
Abstract
Almost all agents that exhibit neuroprotection when administered into the cerebral ventricles are ineffective or much less effective in rescuing damaged neurons when infused into the blood stream. Search for an intravenously infusible drug with a potent neuroprotective action is essential for the treatment of millions of patients suffering from acute brain diseases. Here, we report that postischemic intravenous infusion of a ginseng saponin, ginsenoside Rb1 (gRb1) (C54H92O23, molecular weight 1109.46) to stroke-prone spontaneously hypertensive rats with permanent occlusion of the middle cerebral artery distal to the striate branches significantly ameliorated ischemia-induced place navigation disability and caused an approximately 50% decrease in the volume of the cortical infarct lesion in comparison with vehicle-infused ischemic controls. In subsequent studies that focused on gRb1-induced expression of gene products responsible for neuronal death or survival, we showed that gRb1 stimulated the expression of the mitochondrion-associated antiapoptotic factor Bcl-xL in vitro and in vivo. Moreover, we revealed that a Stat5 responsive element in the bcl-x promoter became active in response to gRb1 treatment. Ginsenoside Rb1 appears to be a promising agent not only for the treatment of cerebral stroke, but also for the treatment of other diseases involving activation of mitochondrial cell death signaling.
Keywords
Introduction
Approximately 4 million people die of cerebral stroke, and more people with stroke become handicapped every year in the world, possibly because of the lack of intravenously infusible drugs with a potent neuroprotective action. Almost all agents that exhibit neuroprotection when administered into the cerebral ventricles are ineffective or much less effective in rescuing damaged neurons when infused into the blood stream (Wu and Pardridge, 1999). This is because such neuroprotective agents do not pass through the blood brain barrier and/or they are likely to be degraded into nonbioactive fragments in the blood stream, mostly by the actions of proteases.
Over the past several decades, biomedical researchers have been making desperate efforts, without any great success, to develop intravenously infusible neuroprotective agents, with great emphasis on glutamate receptor antagonists like MK-801. However, most glutamate receptor antagonists, even though alleviating ischemic neuronal damage, frequently cause serious adverse effects in humans by modulating neural transmission in an adverse manner, and might not be applicable for clinical use. A search for intravenously infusible neuroprotective agents that act selectively on damaged brain tissue without affecting neural transmission appears to be essential for the treatment of cerebral stroke. We speculate that development of an intravenously infusible agent with a potent neuroprotective action would be absolutely of value from the clinical point of view, because such an effective agent would greatly contribute to elucidation of the molecular mechanisms underlying the brain diseases for which the agent is applied, and would facilitate the development of other innovative treatment protocols or drugs over the next 5 to 10 years or beyond.
Ginseng root (Panax ginseng CA MEYER) has been prescribed to patients for thousands of years in Asian countries, without apparent adverse effects. Ginseng root consists of two major ingredients: crude ginseng saponin and crude ginseng nonsaponin fractions. To date, more than 20 saponins have been isolated from ginseng root and identified chemically. They can be classified into three major groups according to their chemical structure: protopanaxadiol, protopanaxatriol and oleanolic acid saponins, of which ginsenoside Rb1 (gRb1), ginsenoside Rg1 and ginsenoside R0 are representative substances, respectively (Shibata et al, 1994).
In the present study, we first showed that postischemic intravenous infusion of gRb1 ameliorated cerebrocortical infarct size, place navigation disability and secondary thalamic degeneration in spontaneously hypertensive rats-stroke prone (SHR-SP), with permanent occlusion of the unilateral middle cerebral artery (MCA) distal to the striate branches. In subsequent in vitro and in vivo experiments that focused on gRb1-induced expression of gene products responsible for neuronal death or survival, we showed that gRb1 stimulated neuronal Bcl-xL protein expression and prevented neuronal apoptosis at concentrations of 1 to 100 fg/ml in vitro, and that postischemic intravenous infusion of gRb1 in MCA-occluded SHR-SP induced Bcl-xL expression.
Materials and methods
All experimental procedures were conducted according to the Guide for Animal Experimentation at Ehime University School of Medicine. Animals were housed in an animal room with a temperature range of 21 to 23°C and a 12-h light/dark cycle (light on: 0700 to 1900 h). Animals were allowed access to food and water ad libitum until the day of the experiment.
Isolation and Purification of Ginsenoside Rb1
Ginsenoside Rb1 was isolated and purified from the crude saponin fraction of the rhizome of Panax Ginseng CA MEYER, Korea Red Ginseng, by repeated-column chromatography on silica gel with CHCl3–MeOH–H2O (65:35:10) and on octadecylsilyl (ODS) silica with MeOH–H2O (1:1–7:3) (Samukawa et al, 1995).
Middle Cerebral Artery Occlusion in Spontaneously Hypertensive Rats-Stroke Prone
SHR-SP aged 16 to 18 weeks were used. Rats were anesthetized with 1.5% halothane in a 4:3 mixture of nitrous oxide and oxygen, and brain temperature was kept at 37.0°C ± 0.2°C during surgery. The left MCA above the rhinal fissure and distal to the striate branches was coagulated and cut (Zhang et al, 2004). An osmotic minipump (Model 2004 or 2001, Alza Corp., Palo Alto, CA, USA) filled with either gRb1 solution or vehicle (saline) was implanted subcutaneously in the back of each animal before MCAO, and a fine silicon tube connected to the minipump was inserted into the left femoral vein after MCAO. After recovery from anesthesia, the animals were maintained in an air-conditioned room at approximately 22°C.
Measurement of Brain Temperature and Blood Pressure
The brain temperature of rats with or without gRb1 treatment was monitored by the combination of a temperature probe (XM-FH-BP 5 mm; Mini-Mitter Co., Sunriver, OR, USA) inserted into the right cerebral hemisphere (1 mm anterior to the bregma, 1.5 mm lateral to the midline) and the telemetry system receiving signals from the probe (DataScience Int., St Paul, MN, USA). The rats had free access to food and water except for the periods of minipump implantation and MCA occlusion. The brain temperature was continuously monitored until the end of the experiment.
The mean arterial blood pressure (MABP) in each animal was measured using a rat tail manometer-tachometer system (MK-1030, Muromachi Co., Tokyo, Japan).
Intravenous Infusion of Ginsenoside Rb1 after Middle Cerebral Artery Occlusion in Spontaneously Hypertensive Rats-Stroke Prone
Ginsenoside Rb1 was dissolved in isotonic saline. Then, 60 μL of gRb1 solution (6 or 60 μg/60 μL) or the same volume of vehicle (saline) was injected into the left femoral vein of rats weighing 250 to 300 g immediately after MCAO, and then gRb1 at a corresponding dose (6 or 60 μg/day, respectively) was continuously infused for 4 weeks through the minipump (n = 5 to 8 per group). Middle cerebral artery-occluded animals infused with saline (n = 8) were used as a control. Mean arterial blood pressure and the brain temperature of SHR-SP before MCAO were 203.1 ± 6.9 mm Hg and 37.0°C ± 0.2°C, respectively. These were not affected by MCAO or gRb1 infusion (data not shown).
Water Maze Test
The rats were subjected to repeated Morris water maze tests at 2 and 4 weeks after MCAO or sham operation. Each test included three trials per day for 4 consecutive days. The rats were allowed to swim until they reached the submerged platform and to stay there for at least 10 secs. In the case that rats could not escape onto the platform within 90 secs, they were placed by hand onto the platform for 15 secs and their escape latency was recorded as 90 secs. The mean latency of finding the invisible platform was measured for individual animals on each day.
Morphologic Study
After water maze tests, the animals were anesthetized with an intraperitoneal injection of chloral hydrate (300 mg/kg) and perfused transcardially with 0.1 mol/L phosphate-buffered saline (pH 7.4), followed by perfusion with 500 mL of 4% paraformaldehyde in 0.1 mol/L phosphate buffer (pH 7.4). The brain was dissected out and fixed in the same fixative. After taking a photograph of each brain, the infarcted region was traced on a sheet. Then the areas of the infarcted region and the left hemisphere of the brain were measured by a computerized processing system. The ratio of the infarcted area to the left hemispheric area was calculated. The brain was embedded in paraffin, and serial coronal sections 5-μm thick were cut and stained with 0.1% cresyl violet.
The area of the thalamus was measured with a planimeter in sections 3.6 mm posterior to the bregma, and the ratio of the thalamic area on the MCA-occluded side to that on the contralateral side was calculated. Neuronal density per 0.099 mm2 in the ventroposterior (VP) thalamic nucleus was evaluated in each animal. The ratio of the thalamic area and neuronal density in the VP thalamic nucleus were evaluated by an investigator without any prior information.
To investigate whether cerebrovascular networks were reorganized in the putative ischemic penumbra, paraffin sections from gRb1-treated brain around the level 2.8 mm posterior to the bregma were examined with Nomarski optics (n = 5 per group). Four photomicrographs were taken from the parietal cortex of each cerebral hemisphere, and the vascular area per 1.27 mm2 in the rescued parietal cortex on the lesioned side was compared with that in the counterpart on the contralateral side. Blood vessels identified in the photomicrographs were traced with black ink, and the vascular area was measured using NIH image software.
Delayed Intravenous Infusion of Ginsenoside Rb1 after Middle Cerebral Artery Occlusion in Spontaneously Hypertensive Rats-Stroke Prone
Ginsenoside Rb1 was dissolved in isotonic saline. Then, 60 μL gRb1 solution (6, 60, 3000 or 12,000 μg/60 μL) or the same volume of vehicle (saline) was injected into the left femoral vein of rats weighing approximately 300 g at 2 h after MCAO, and then gRb1 at a corresponding dose (6, 60, 3000 or 12,000 μg/day, respectively) was continuously infused through the minipump (n = 7 per group). The dose of continuous gRb1 infusion was calculated as 20, 200 μg/kg per day, 10 or 40 mg/kg per day, respectively. Middle cerebral artery-occluded animals infused with saline (n = 7) were used as a control. At 1 day after MCAO, animals were killed with an overdose of pentobarbital and decapitated. The brain was removed and sectioned coronally into seven 2-mm slices. Then the brain slices were subjected to 2,3,5-triphenyltetrazolium chloride (TTC) staining as described below.
