscope equipped with an image analyzing system (Keyence, Osaka, Japan). To reduce artifacts from brain edema, we applied the indirect measurement method based on the contralateral brain volume.23–25 Infarct, cleaned, and non-infarct volumes were calculated as a percentage of the contralateral hemisphere. To determine the size of the cleaned area (%) we used the following formula: (contralateral hemisphere volume − ipsilateral hemisphere volume) × 100/contralateral hemisphere volume.
To assess the effects of GcMAF against brain damage in rats subjected to 2-h MCAO-reperfusion (MCAO-R), rats were randomly allocated into two groups receiving intra-peritoneal injection of GcMAF (40 ng/kg/day) or saline (vehicle control, VC) for 7 days on days 0–6 or days 7–14 and sacrificed on day 7 or day 14 (GcMAF, n = 7; VC, n = 7, each). To examine the phagocytic effects of macrophages after cerebral ischemia, rats subjected to 2-h MCAO-R were injected intraperitoneally with 10 ml/kg of clodronate-liposomes (CLOD) (Clophosome®-A; Tribioscience, Sunnyvale, CA, USA) according to the instructions from the manufacturer (0.2 ml/20–25 g) on day 7 and then received injections of GcMAF (40 ng/kg/day) or saline (CLOD, n = 7; CLOD plus GcMAF, n = 7) and underwent determination of mRNA levels in the peri-infarct area. Another set of rats in each group was assessed immunohistochemically (n = 4)
According to neurological scoring as described by Huang et al. 26 and Yang et al., 27 we recorded a score from 0 to 12 after successful MCAO-R. Neurological scores for the other MCAO-R rats were recorded after treatment during days 7–28 and rats were sacrificed on day 28 (GcMAF, n = 7; VC, n = 7) for immunohistochemical assessment. Neurological deficits were assessed by an examiner blinded to the treatment rats had received.
Preparation of GcMAF
GcMAF and biotin-labeled GcMAF were prepared by Uto et al. 18 at the Faculty of Bioscience and Bioindustry, Tokushima University and intraperitoneally injected every day at a dose of 40 ng/kg/day on days 0–6, days 7–14 or days 7–28. To examine the distribution of intraperitoneally (ip) injected GcMAF in the brain, rats were injected daily with biotin-labeled GcMAF (40 ng/kg/day) starting on the day following 2-h MCAO-R. The dose of GcMAF was determined based on the results of a previous study into the immunomodulatory effects of GcMAF. 28 In that study, GcMAF at doses between 4 ng/kg and 4 μg/kg/day was injected ip for 14 days in mice. However, no dose-dependent efficacy of GcMAF was observed. 28 In another study, 29 the macrophage-activating effects of GcMAF were confirmed at a dose of 40 ng/kg/day as well as in our preliminary study. We then settled on use of the same dose of GcMAF for the present study.
Immunohistochemistry
Rat brains were transcardially perfused with 4% paraformaldehyde in PBS on ice. After fixing, 10-μm-thick frozen sections were mounted on Matsunami adhesive silane-coated glass slides (Matsunami Glass, Tokyo, Japan). After 30 min of blocking with serum-free protein block (Dako Cytomation, Agilent, Tokyo, Japan), slides were incubated with primary antibodies diluted with Canget signal immunostain (Toyobo, Osaka, Japan).
Antibodies were mouse monoclonal antibody against CD16 (ab203883; abcam, Cambridge, UK), CD163 (ab182422; abcam), rabbit monoclonal against CD36 (ab133625; abcam) and CD68 (ab125157; abcam), rabbit polyclonal against neuronal nucleus (NeuN) (ab104225; abcam) and mouse monoclonal antibody against nestin (ab11306; abcam). Antibody against vascular endothelial growth factor (VEGF) (SC-7269; Santa Cruz Biotechnology) was also used, as was rabbit polyclonal antibody against MAP2 (SC-20172; Santa Cruz Biotechnology), goat polyclonal antibody against glial fibrillary acidic protein (GFAP) (SC-6170; Santa Cruz Biotechnology), and CD36 (SC-7641; Santa Cruz Biotechnology). Sheep polyclonal antibody was used against bromodeoxyuridine (BrdU) (abcam) dissolved in 0.9% saline (concentration, 100 mg/ml). To label proliferating cells in the brain, rats received intraperitoneal injections of BrdU (50 mg/kg) at 48, 24, 12, and 4 h before sacrifice. To study progenitor cell proliferation, sacrifice was performed 4 h after the fourth BrdU injection on day 28. To examine the specificity of immunoreactivity, the primary antibody was omitted to provide a nonspecific control. Visualization was achieved with Alexa Fluor 594 donkey anti-mouse immunoglobulin (IgG), 594 rabbit anti-mouse IgG, 594 donkey anti-rabbit IgG, 594 donkey anti-goat IgG, 488 goat anti-rabbit-, 488 donkey anti-goat-, or 488 donkey anti-sheep IgG (Molecular Probes, Eugene, OR). The positive area was assessed under a BZ-X710 microscope equipped with an image analyzing system (Keyence).