2,3,5-Triphenyltetrazolium Chloride Staining
The brain slices were immediately stained with 2% TTC (Sigma, St Louis, MO, USA) and incubated at 37°C for 30 mins. The border between infarcted and noninfarcted tissue was outlined with an image analysis system, and the area of infarction was measured by subtracting the area of the nonlesioned ipsilateral hemisphere from that of the contralateral hemisphere (Swanson et al, 1990). The volume of infarction was calculated by integration of the lesion areas at all equidistant levels of the forebrain.
Three-Min Ischemia in Gerbils
Male Mongolian gerbils weighing 70 to 80 g (approximately 12 weeks of age) were used. The animals were anesthetized with 1.5% halothane in a 4:3 mixture of nitrous oxide and oxygen and placed in a stereotaxic apparatus. An osmotic minipump (Alza model 2001) filled with gRb1 solution or vehicle (saline) alone was implanted subcutaneously in the back of each animal, and a needle connected to the minipump was placed in the left lateral ventricle at a point 1.5 mm anterior, 1.0 mm lateral and 2.7 mm ventral to the bregma. Three-minute occlusion of both common carotid arteries was performed as described previously (Morita et al, 2001).
Intracerebroventricular Infusion of Ginsenoside Rb1 in Gerbils
Immediately after 3-min forebrain ischemia, 2 μL saline containing 2.5 or 25 ng gRb1 was injected into the left lateral ventricle through a Hamilton syringe, and then gRb1 (60 or 600 ng/day) was continuously infused for 7 days into the left lateral ventricle (n = 8 in each group). Control gerbils received the same volume of saline infusion after 3-min forebrain ischemia (n = 6).
In situ Detection of DNA Fragmentation (Terminal Deoxynucleotidyl Transferase-Mediated Deoxyuridine Triphosphate Nick End-Labeling (TUNEL) Staining)
After 3-min transient forebrain ischemia, gerbils were infused with 0, 60 or 600 ng/day gRb1 (n = 6 to 8 in each group). At 7 days after the ischemic insult, animals were perfused with 4% paraformaldehyde in 0.1 mol/L phosphate buffer (pH 7.4) under pentobarbital anesthesia. Paraffin sections of the brain from each animal were processed for TUNEL staining (ApopTag, Intergen, NY, USA). All TUNEL-positive neurons along a 1-mm linear length of the CA1 field in two serial coronal sections were counted, and the mean number of positive neurons was calculated in each animal.
Cortical Neuron Cultures
The cerebral cortices of 17-day-old rat embryos were aseptically dissected out. Cortical neurons were dissociated from the tissues as described elsewhere (Toku et al, 1998). The dissociated cells were seeded on 24-well plastic plates (Corning, New York, NY, USA) coated with poly-L-lysine, at a density of 2.5 × 105 cells/cm2. The purity of the cultured neuron was confirmed by Western blot analysis with the antibodies against MAP2, GFAP and ED1 (Sugishita et al, 2001).
Sodium Nitroprusside (SNP) Treatment of Cultured Cortical Neurons
To induce apoptotic cell death, rat cortical neurons were exposed to an NO donor, 100 μmol/L SNP (Wako, Osaka, Japan), for 10 mins (Toku et al, 1998). Cortical neurons were cultured for 3 or 4 days in vitro, and then further cultured in the presence of 0 to 105 fg/ml gRb1 for 24 h, followed by SNP treatment. After treatment, neurons were cultured for 16 h with 0 to 105 fg/ml gRb1. Such low concentrations of gRb1 were chosen in this experimental design on the basis of repeated preliminary studies with a wide range of gRb1 concentrations.
Alamar Blue Assay
To evaluate the neuroprotective effect of gRb1, survival of cultured cortical neurons was assessed, using Alamar blue (Alamar Biosciences, Sacramento, CA, USA) as a redox indicator (Morita et al, 2001).
Lipid Peroxidation Assay
Cortical neurons of 17-day-old rat embryos were cultured at a density of 8.0 × 105 cells/cm2. On the fourth day of culture, a freshly prepared solution containing 100 nmol/L FeSO4 and 1 mmol/L ascorbic acid was added to the medium, DMEM, to which 0.1 to 1000 fg/ml gRb1 had been added on the second day of culture, and the neurons were maintained for 2 h at 37°C. Then, the relative level of lipid peroxidation in the cultured neurons was quantified by using 2-thiobarbituric acid fixation and subsequent measurement of light absorbance at 535 nm (Peng et al, 1998). Thiobarbituric acid reactive substances (TBARS) levels were expressed as malonyldialdehyde (MDA) equivalent (nmol/mL).
1,1-Diphenyl-2-picrylhydrazyl (DPPH) Radical Scavenging Assay
The activity of gRb1 to scavenge stable DPPH (Sigma Chemical Co., St Louis, MO, USA) radical was measured as described elsewhere (Mellors and Tappel, 1966). Briefly, DPPH radical was dissolved in ethanol to a final concentration of 500 μmol/L. Reactions were initiated by adding 100 μL of the freshly prepared DPPH radical solution to 100 μL gRbl solution at various concentrations (0–100 mg/mL). The reactions were incubated at room temperature for 25 mins, and the absorbance at 517 nm was measured using a plate reader (model 550, Bio-rad Lab., Hercules, CA, USA).
Construction of Bcl-X Promoter Plasmids
The murine Bcl-X promoter fragment between positions −638 and −95 relative to the translation start site was prepared by polymerase chain reaction (PCR) with primers 5′-CCACGAGCTCGATCTGGTCGATGGAG GAAC-3′ and 5′-AAACACCTGCTCACTTACTGGGTC-3′. This segment, which contains the Stat5 response element (STRE), was digested by SacI and BamHI, and inserted between the SacI and BamHI sites of the pGL2-basic vector (Promega, Madison, WI, USA) to make the bcl-x-wt plasmid. A mutation in STRE was generated by PCR using a sense primer (5′-AGGCATTGAGGATAAAAGGG-3′) and an antisense primer (5′-CCCTTTTATCCTCAATGCCT-3′) carrying mutations, to make the bcl-x-mut plasmid. Another promoter fragment between positions −1217 and −592, which does not contain STRE, was also prepared by PCR with primers 5-CGGCCCTCGAGCCCTG CAGGGGGCT-3 and 5-AATTGCGAAGCTTAGGAACCTG CC-3, and inserted between the XhoI and HindIII sites of the pGL2-basic vector to make the bcl-x-0.6L plasmid (Silva et al, 1999).
Transient Transfection and Promoter Assays
Primary cultured neurons were seeded on 24-well PLL-coated plastic plates. On the third day of culture, the cells were cotransfected with 0.4 μg reporter plasmid and 0.1 μg pRL-TK (Promega) internal control plasmid using Lipofectamine Plus Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. After transfection, the cells were incubated at 37°C for 16 h. On the next day, gRb1 was added to the medium at a concentration of 100 fg/mL, and neurons were incubated for a further 24 h. The activities of firefly luciferase from bcl-x promoter-luciferase plasmids and renilla luciferase from pRL-TK plasmid in the cell extracts were evaluated using a Dualluciferase assay kit (Promega) with a luminometer (TD-20/20; Turner Designs Inc., Sunnyvale, CA, USA) according to the manufacturer's protocol.
Analysis of bcl-xL mRNA Expression
Total RNA was extracted using Isogen (Nippon Gene, Tokyo, Japan), and treated with DNase. Oligo dT primers together with total RNA and Moloney murine leukemia virus reverse transcriptase (GibcoBRL) were used to obtain single-strand DNA. Polymerase chain reaction was conducted using Takara Taq polymerase (Takara, Tokyo, Japan). The following conditions were used for PCR amplification: cDNA products of the reverse transcription reaction were denatured for 2 mins at 94°C before 20 cycles (for β-actin) or 25 cycles (for bcl-xL) at 94°C for 1.5 mins, 55°C for 1.5 mins and at 72°C for 2 mins. The reverse transcriptase-polymerase chain reaction (RT-PCR) products were separated on 3% agarose gel and visualized with ethidium bromide. The following pairs of oligonucleotides corresponding to certain sequences within the coding regions of the bcl-xL and /i-actin genes were used as primers: rat bcl-xL primers, 5′ primer (5′-GTAGTGAATGA ACTCTTTCGGGAT-3′), 3′primer (3′-CCAGCCGCCGTTCT CCTGGATCCA-3′), rat β-actin primers, 5′ primer (5′-AGA AGAGCTATGAGCTGCCTGACG-3′) and 3′ primer (5′-TAC TTGCGCTCAGGAGGAGCAATG-3′). Polymerase chain reaction was performed five times for each sample.
Analysis of Bcl-xL Protein Expression
Homogenates were solubilized in Laemmli's sample solution containing 2% sodium dodecyl sulfate. An equal amount of protein (15 μg) from the homogenates was electrophoresed and processed for Bcl-xL immunoblot analysis using a monoclonal antibody against Bcl-xL protein (Transduction Laboratories Inc., Lexington, KY, USA). For semiquantitative evaluation, the immunoreactive bands were subjected to densitometric analysis.
Statistics
All values are presented as mean value ±s.d. Bcl-xL immunoreactivity in vehicle- and gRb1-vtreated gerbils was analyzed statistically by two-tailed Mann-Whitney U-test. All other statistical significance was tested by one-way ANOVA followed by Bonferroni's multiple comparison test. A P-value less than 0.05 was considered to be statistically significant.
Results
Purity of Ginsenoside Rb1
The purity of gRb1 used in this study was more than 99.999%, as determined by high-performance liquid chromatography (Figure 1). Approximately 5 g of gRb1 was isolated and purified from 10 kg of Korea Red Ginseng, 6-year root.
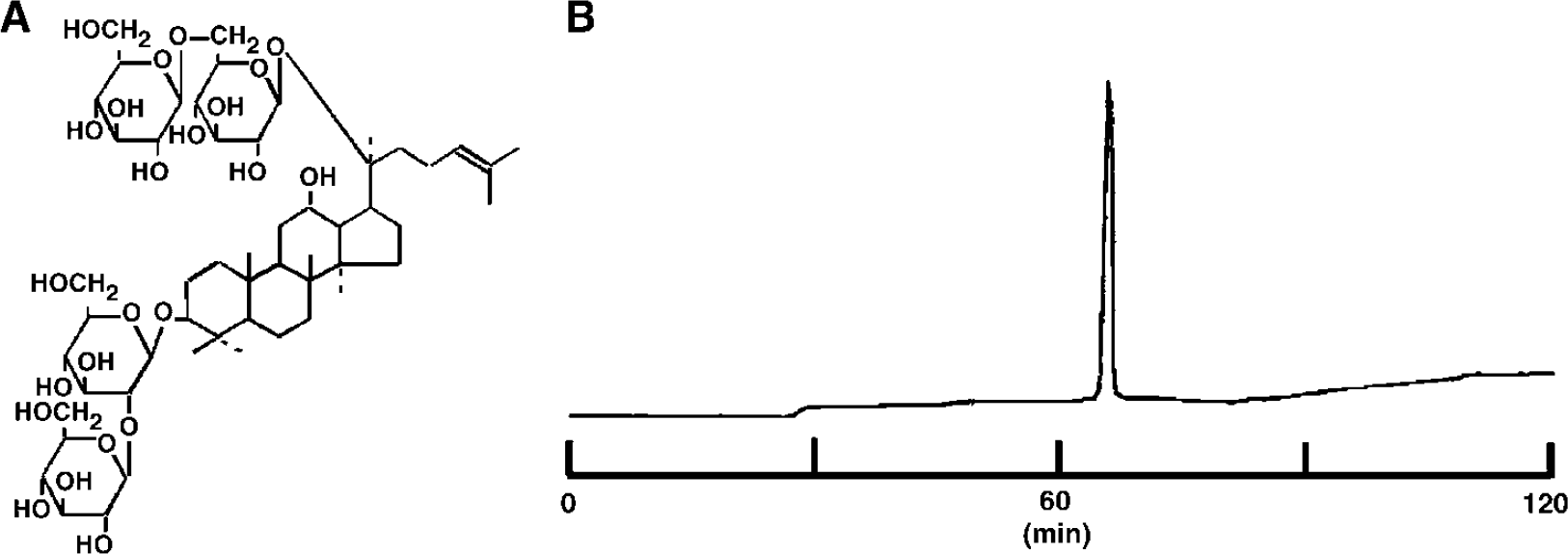
(
Intravenous Infusion of Ginsenoside Rb1 Ameliorates Cortical Infarct Size, Place Navigation Disability and Secondary Thalamic Degeneration in Middle Cerebral Artery-Occluded Spontaneously Hypertensive Rats-Stroke Prone
We first investigated the effects of postischemic intravenous infusion of gRb1 on cortical infarct size, place-navigation disability and secondary thalamic degeneration in SHR-SP, with permanent occlusion of the left MCA above the rhinal fissure and distal to the striate branches. This MCA-occluded rat model has more severe neuronal damage than the gerbil forebrain ischemia model, and more closely mimics clinical infarction than do gerbils with transient forebrain ischemia (Coyle and Jokelainen, 1983; Igase et al, 1999; Tamura et al, 1981). All animals survived until the end of the experiment and exhibited cerebral infarction. The left cerebral hemisphere of MCA-occluded rats treated with vehicle (saline) alone for 28 days exhibited conspicuous cortical infarction (Figure 2A). When 6 or 60 μg/day gRb1 was intravenously infused after permanent MCAO for 28 days, the infarct was markedly reduced in size (Figure 2B). It is possible that intravenous infusion of gRb1 rescued many neurons in the ischemic penumbra. The proportion of infarct size in the group treated with gRb1 (6 or 60 μg/day) was significantly smaller than that in the vehicle-treated group (Figure 2C). Nissl staining of saline-infused brain showed shrinkage and loss of neurons in the VP thalamic nucleus in comparison with thalamic neurons in sham-operated animals, whereas in gRb1-treated ischemic brain, the majority of thalamic neurons appeared to be intact (Figures 2D to 2F). There was a significant difference in neuron number in the VP thalamic nucleus between the saline-infused ischemic group and the gRb1-treated ischemic group (Table 1). As shown in Figure 2G and 2H, postischemic intravenous infusion of gRb1 at a dose of 6 or 60 μg/day significantly decreased the escape latency on repeated trials of the Morris water maze test, especially on the fourth trial day at 2 weeks after MCAO and on the first, third and fourth trial days at 4 weeks after MCAO. There was no significant difference in swimming speed among the experimental groups (data not shown). Moreover, intravenous gRb1 infusion did not affect cerebral blood flow, as monitored by laser Doppler flowmetry (Omega flow FLO-N1, Neuroscience Inc., Tokyo, Japan) (data not shown). These findings suggest that intravenous infusion of gRb1 after permanent MCAO prevents place navigation disability, cortical infarction and secondary thalamic degeneration in SHR-SP. Surprisingly, the neuroprotective effect of intravenously infused gRb1 at a dose of 6 μg/day was much more potent than that of intracerebroventricularly infused gRb1 at the same dose, and it was as potent as the neuroprotective effect of intracerebroventricularly infused gRb1 at a dose of 0.6 μg/day. Moreover, intravenous infusion of gRb1 ameliorated ischemia-induced neuronal damage over a wider dose range than intracerebroventricular infusion of this agent (Zhang et al, 1998). The neuroprotective effects of gRb1 appeared to be ascribable, at least in part, to prevention of neuronal apoptosis, which is known to occur in the ischemic penumbra (Ferrer and Planas, 2003).
Left-to-right ratio of thalamic area and VP thalamic neuron number
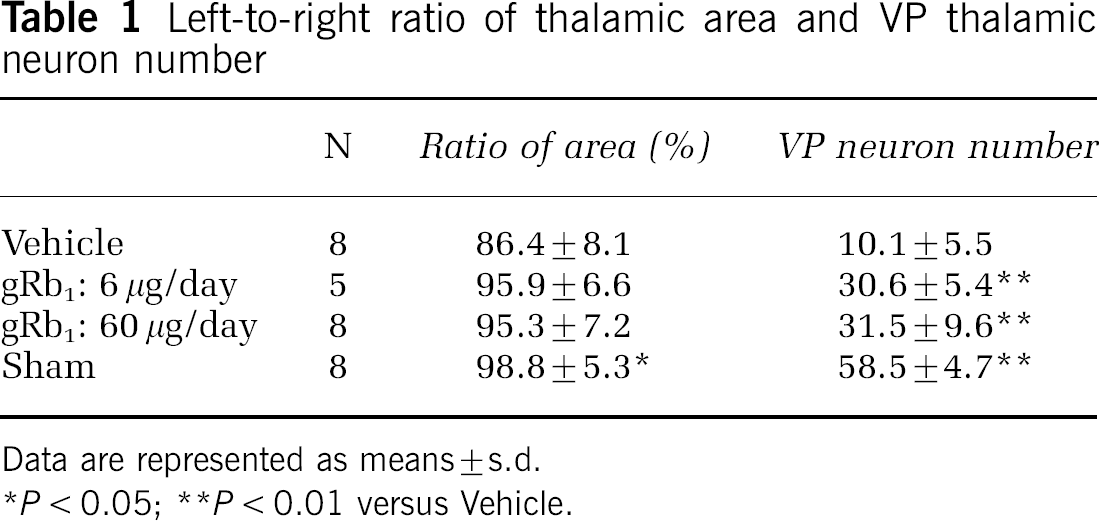
Data are represented as means ± s.d.
P < 0.05
P < 0.01 versus Vehicle.
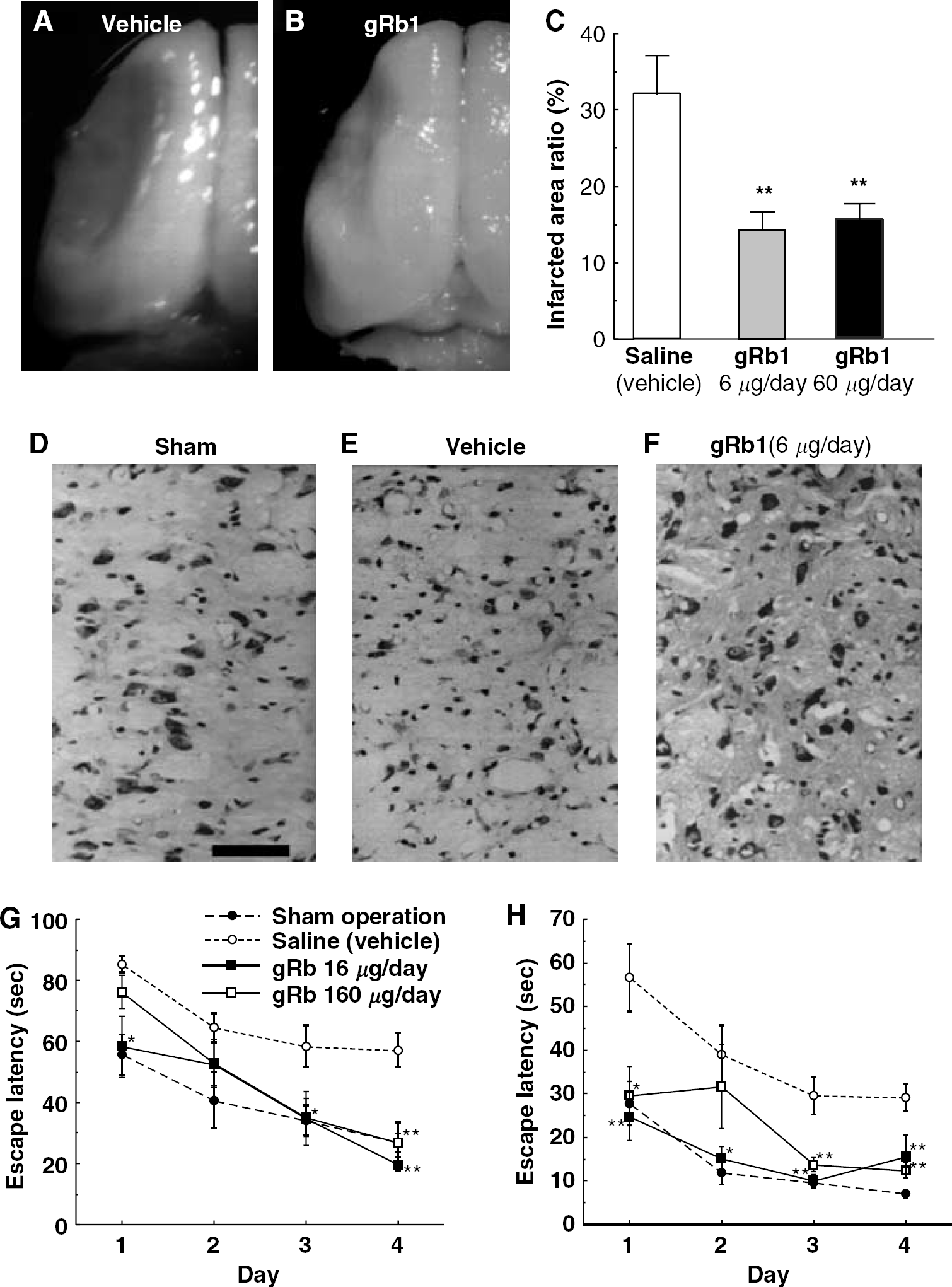
Intravenous infusion of gRb1 ameliorates place navigation disability, cortical infarct size, and secondary thalamic degeneration in SHR-SP with permanent MCAO. (
Intravenous Infusion of Ginsenoside Rb1 Results in Reorganization of Disrupted Cerebrovascular Networks in Ischemic Penumbra
We next investigated whether the cerebrovascular networks disrupted by permanent MCAO were reorganized in the putative ischemic penumbra, which was rescued by intravenous infusion of gRb1. Paraffin sections from gRb1-treated brains around the level 2.8 mm posterior to the bregma were examined with Nomarski optics (Figure 3A), and vascular (blood vessel) area per 1.27 mm2 in the rescued parietal cortex 300 μm away from the site of marked gliosis in the periphery of the cortical infarct was compared with that in the counterpart on the nonlesioned (contralateral) side (Figures 3B and 3C). As shown in Table 2, there was no significant difference in vascular area between the sides. Although clear demarcation of penumbra was not defined in this study, it is likely that 28-day intravenous infusion of gRb1 after permanent MCAO resulted in almost complete reorganization of cerebrovascular networks in the putative ischemic penumbra rescued. For these reasons, treatment of cerebral infarction with intravenous infusion of gRb1 appeared to be complete by 28 days after permanent MCAO, and, hopefully, termination of gRb1 treatment at this time would no longer lead to aggravation of neuronal damage in the MCA-occluded brain.