RNA isolation and quantitative real-time polymerase chain reaction (qRT-PCR)214
Total RNA was extracted with MagNA Pure Compact (Roche Diagnostics, Tokyo, Japan). For the reverse transcription of total RNA to cDNA we used a transcriptor first-strand cDNA synthesis kit (Roche Diagnostics). For each sample, qRT-PCR was performed in a Light Cycler 2.0 system (Roche Diagnostics). Light Cycler Fast Start DNA master and SYBR green I (Roche Diagnostics) were used for IL-6, IL-1β, IL-4, Arg-1 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The primers were: forward (F), 5′-TCT CAG GGA GAT CTT GGA AAT G-3′; reverse (R), 5′-TAG AAA CGG AAC TCC AGA C-3′ for rat IL-6; (F), 5′-TGC AGG CTT CGA GAT GAA C-3′, (R), 5′-AGC TCA TGG AGA ATA CCA CTT G-3′ for rat IL-1β; F, 5′-CAC CGA GAT GTT TGT ACC AGA-3′, R, 5′-TCA AGC ACG GAG GTA CAT CA-3′ for rat IL-4; F, 5′-TGT GGG AAA AGC CAA TGA AC-3′, R, 5′-AGA TGC TTC CAA TTG CCA TA-3′ for rat Arg-1; and F, 5′-TAC ACT GAG GAC CAG GTT G-3′, R, 5′-CCC TGT TGC TGT AGC CAT A-3′ for rat GAPDH. PCR conditions were: 95°C for 10 min followed by 40 cycles of 95°C for 10 s, 60°C for 10 s, and 72°C for 8 s. Results were normalized to the expression of GAPDH mRNA.
Statistical analysis
MCAO-R rats were randomly divided into groups for determination of the cortical infarct area, immunohistochemical (IHC) analysis, and quantification of mRNA levels. Sham rats were sacrificed on day 1 and samples were collected for each analysis of IHC and mRNA level. The mRNA level was normalized to the level of GAPDH mRNA. All data are presented as the mean ± standard deviation (SD). Samples sizes were decided based on previous studies.23,24 Differences between two groups were examined using Student’s t-test and multiple comparisons between more than three groups were analyzed using Scheffe’s test. For testing the normality of data, Shapiro-Wilk test was used. Values of p < 0.05 were regarded as indicating a significant difference. Statistical analyses were conducted using JMP version 13.2 software (SAS Institute Inc., Cary, NC, USA).
Results
Cortical infarct area accompanied by an increase in M1-type macrophages and inflammatory cytokines in the acute phase of post-ischemia
In rats subjected to 2-h MCAO-R (Figure 1(a)), infarct areas in the cortex and basal ganglia declined consistently from day 1 to day 7 and remained almost the same size on days 7 and 14 (Figure 1(b)). On post-ischemia day 3, CD16+ cells were highly expressed in the cortex, but not in the basal ganglia compared to the Sham group without ischemia, then decreased to almost the same level as Sham rats by day 7. Expression of CD163+ cells was low for 7 days after MCAO-R (Figure 1(c)). We then focused on the cortex to assess the role of macrophages. Consistent with the elevated expression of CD16+ cells, mRNA levels of IL-6 and IL-1β in the cortex were significantly higher in MCAO-R rats than in sham-operated rats on day 1; then decreased to baseline on day 7 as in sham-operated rats (Figure 1(d)). However, mRNA levels of Arg-1 and IL-4 showed no significant difference between Sham and MCAO-R rats during the 7 days (Figure 1(d)).
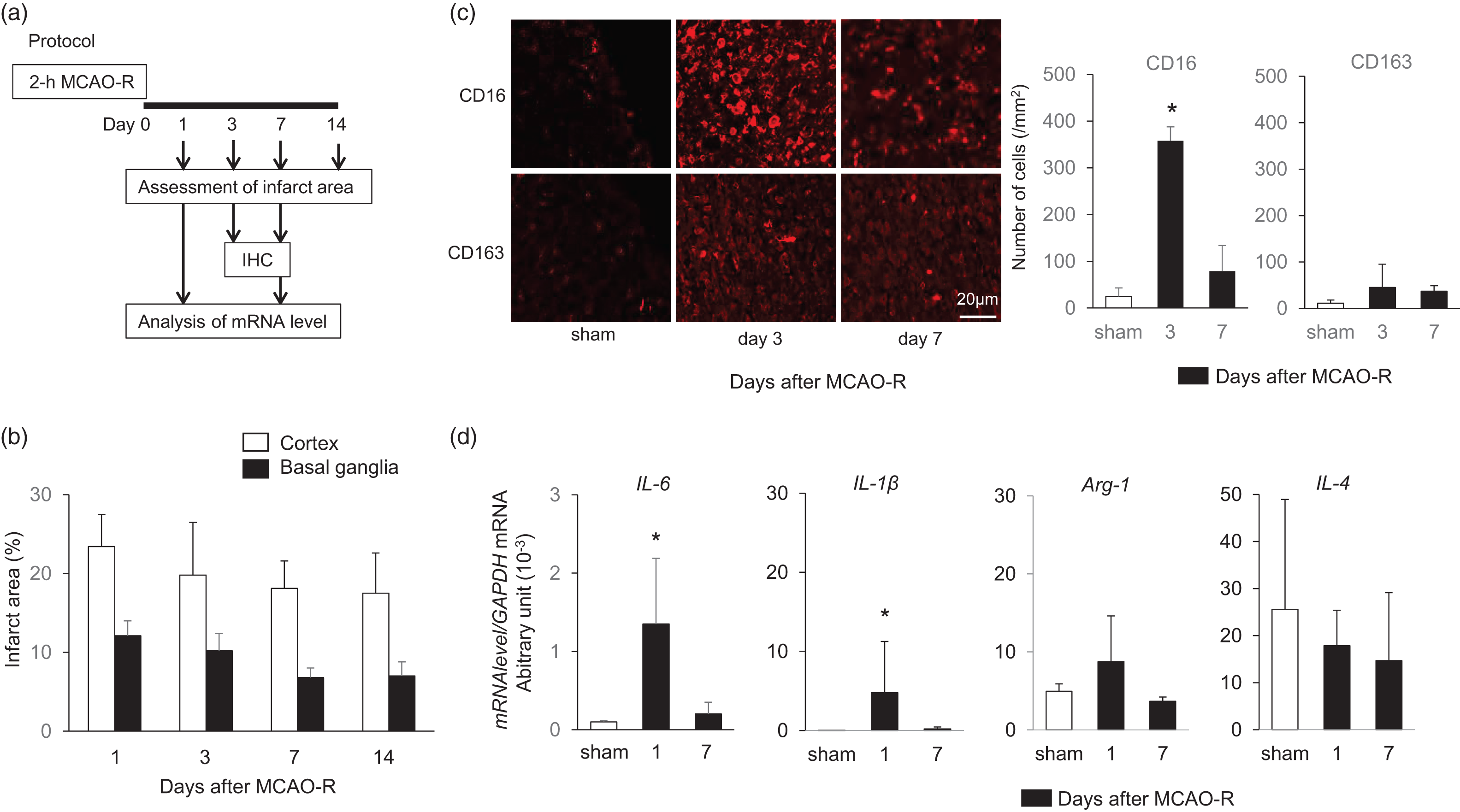
High expression of CD16+ but not CD163+ cells associated with elevation of cytokines IL-6 and IL-1β in the acute phase after cerebral ischemia-reperfusion. (a) Experimental protocol. Rats were subjected to middle cerebral artery occlusion for 2-h and reperfusion (MCAO-R) under anesthesia and compared to sham-operated rats. (b) Changes of infarct area in cortex and basal ganglia on days 1, 3, 7, and 14 in rats subjected to MCAO-R (n = 7 each). (c) Expression of CD16+ and CD163+ cells 3 and 7 days in rats subjected to MCAO-R compared to sham-operated rats (n = 4). (d) Changes in mRNA levels of IL-6, IL-1β, Arg-1, and IL-4 normalized by GAPDH mRNA level on days 1 and 7 after MCAO-R (n = 7). Data are given as mean ± SD.
Taken together, the expansion of brain damage after MCAO-R accompanied by transient increases in levels of pro-inflammatory cytokines may be associated with higher expression of CD16+ M1-type macrophages in the acute phase.
Treatment with GcMAF in the acute phase of post-ischemia led to increases in both M1- and M2-type macrophages