Ratio of cerebrovascular area to 1.27 mm2 brain area
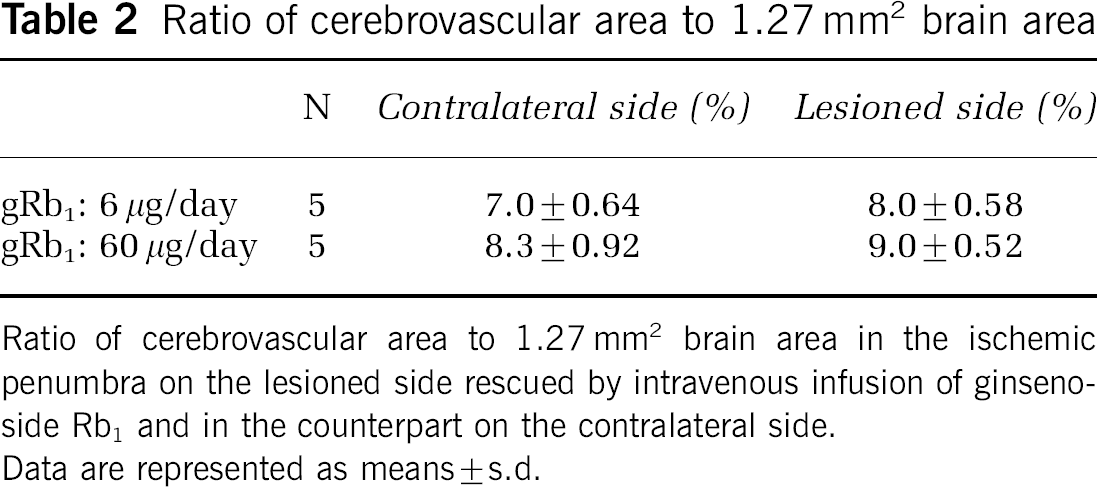
Ratio of cerebrovascular area to 1.27 mm2 brain area in the ischemic penumbra on the lesioned side rescued by intravenous infusion of ginsenoside Rb1 and in the counterpart on the contralateral side.
Data are represented as means ± s.d.
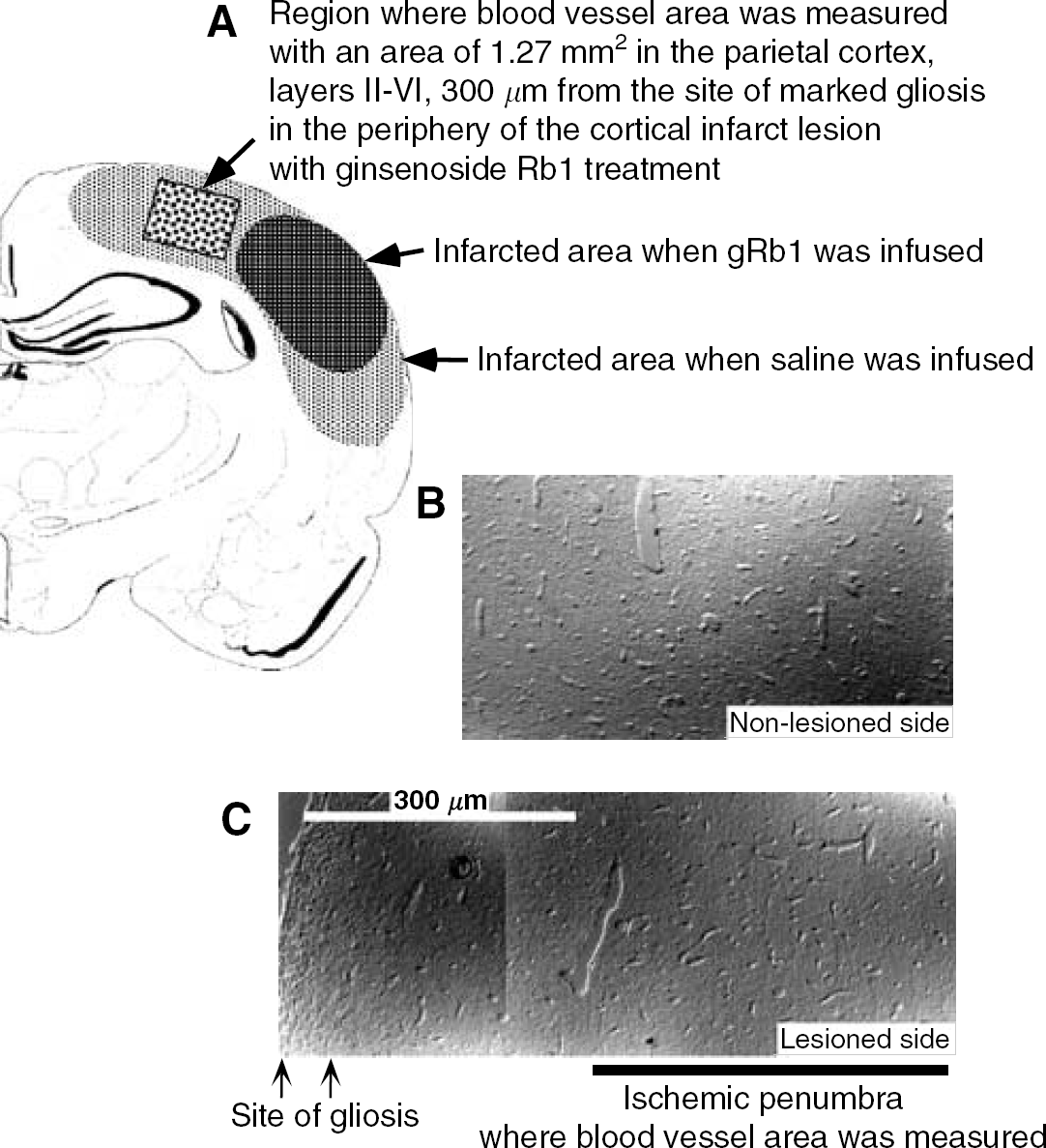
Cerebrovascular networks disrupted by permanent MCAO are reorganized in the putative ischemic penumbra, which was rescued by intravenous infusion of gRb1. (
Assessment of Therapeutic Time Window for Neuroprotective Effect of Ginsenoside Rb1
After confirming the reorganization of cerebrovascular networks, we investigated the effect of gRb1 in the situation where its first administration was delayed for 2 h after MCAO. As shown in Figure 4, a significant reduction in total infarct volume was observed when the treatment was delayed until 2 h after MCAO and gRb1 was administered at low doses (6 or 60 μg/day). However, this protective effect of gRb1 was not evident when administered at high doses (3 or 12 mg/day). In addition, as shown in Table 3, there were no significant differences in MABP and brain temperature among three groups (0, 6 μg/day, 12 mg/day).