Next, to examine the role of M2-type macrophage activation in the acute phase after MCAO-R, we used GcMAF, an activator of M2-type macrophages (Figure 2(a)). Unfortunately, treatment with GcMAF in the early phase after MCAO-R seemed to enlarge the infarct area on days 1, 3 and 7 (Figure 2(b)), which significantly increased CD16+ M1-type macrophages as well as CD163+ M2-type macrophages, compared to VC in the penumbra adjacent to the infarct area (Figure 2(c)). Both CD16+ and CD163+ macrophages co-localized with phagocytes CD36+ cells (Figure 2(d)). Under conditions of high mRNA levels of IL-6 and IL-1β (Figure 1(d)) in the acute phase, CD163+ M2-type macrophages induced by GcMAF may transform phenotypes, resulting in an elevation of CD16+ M1-type macrophages and leading to expansion of the cerebral infarct area (Figure 2(e)). These findings seem to be consistent with results from other studies. 5 . 7
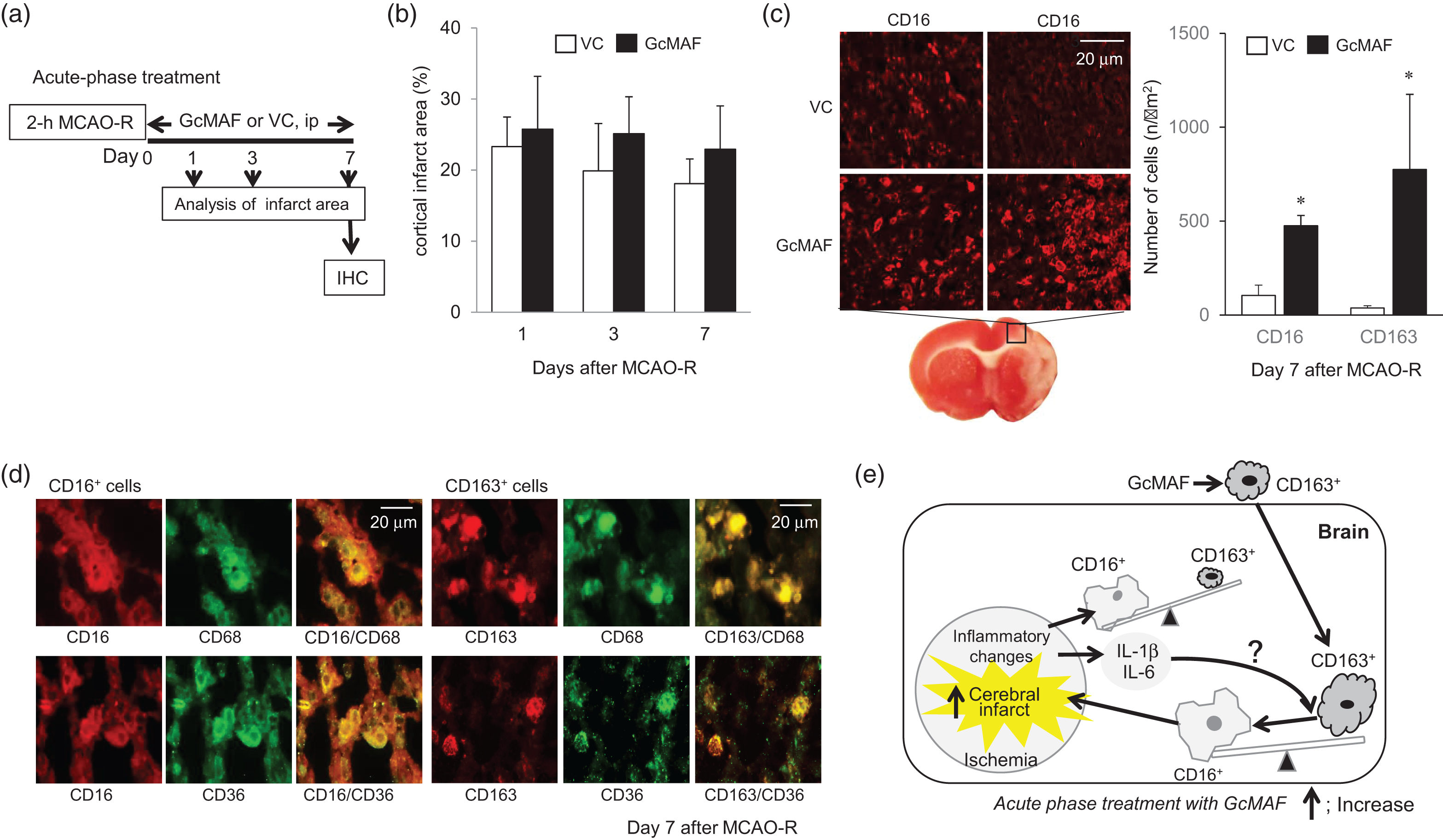
Deleterious effects of M2-type macrophage activation in the acute phase after cerebral ischemia-reperfusion. (a) Experimental protocol. Each cortical infarct area was determined on days 1, 3, and 7 after acute-phase treatment with GcMAF (Days 0–6) and compared to vehicle control. (b) Changes in infarct area shown as a percentage of the contralateral hemisphere in VC and GcMAF groups. Data are given as mean ± SD (each, n = 7). (c) Expression of CD16+ and CD163+ cells on day 7 after treatment with VC and GcMAF. (d) Immunohistochemical staining of CD16+ and CD163+ cells co-localized with CD68+ monocyte/macrophages and CD36+ phagocytic cells (n = 4). (e) Schematic diagram; deleterious effects of treatment with GcMAF in the acute-phase of post-ischemia. Data are given as mean ± SD.
Clearance of the infarct area associated with M2-type macrophage activation by GcMAF in the subacute phase of post-ischemia
We further examined the effects of cerebral infarction on day 14 after treatment with GcMAF during days 7–13 (subacute phase) after ischemia induction using serial slices (Figure 3(a)). No difference was seen in size of the non-infarcted area (Figure 3(b)), while the disappeared area was significantly larger in all GcMAF-treated rats than in VC rats, resulting in significantly smaller cortical infarct area in all GcMAF-treated rats than in VC rats (Figure 3(b)). Immunohistochemically, we found that expression levels of CD16+ cells were low and no difference was evident between VC and GcMAF treatment groups (Figure 3(c)). On the other hand, CD163+ cells were highly expressed on day 14 after GcMAF treatment during the subacute phase (day 7–13) and accompanied the elevation of CD36+ phagocytic cells (Figure 3(c)). The disappeared cortical infarct area seemed to be associated with phagocytosis via the increased M2-type macrophages resulting from GcMAF administration.
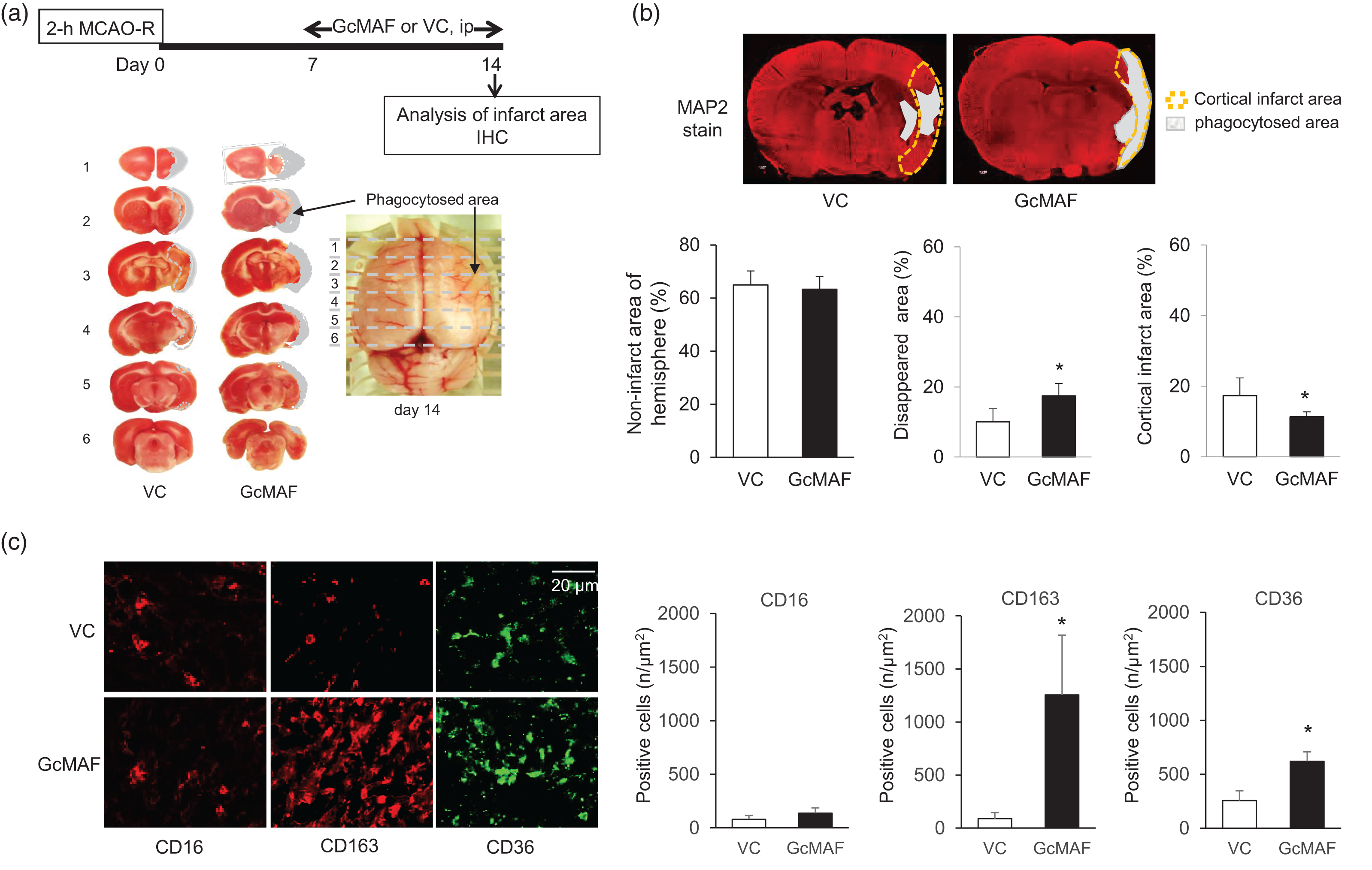
Effects of M2-type macrophage activation during the subacute phase (days 7–13) after MCAO-R. (a) Experimental protocol and photographs of brain slices treated with GcMAF on day 14 after MCAO-R. (b) Non-infarct area, cortical infarct area and phagocytosed area normalized as a percentage of the contralateral hemisphere. Each data indicates mean ± SD (each, n = 7). (c) Representative expression of CD16+ M1- and CD163+ M2-type macrophages and CD36+ cells assessed immunohistochemically in the peri-infarct region on day 14 of post-ischemia (n = 4). Data are given as mean ± SD.
Phagocytosis activated by GcMAF in the infarct area is attributable to M2 macrophages
To further confirm the phagocytosis by activation of M2 macrophages on the infarct area, we administered CLOD one day before the delivery of GcMAF in the subacute phase after ischemia induction. CLOD ingested macrophages via endocytosis, fusing with the lysosomes, thereby disrupting and killing macrophages.30,31 Based on the report that CLOD encapsulated in liposomes at a concentration of 7 mg/ml depletes 90% of macrophages within 24–36 h after systemic administration,32,33 CLOD was injected 1 day before treatment with GcMAF (Figure 4(a)).
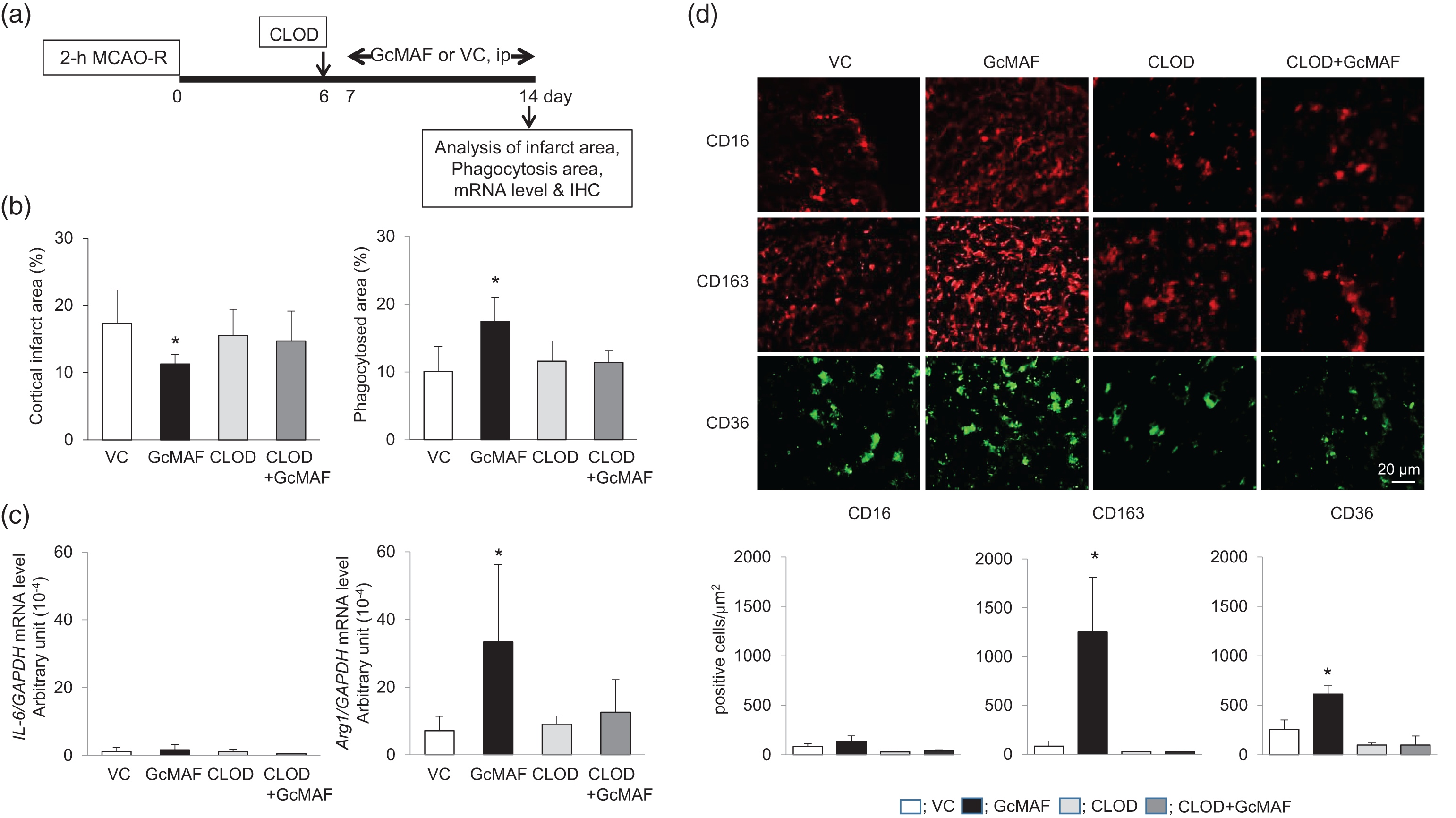