Physiological parameters
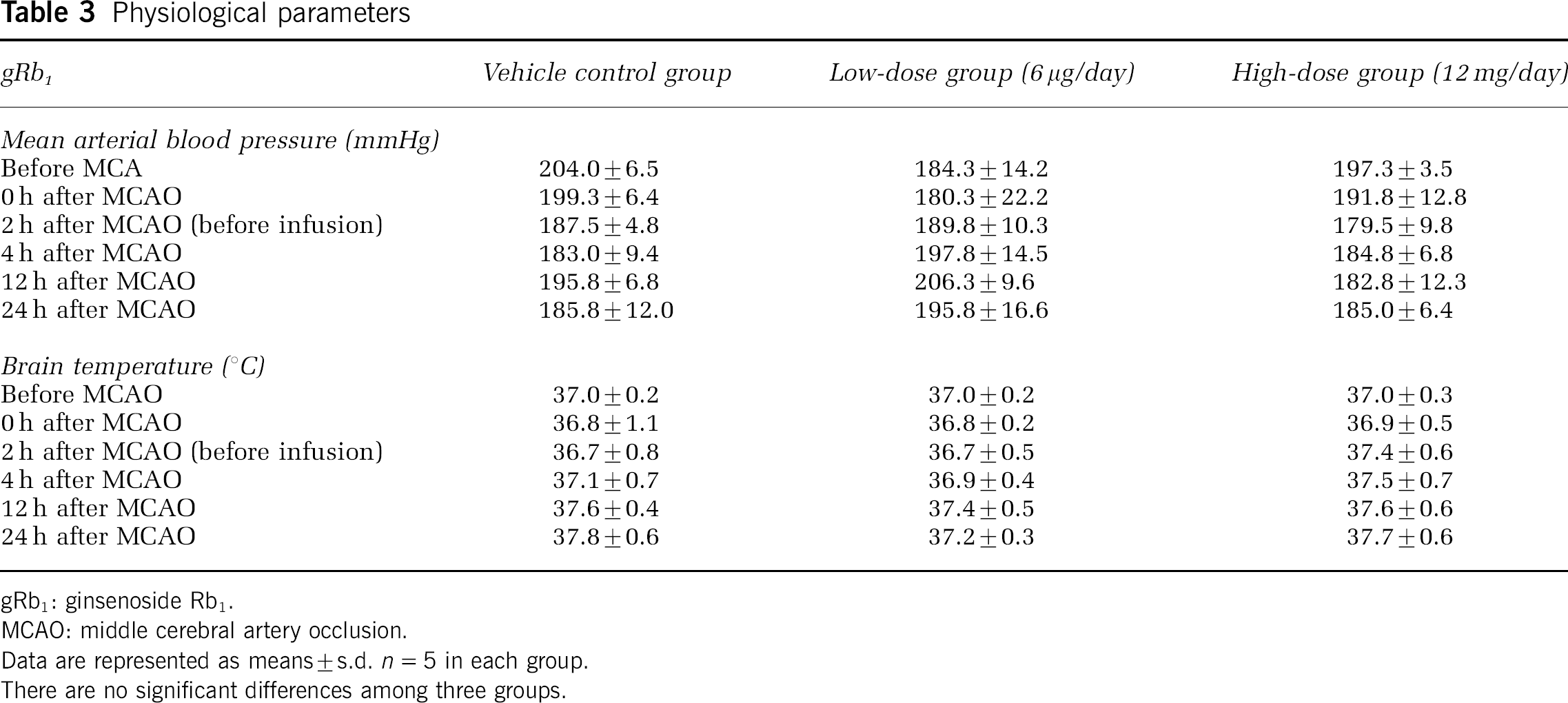
gRb1: ginsenoside Rb1.
MCAO: middle cerebral artery occlusion.
Data are represented as means ± s.d. n = 5 in each group.
There are no significant differences among three groups.
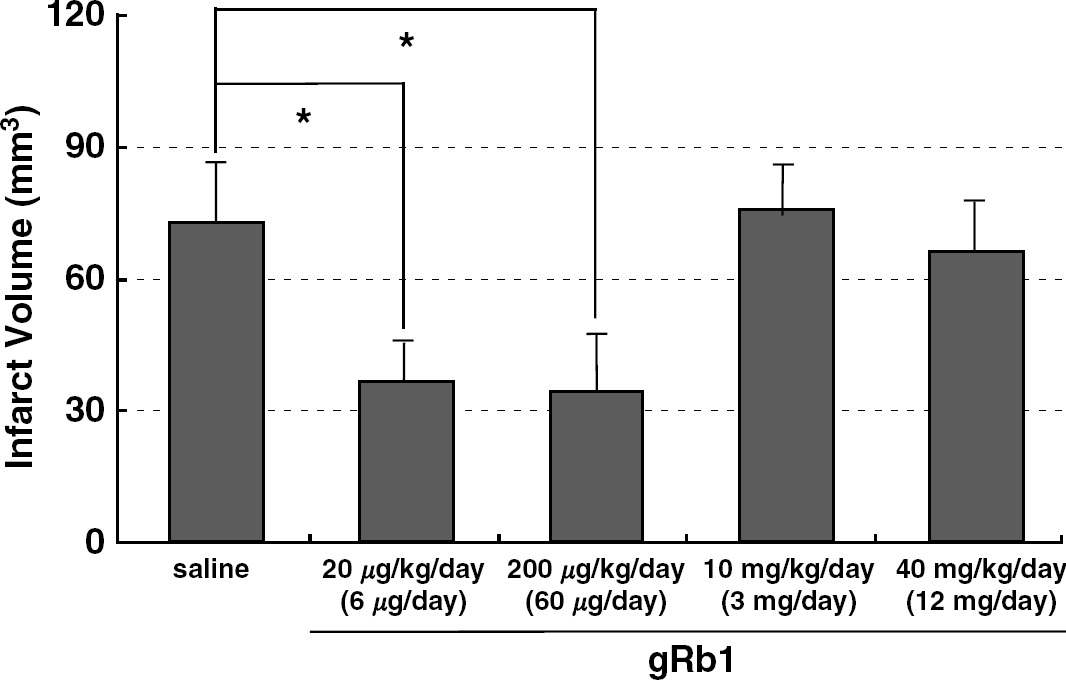
Effect of gRb1 administered at 2 h after permanent MCAO on total infarct volume in SHR-SP. Vehicle or gRb1 was administered at a predetermined dose (6, 60, 3000 or 12,000 μg/day). Note that gRb1 at doses of 6 and 60 μg/day significantly reduced the total infarct volume. N = 7 per group.
Ginsenoside Rb1 Prevents Apoptotic Cell Death of Neurons In Vitro
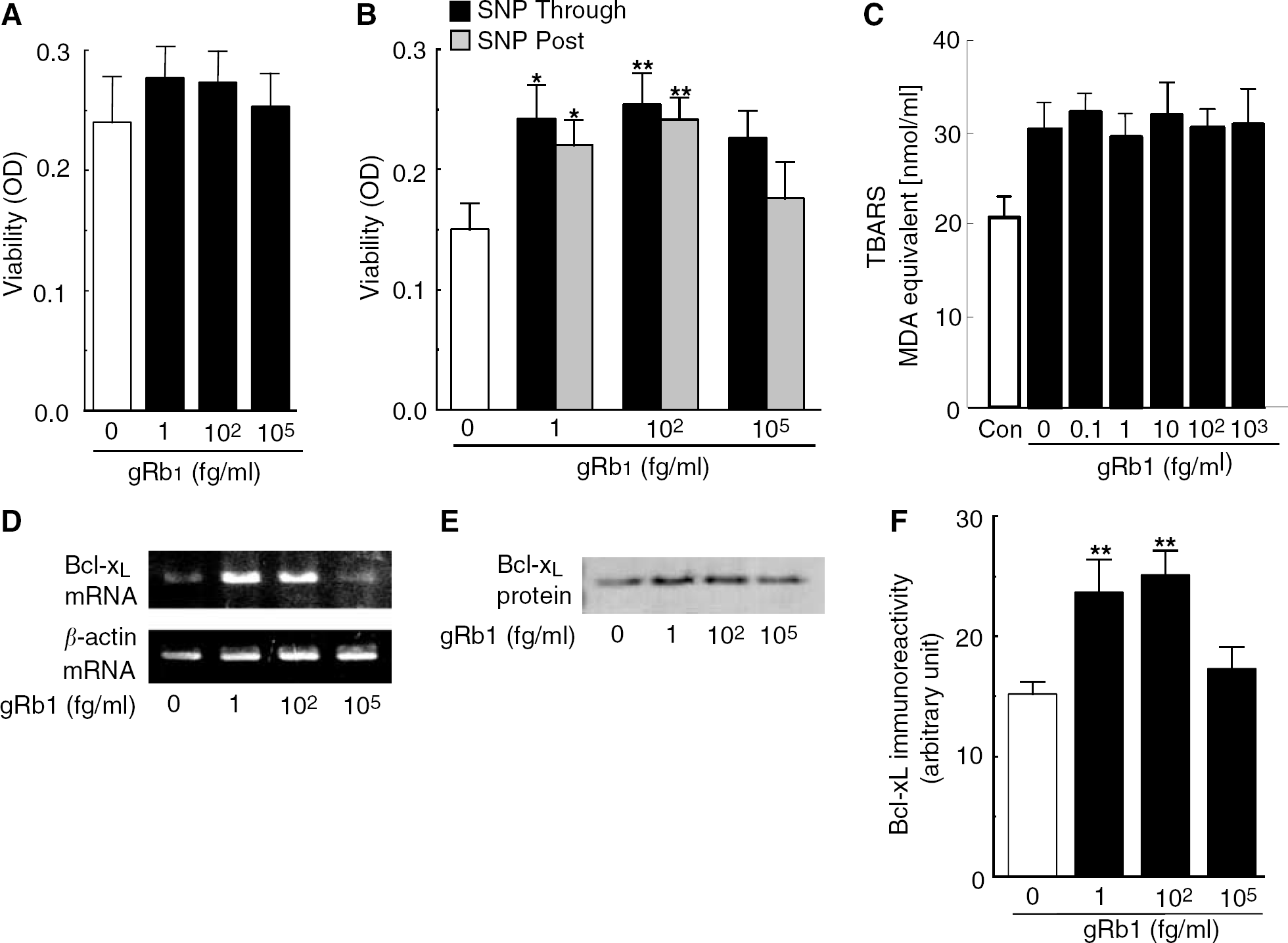
To confirm the antiapoptotic action of gRb1, we investigated whether gRb1 prevented neuronal apoptosis, which was induced by 10-min exposure of cultured neurons to the NO donor SNP, using alamar blue assay (Toku et al, 1998). Ginsenoside Rb1 at concentrations of 1 to 105 fg/mL did not affect the viability of control cultured cortical neurons without SNP treatment (Figure 5A). When cortical neurons were exposed to 100 μmol/L SNP for 10 mins, many of them underwent apoptosis within 16 h (Figure 5B), as described by Toku et al (1998). However, when cortical neurons were incubated with 1 or 100 fg/mL gRb1 before, during and after SNP treatment (indicated as ‘Through’ in Figure 5B), cell viability was maintained at a level close to that of control cortical neurons not exposed to SNP (Figure 5A). Furthermore, 1 or 100 fg/mL gRb1 was similarly effective in rescuing cortical neurons when added to neuronal cultures only after SNP treatment (indicated as ‘Post’ in Figure 5B). The antiapoptotic action of gRb1, which was added to the cultured neurons after SNP treatment, implies that gRb1 activates intracellular signaling to promote neuronal survival rather than acting as a scavenger of NO per se. This speculation was further reinforced by the finding that gRb1 at concentrations of 1 to 1000 fg/mL did not significantly attenuate membrane lipid peroxidation induced by the hydroxylradical-promoting agent (FeSO4, although all values of TBARS were significantly increased after FeSO4 treatment (Figure 5C). Furthermore, as shown in Table 4, gRb1 did not exhibit DPPH radical scavenging activity at concentrations lower than 100 mg/mL. Thus, gRb1 added to cultured neurons either before or after SNP treatment prevents neurons from undergoing apoptosis, and the present in vitro experiments do not support the previous notion that gRb1 protects ischemic neurons by suppressing oxygen-free radical-induced lipid peroxidation (Lim et al, 1997).