Effect of treatment with GcMAF in the subacute phase of post-ischemia with or without clodronate-liposome (CLOD) pre-treatment. (a) Experimental protocol. CLOD (10 ml/kg, intraperitoneal) was injected one day before treatment with GcMAF during days 7–13 and each assessment was performed on day 14. (b) Cortical infarct area and phagocytosis area on day 14 shown as percentages of the contralateral hemisphere (n = 7). (c) The mRNA levels of IL-6 and arginase-1 (Arg-1) determined in the penumbra on day 14. Data are given as mean ± SD (each group, n = 7). (d) Immunohistochemical expressions of CD16
Injection of CLOD abrogated the GcMAF-induced decrease in the cortical infarct area and the increase in the phagocytosed area (Figure 4(b)) without affecting the non-infarct area (data not shown). The IL-6 mRNA level was low and unaffected by CLOD, whereas the mRNA level of Arg-1 was significantly higher in GcMAF-treated rats than in VC rats; this increase was abolished by CLOD (Figure 4(c)). Immunohistochemically, CLOD abolished the GcMAF-induced increase in CD163+ M2-type macrophages and CD36+ cells without affecting CD16+ M1-type macrophages (Figure 4(d)). Together, these findings demonstrated the clearance of cortical infarct area and the elevation of Arg-1 mRNA level via activation of M2-type macrophages increased by GcMAF.
Expression of neurogenesis- and angiogenesis-related molecules in the peri-infarct area associated with M2-type macrophage activation
Finally, to assess whether activation of M2-type macrophages by GcMAF is associated with expression of neurogenesis- and angiogenesis-related molecules, we evaluated the expression of MAP2, BrdU, and nestin, a neuronal stem cell marker in rats treated with GcMAF during days 7–28 after ischemia induction. In the cortical peri-infarct area, expressions of MAP2, BrdU, and nestin were significantly higher in GcMAF-treated rats than in VC rats (Figure 5(a)). The appearance of neurovascular unit constituents involving VEGF, GFAP and the neuronal cell marker NeuN was also observed in the cortical peri-infarct area of GcMAF-treated rats, but not VC rats (Figure 5(b)). However, we could not observe significant neurological improvements in GcMAF-treated rats on day 28 (data not shown). Assessment after a longer duration of treatment with GcMAF may be required.