Effects of ginsenoside Rb1 (gRb1) on DPPH radical
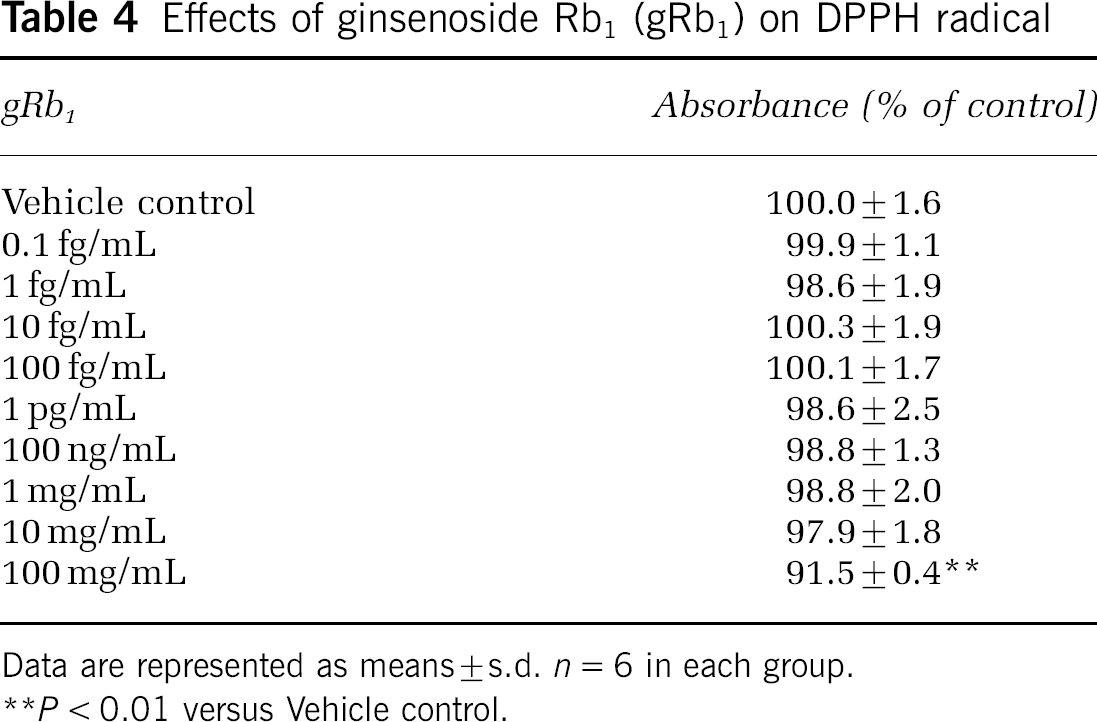
Data are represented as means ± s.d. n = 6 in each group.
P < 0.01 versus Vehicle control.
Prevention of NO-induced neuronal apoptosis by gRb1 through upregulation of Bcl-xL expression. (
Ginsenoside Rb1 Upregulates Expression of Antiapoptotic Factor Bcl-xL In Vitro
To gain an insight into the molecular mechanisms underlying gRb1-mediated neuronal survival, we investigated gRb1-induced changes in the expression of Bcl-2, Bcl-w, Bcl-xL, Bcl-XS, apoptosis activating factor-1 (Apaf1), Akt, Bax, Bak, Bad, Fas, Fas ligand, P53, P21 and GADD45 mRNA in neurons, using RT-PCR. Among the apoptosis-regulating factors examined, Bcl-xL mRNA expression was markedly upregulated by treatment with gRb1 at concentrations of 1 to 100 fg/mL, which were the optimal concentrations for gRb1 to exhibit an antiapoptotic action. Reverse transcriptase-polymerase chain reaction using specific primers that amplify a 189-bp fragment of rat Bcl-xL mRNA detected a PCR product of the expected size, and its identity was confirmed by direct sequencing. As shown in Figure 5D, cortical neurons treated for 24 h with 1 or 100 fg/mL gRb1 showed an increase in Bc1-xL mRNA expression compared with the gene expression of control cortical neurons. Densitometric analysis showed that neurons cultured in the presence of 1 or 100 fg/mL gRb1 expressed approximately 6 times as much Bcl-xL mRNA as that expressed in neurons without gRb1 treatment. Neurons cultured with 105 fg/mL gRb1 did not exhibit any detectable change in Bcl-xL mRNA expression. In addition, we conducted Western blot using an antibody against Bcl-xL protein. Bcl-xL protein with a molecular mass of approximately 29 kDa was constitutively expressed in cultured neurons (Figure 5E). Consistent with the increase in Bcl-xL mRNA expression, Bcl-xL protein expression was clearly increased in neurons cultured for 48 h in the presence of 1 or 100 fg/mL gRb1 (Figure 5E). Densitometric analysis of the immunoreactive bands revealed that gRb1 treatment caused a 50% increase in Bcl-xL protein expression in cortical neuron cultures (Figure 5F). These findings suggest that gRb1 prevents neuronal apoptosis through upregulation of the antiapoptotic factor, Bcl-xL. To our knowledge, gRb1 is the only nonpeptide agent that facilitates neuron survival by upregulating Bcl-xL protein expression.
Induction of Bcl-xL mRNA by Ginsenoside Rb1 is Stat5 Dependent
To determine whether induction of Bcl-xL mRNA by gRb1 is actually dependent on activation of the transcription factor signal transducer and activator of transcription 5 (Stat5), we constructed bcl-x promoter-luciferase plasmids carrying a wild-type STRE (bcl-x-wt) or a mutant STRE (bcl-x-mut), and a plasmid that does not carry STRE (bcl-x-0.6L) (Figure 6A). Primary cultured neurons were transfected with these promoter-reporter plasmids and incubated for 24 h in the presence or absence of gRb1 (100 fg/mL). Then, luciferase activity in the cell lysate was assayed. Consequently, luciferase activity in gRb1-treated neurons was significantly higher than that in untreated neurons when the cells were transfected with bcl-x-wt plasmid (2.7-fold; P < 0.01; Figure 6B). When bcl-x-mut or bcl-x-0.6L plasmid was used for transfection, there was no significant difference between gRb1-treated neurons and untreated neurons (Figure 6B). These findings indicate that Stat5 responsive element on the bcl-xL promoter is responsible for the induction of bcl-xL mRNA by gRb1 in cultured neurons.
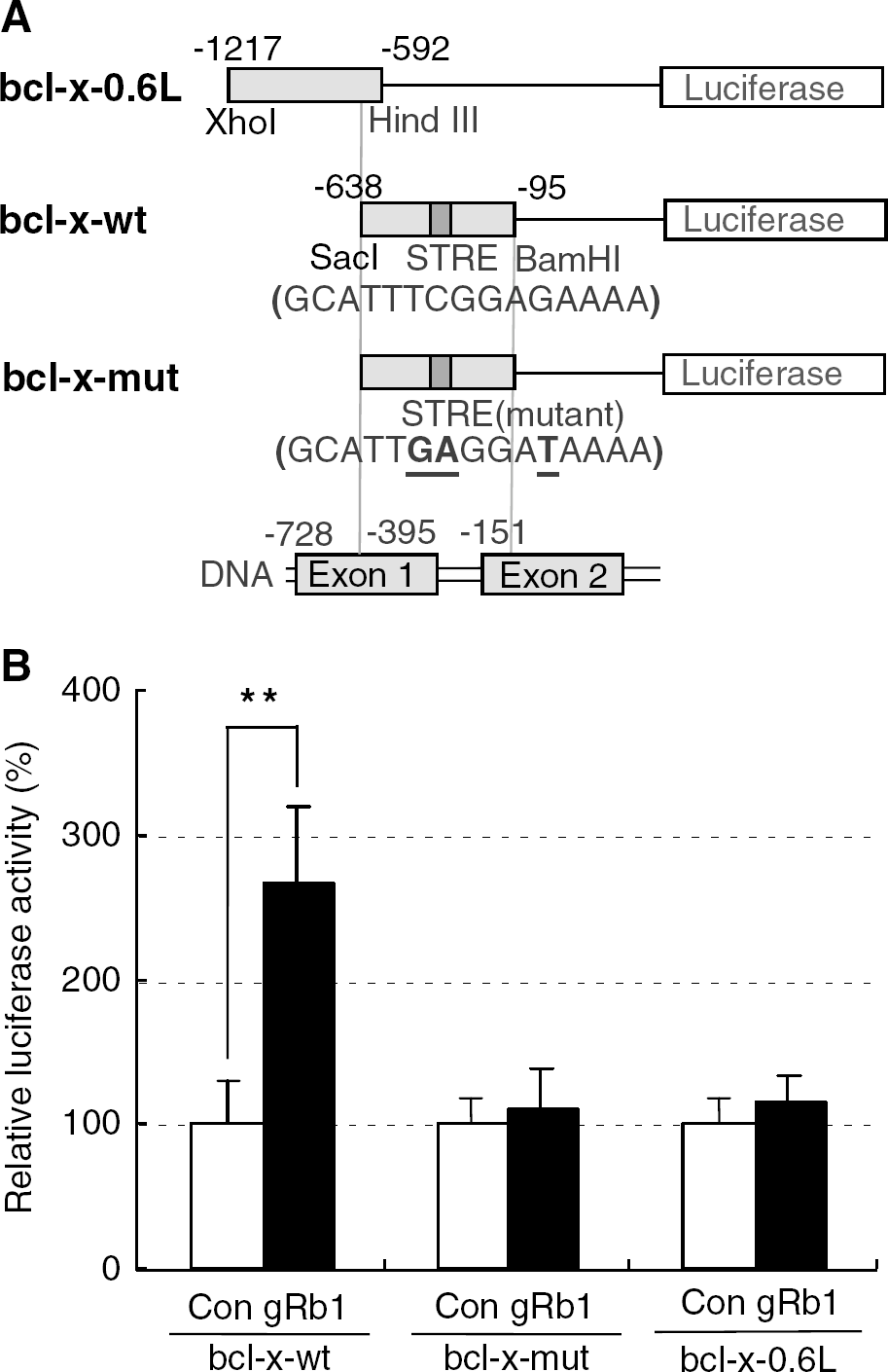
Transcriptional activation of luciferase reporter construct driven by bcl-x promoter in response to gRb1 treatment. (
Ginsenoside Rb1 Prevents Delayed Neuronal Death and Induces Bcl-xL Protein In Vivo
We next attempted to confirm that gRb1 prevents delayed neuronal death and upregulates Bcl-xL protein expression in vivo. We used a gerbil model with transient forebrain ischemia of 3-min duration. Gerbils are known to show a loss of nearly one half of hippocampal CA1 neurons 7 days after 3-min ischemia (Morita et al, 2001; Sakanaka et al, 1998), and, moreover, many of the surviving CA1 neurons in the same period exhibit TUNEL-positive reactions. We investigated whether intracerebroventricular gRb1 infusion (60 or 600 ng/day), starting after 3-min forebrain ischemia, reduced the number of TUNEL-positive neurons in the hippocampal CA1 field. As described before (Morita et al, 2001; Wen et al, 1998), many TUNEL-positive neurons were present in the hippocampal CA1 field of 3-min ischemic gerbils with saline infusion 7 days after the ischemic insult (Figure 7A), suggesting that irreversible neuronal degeneration was in progress during this period. Intracerebroventricular infusion of gRb1 caused a significant decrease in the number of TUNEL-positive neurons in a dose-dependent manner (Figures 7B to 7D). Moreover, the CA1 field of gRb1 (600 ng/day)-infused gerbils, when investigated with Western blot, contained a larger amount of Bcl-xL protein than that of vehicle (saline)-treated gerbils at 2 days after 3-min forebrain ischemia (Figures 7E and 7F). These findings suggest that intracerebroventricular infusion of gRb1 prevents delayed neuronal death in vivo through upregulation of Bcl-xL protein expression.
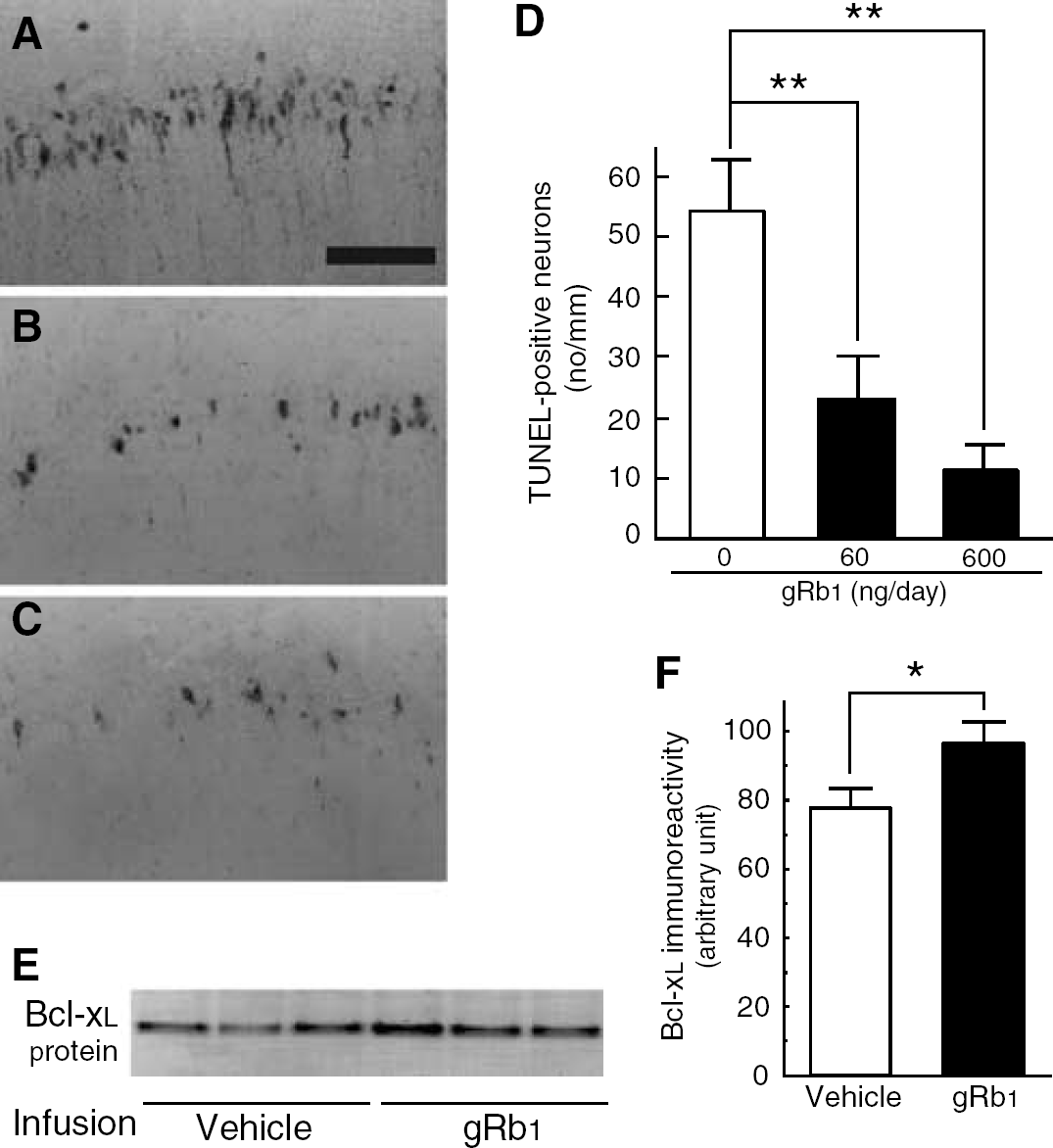
Intracerebroventricular infusion of gRb1 prevents apoptotic neuron death in hippocampal CA1 field of gerbils with transient forebrain ischemia of 3-min duration. (
Intravenous Infusion of Ginsenoside Rb1 Upregulates Bcl-xL mRNA Expression in Middle Cerebral Artery-Occluded Brain
To confirm that intravenous infusion of gRb1 induces Bcl-xL expression in the ischemic brain, we next investigated Bcl-xL mRNA expression within the cerebral cortex on the lesion side at 12 h after gRb1 (6 or 60 μg/day) or vehicle (saline) infusion in MCA-occluded rats, using RT-PCR under semiquantitative conditions. The PCR product showed the expected size, and its identity was confirmed by direct sequencing. The lesioned cerebral cortex of MCA-occluded rats with intravenous infusion of gRb1 exhibited higher Bcl-xL mRNA expression than that of rats with vehicle infusion (Figure 8A). As shown in Figure 8B, Bcl-xL mRNA expression in the gRb1-treated group was significantly (approximately 3 times) higher than that in the untreated group. These data clearly show that intravenous infusion of gRb1 upregulated Bcl-xL mRNA expression in the MCA-occluded rat brain.
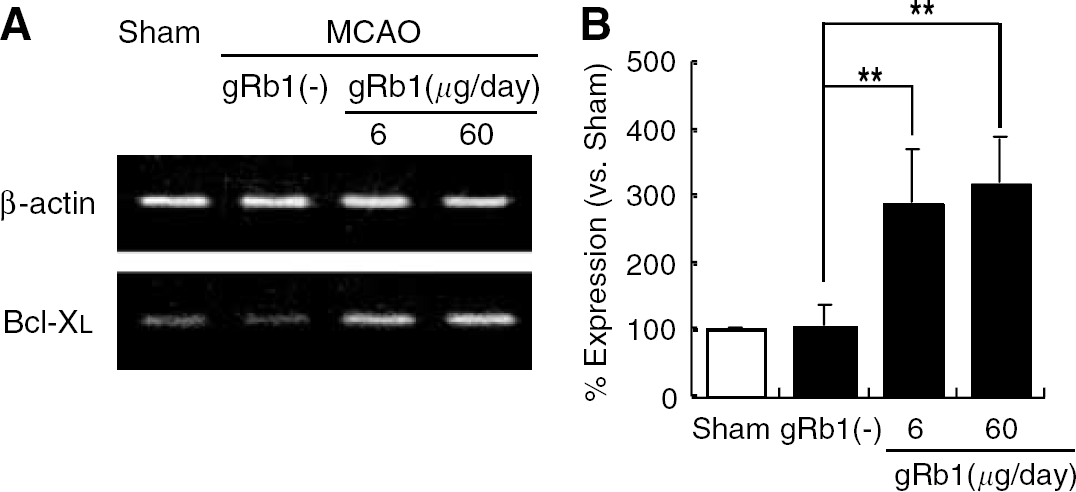
Intravenous infusion of gRb1 upregulates Bcl-xL mRNA expression in the ischemic cortex of the MCA-occluded rat. (
Discussion
In the present study, we showed that gRb1 upregulated the expression of Bcl-xL, both in vivo and in vitro. Moreover, we revealed that a Stat5 responsive element in the bcl-x promoter became active in response to gRb1. This was confirmed by the findings that mutagenesis or deletion of this sequence motif abrogated its promoter activity. Bcl-xL protein can act upstream of mitochondria in the initiation of apoptosis and inhibit the mitochondrial cell death signaling pathway, suggesting that Bcl-xL is situated in the center of neuron survival signaling via mitochondria. Therefore, we speculate that upregulation of Bcl-xL protein by gRb1 is a safer and more practical way of neuroprotection than caspase inhibition, if the mitochondrial cell death signaling pathway becomes irreversible after liberation of Apaf1 from Bcl-xL. In fact, our study revealed that gRb1 rescued cortical neurons in the ischemic penumbra and reduced the volume of the cortical infarct by approximately 50%, without any noticeable adverse effects. In addition, we showed the reorganization of vascular networks in the ischemic penumbra after the treatment of gRb1. Although clear demarcation of ischemic penumbra was not defined in this study, it is likely that the reorganization of vascular networks in the putative ischemic penumbra also plays an essential role in the effects of gRb1 on neural injury. Further investigation must be required to clarify the neuroprotective machanism of gRb1.