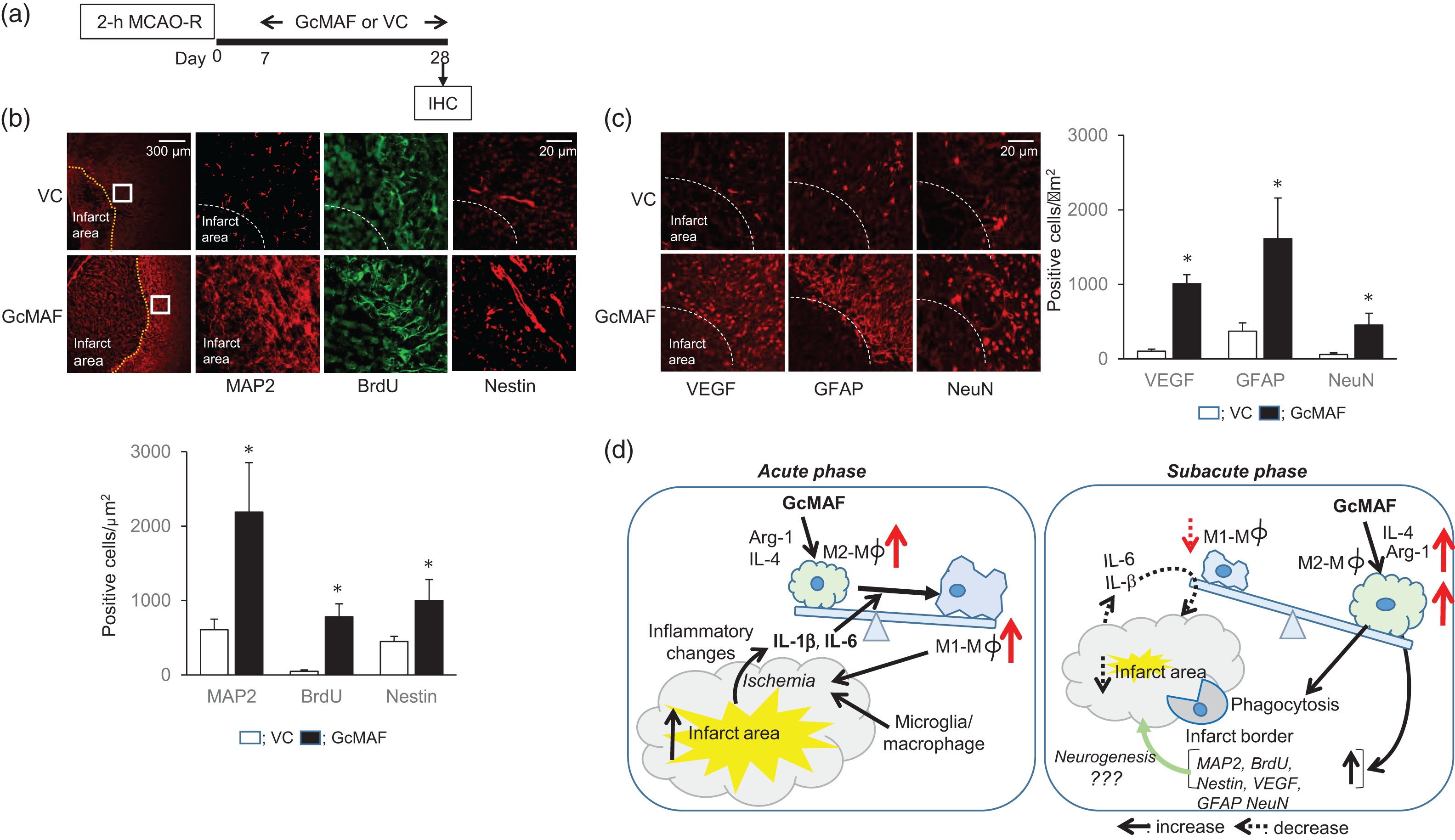
Expression of neuro- and angiogenesis-related molecules after treatment with GcMAF in the subacute to late phase of post-ischemia. (a) Experimental protocol. GcMAF or VC was injected during days 7–27 of post-MCAO-R and rats were sacrificed on day 28. (b) Immunohistochemical assessment of expressions of MAP2, BrdU, and nestin in the ischemic penumbra of GcMAF-treated rats compared to VC. (c) Expressions of VEGF, GFAP, and NeuN in the ischemic penumbra of GcMAF-treated rats compared to VC rats. Data are given as mean ± SD (each group n = 4,
Discussion
Several studies have reported that activation of M2-type macrophages is protective against cerebral ischemia. However, we first demonstrated that the significance of activating M2-type macrophages differed between acute and subacute phases after cerebral ischemia in rats. Importantly, under conditions of the predominant expression of M1 macrophages with the elevation of IL-6 and IL-β in the acute phase of post-ischemia, specific activation of M2 macrophages by GcMAF did not exert beneficial effects. Unfortunately, GcMAF augmented the inflammatory response, thereby expanding the cortical infarct area. Under the predominant conditions of M1 macrophages accompanied by increased IL-1β and IL-6 cytokines, treatment with GcMAF increased pro-inflammatory M1-type macrophages as well as anti-inflammatory M2 type-macrophages (Figure 2(e)). On the other hand, treatment with GcMAF in the subacute phase of post-ischemia was associated with “cleaning” of the infarcted area by phagocytosis through the activation of M2-type macrophages without affecting M1-type macrophages, (Figures 3(b) and 4(d)), leading to high expression of neurogenesis- and angiogenesis-related molecules in the late phase (Figure 5(b) and (c)). Our findings suggest that phase-specific activation of M2 macrophages post-ischemia may be crucial to the induction of beneficial effects. The role of M2-type macrophage-specific activation after cerebral ischemia may differ depending on the post-ischemic phase. We therefore emphasize that the timing leading to activation of M2-type macrophages should be taken into consideration.
Microglia/macrophages are activated within minutes of ischemic onset and induce the production of inflammatory cytokines, including IL-1β and TNF-α. 31 After the rapid activation of resident microglial cells, blood-derived macrophages are recruited into ischemic brain tissue and rapidly mobilized to the site of injury, where they initiate the release of effector molecules and recruit other immune cells.1,16 Since mRNA levels of IL-6 and IL-1β were higher on day 1 than on day 7 after cerebral ischemia, these increases may be attributable to the high expression of M1 macrophages in the present study. In association with the up-regulation of IL-6 and IL-1β, expression of M1 macrophages increased on day 1 and reached baseline levels on day 7. In the early post-ischemic phase, expression of CD16+ M1 macrophages was dominant. As this phenomenon coincided with expansion of the ischemic penumbra in the presence of increased cytokines, the presence of M1 macrophages may be associated with the detrimental effects observed in the early post-ischemic phase. Hu et al. 5 reported that mRNA levels of IL-6 gradually increased in the ischemic core over the course of 14 days. A more recent study documented post-ischemic changes in IL-1β and TNF-α via modulating microglial activation and M1 polarization.29,30 These findings partly coincide with ours. Since GcMAF had no beneficial effects under the condition of high expression of M1-type macrophages with IL-6 and IL-1β, the timing of M2-type macrophage activation should be re-considered.
Despite the decrease in mRNA levels of IL-1β and IL-6 on day 7 after ischemia induction, brain Arg-1 and IL-4 mRNA levels were retained during observation periods for 14 days. Subacute-late phase treatment with GcMAF produced an M2-type macrophage-dominant condition without affecting M1-type macrophages, and an increase in “clearance” through phagocytosis of the infarcted area. We confirmed that the effects were abrogated by CLOD. 33 Since CD163+ M2-type macrophages induced by GcMAF were co-localized with CD36+ cells, but not Iba-1+ cells (data not shown), the delivered GcMAF may play a role in the activation of M2-type monocytes/macrophages derived from peripherally to perform phagocytosis.
Phagocytosis promotes the secretion of anti-inflammatory mediators, and thus may contribute to the creation of favorable post-stroke conditions for brain recovery. 15 The M2 macrophage-related post-ischemic production of TGF-β, IL-10, and Arg-1 may facilitate tissue repair by promoting the resolution of inflammation and provision of direct cytoprotective effects to surviving cells in the ischemic territory.1,15 These earlier findings support our observation that the increase in Arg-1 elicited by GcMAF was associated with the expression of survival-related molecules in the peri-infarct area.
Other studies34,35 have also suggested that M2-type macrophages play a crucial role in resolving the inflammatory response and participate in debris scavenging, tissue remodeling, and angiogenesis. Consistent with these findings, when M2-type macrophages dominated in the subacute-phase of post-ischemia, phagocytosis may accelerate the removal of debris including apoptotic or necrotic cells without affecting the non-infarct area in this study. In addition, the increase in M2-like macrophages was associated with expression of survival-related molecules in the peri-infarct area. Activation of M2-type macrophages by subacute- to late-phase treatment with GcMAF may thus have beneficial effects.35,36
Study limitations
This study shows some limitations. How subacute-to late-phase treatment with GcMAF activates M2 macrophages remains unknown. GcMAF is thought to be an activator of macrophage phagocytosis, but does not switch the cell polarization to M1-like macrophages. Uto et al. 18 reported that in vivo and in vitro, degalactosylated/desialylated bovine colostrum induces macrophage phagocytic activity without affecting the production of inflammatory cytokines such as TNF-α and IL-1β. Desestret et al. 35 showed that in co-cultures of bone-marrow-derived macrophages and in hippocampal slices subjected to oxygen and glucose deprivation, as a condition mimicking cerebral ischemic conditions, M2-type macrophages provided potent protection against neuronal cell loss. When they evaluated the possibility of M2-type macrophage cell therapy for stroke, treatment in the acute phase (day 4) failed to improve stroke outcomes. In IL-4-knockout mice with experimental brain ischemia, M2 macrophages were reduced, and treatment with IL-4 resulted in neurological recovery from day 14 through M2-like macrophages. However, IL-4 injection into wild-type mice had no favorable effects. 11 We found that M1 macrophages remained potent even 3 days after ischemia induction (data not shown) and the level of IL-4 mRNA was retained, suggesting that the timing of treatment with M2 macrophages is key to successful cell therapy and that a reduction in the number of M1 macrophages in the early phase (within 7 days) is needed to obtain the beneficial effects of M2-like macrophage cell therapy.
Since no dose-dependent efficacy of GcMAF was observed between 4 ng/kg and 4 µg/kg, 28 the GcMAF dose, 40 ng/kg in the present experimental study was delivered based on earlier 29 and our preliminary studies. In mouse and in vitro studies, GcMAF extracted from human plasma promoted phagocytic activity via macrophage activation,17–19 but phenotypes of the involved macrophages were not identified and we do not yet know the detailed mechanisms by which GcMAF activates M2 macrophages.
Iadecola and Anrather 2 proposed a process orchestrated by “find me” and “eat me” signals. The “find me” signal involves purines released from injured cells and chemokines that attract microglia and macrophages to the site of injury. The possible mechanisms underlying phagocytosis by M2-like macrophages remain to be elucidated.
Conclusion
We offer the first documentation that in the subacute to late post-ischemic phase, but not in the acute phase, the increase in M2-type macrophages elicited by treatment with GcMAF was associated with promotion of phagocytosis in the infarct area without affecting non-infarct area and high expression of survival-related molecules in the peri-infarct area. Our findings may facilitate the development of new therapeutic strategies in a phase-dependent manner for post-stroke individuals.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding for this work was provided by Grants-in-Aid for Scientific Research (15K10306 to KY; 17K10833 to TK; 26861153 to KN) from the Japan Society for the Promotion of Science and a Grant-in-Aid for the Strategic Young Researcher Overseas Visits Program for Accelerating Brain Circulation from the Japan Society for the Promotion of Science (24S2407).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
Conception and design of study: YK, YU
Acquisition of data: YK, TK, MS, KS, TM
Analysis and/or interpretation of data: KS, KY
Drafting the manuscript: YK, HK, KTK
Revising the manuscript critically for important intellectual content: KTK, YU, YT
Approval of the version of the manuscript to be published: YU, YT
All authors read and approved the final manuscript.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.