Previously, we showed that gRb1 significantly prevents delayed neuronal death in the hippocampal CA1 field of the gerbil when injected intraperitoneally for 7 days before transient forebrain ischemia (Lim et al, 1997; Wen et al, 1996). However, intraperitoneal injections of gRb1, starting immediately after transient forebrain ischemia, do not rescue hippocampal CA1 neurons from delayed neuronal death (Lim et al, 1997; Wen et al, 1996). Presumably, it takes a considerable time for intraperitoneally injected gRb1 to reach the brain, and thus, postischemic gRb1 injections into the peritoneal cavity cannot protect ischemic hippocampal CA1 neurons in the critical periods when neuronal death or survival is determined. In support of this speculation, intracerebroventricular infusion of gRb1 starting immediately after transient forebrain ischemia rescued a significant number of hippocampal CA1 neurons from lethal damage (Lim et al, 1997). Using SHR-SP with permanent MCAO, we previously showed that intracerebroventricular gRb1 infusion starting just after permanent MCAO ameliorates ischemia-induced place navigation disability and cortical infarction (Zhang et al, 1998). These findings suggest that centrally, but not peripherally, infused gRb1 exerts a curative effect on ischemic neuronal damage. Before conducting the current experiments, we speculated that it is extremely difficult to apply gRb1 for the treatment of cerebral infarction, because (1) the effective dose range of gRb1 infused intracerebroventricularly after MCAO is quite narrow; only 0.6 μg/day (but neither 6 nor 0.06 μg/day) gRb1 is effective in ameliorating cortical infarct size and place navigation disability in MCA-occluded rats, and (2) intracerebroventricular infusion of gRb1 is not a practical way for drug delivery in clinical medicine. In the present study, we showed that postischemic intravenous infusion of gRb1 ameliorates, in a wide dose range (6 to 60 μg/day), place navigation disability and cortical infarct size in SHR-SP with permanent occlusion of the MCA distal to the striate branches. It is not always easy to state with certainty why intravenous but not intracerebroventricular infusion of gRb1 exhibits a wide range of effective doses. A possible explanation for this is that the blood brain barrier regulates the central adsorption of gRb1 from the blood stream, so that an optimal amount of gRb1 is transported into the ischemic brain. Presumably, the high stability of gRb1, having both hydrophilic and hydrophobic chemical structures, appears to enable such elegant regulation. Since gRb1 has no site for hapten binding, it is difficult to raise antibodies against gRb1. Moreover, labeling of gRb1 with radioisotopes is not possible at this time, because it cannot be artificially synthesized. Nevertheless, a specific antibody with high sensitivity to gRb1, if produced in the future, would clarify the central distribution and levels of intravenously infused gRb1 in vivo.
To clarify the neuroprotective mechanisms of gRb1, we focused on upregulation of Bcl-xL gene expression by gRb1, because (1) the Bcl-xL gene product is a mitochondrion-associated protein that acts as a key molecule facilitating neuron survival (Polster and Fiskum, 2004); (2) it is abundant in mature neurons of the adult brain in which Bcl-2 expression declines to very low levels (Gonzalez-Garcia et al, 1995); (3) the expression of Bcl-xL protein is markedly increased in neurons close to the ischemic penumbra (Isenmann et al, 1998; Wen et al, 1998); (4) the size of infarcts resulting from permanent occlusion of the MCA was not affected by overexpression of Bcl-2, but was reduced by overexpression of Bcl-xL (Wiessner et al, 1999); and (5) IL-3 and erythropoietin, which have protective actions similar to those of gRb1 on the ischemic hippocampus and on cultured neurons treated with FeSO4, are known to rescue damaged neurons through upregulation of Bcl-xL gene product (Wen et al, 2002, 1998).
Bcl-xL protein is known to suppress activation of procaspase 9 by forming a complex with Apaf1 and to prevent the release of cytochrome c from mitochondria, thereby maintaining cell viability and cell survival. Bcl-xL protein is also known to prevent the release of second mitochondrial activation of caspases (Smac)/direct inhibitor of apoptosis binding protein with low pI (DIABLO) from mitochondria. Second mitochondrial activation of caspases /DIABLO antagonizes X-linked inhibitor of apoptosis protein (XIAP), which blocks the second proteolytic step for full caspase-3 activation (Sprick and Walczak, 2004). Furthermore, the redistribution of apoptosis-inducing factor (AIF) from mitochondria to the nucleus is prevented by Bcl-xL, leading to inhibition of cell death signaling through a caspase-independent pathway (Ferrer and Planas, 2003). In the ischemic hippocampus, mismatch between Bcl-xL mRNA and protein expression is noted in the early postischemic period (Wen et al, 1998), and insufficient translation of Bcl-xL protein in response to the ischemia-induced increase in Bcl-xL mRNA appears to liberate Apaf1 and cytochrome c, which form a complex with procaspase 9, leading to activation of procaspase 9 and caspase 9, and then to activation of the cell executioner, caspase 3 (Love, 2003). Moreover, neuronal death after mild focal ischemia in mice has been shown to be alleviated by intracerebroventricular injection of caspase inhibitors (Endres et al, 1998). It has been reported that a novel small peptidomimetic oxoazepinoindoline caspase inhibitor that crosses the blood–brain barrier reduced the infarct size after both transient and permanent focal brain ischemia, even when administered intravenously (Deckwerth et al, 2001). These findings are in favor of the notion that the mitochondrial cell death signaling pathway is involved in the occurrence of ischemic neuronal death, particularly within the hippocampal CA1 field and ischemic penumbra. Although the most effective way to treat ischemic neuronal damage might be to suppress caspase activity in damaged neurons, the neuroprotective effects of caspase inhibitors have been verified only up to 7 days after mild focal ischemia in mice (Endres et al, 1998). In other words, it cannot be ascertained that the temporary suppression of caspase activity by intracerebroventricular infusion of caspase inhibitors eventually rescues ischemic neurons that are committed to die by caspase activation; the damaged neurons may undergo delayed degeneration as the caspase inhibitors are degraded. In fact, one note of caution comes from the observation that intracerebroventricular infusion of a caspase inhibitor prevented delayed loss of neurons from the CA1 field of the hippocampus after ischemia, but did not prevent impairment of induction of long-term potentiation (Gillardon et al, 1999). The implication is that interference with caspase could lead to the preservation of damaged, malfunctioning neurons.
There have been many in vitro studies dealing with the PI3K-Akt-Bad pathway affecting the antiapoptotic function of Bcl-xL (Love, 2003). The level of activated Akt increased within hours after the onset of ischemia and subsequently declined (Noshita et al, 2001). Infarct volume after MCAO was significantly reduced by overexpression of the active form of Akt under the control of the damage-induced neuronal endopeptidase (DINE) promoter (Ohba et al, 2004). However, phosphorylation of Bad was not detected after cerebral ischemia, despite an increase in the level of phosphorylated Akt (Friguls et al, 2001). The precise mechanism of the PI3K-Akt-Bad pathway after ischemia remains unclear. In addition to the mitochondrial cell death signaling pathway, the death receptor-mediated signaling pathway might also be involved in neuronal cell death after ischemia. One of the most investigated death receptors after cerebral ischemia is CD95/Fas. Although early studies showed that CD95/Fas was not expressed in the adult brain (Siesjo et al, 1999), recent studies showed that CD95/Fas was expressed in the brain after cerebral ischemia (Jin et al, 2001; Padosch et al, 2003). Infarct size resulting from focal cerebral ischemia is greatly reduced in lymphoproliferation mutant (lpr) mice expressing dysfunctional Fas (Martin-Villalba et al, 1999). At present, we do not know much about the interactions between gRb1 and CD95/Fas or Akt. Thus, it is expected that further studies on the intracellular survival signals stimulated by the potent neuroprotectant, gRb1, will lead to elucidation of the molecular mechanisms underlying ischemic neuronal death.
Another advantage of gRb1 might be that it does not affect cell viability when added to control neuronal cultures, despite its potent antiapoptotic action on SNP-treated neurons. Furthermore, intracerebroventricularly infused gRb1 has been shown to exert no effects on cerebral blood flow, brain temperature or MABP (Lim et al, 1997; Zhang et al, 1998), and intravenous gRb1 infusion did not affect brain temperature or MABP in the present study. These findings suggest that gRb1 rescues ischemic neurons without affecting physiological parameters or possibly neural transmission. Since ginseng has been taken by many people in Asian countries for thousands of years, gRb1 as an ingredient of ginseng is expected to cause few adverse effects in humans. It is tempting to speculate that intravenous infusion of gRb1 could be applied in patients with acute cerebral stoke in the ambulance before diagnosis of the type of stroke by computed tomography. Ginsenoside Rb1, which upregulates Bcl-xL at extremely low concentrations, might also be used for the treatment of neurodegenerative diseases, ischemia–reperfusion injury, graft-versus-host disease and autoimmune disorders involving activation of mitochondrial cell death signaling.
In conclusion, the present experimental results introduce a new concept of gRb1-mediated general cytoprotection to biomedical research. The precise intracellular mechanism(s) by which gRb1 induces Bcl-xL protein production remains to be determined, but the present study seems to open a new horizon in research on intravenously infusible neuroprotective agents that may rescue millions of patients with diseases of the brain, generation after generation.
Footnotes
Acknowledgements
We are grateful to Drs T Suzuki and Y Ohta for providing stroke-prone spontaneously hypertensive rats and to Drs K Ikoma and H Matsuda for their encouragement and valuable suggestions throughout this work. We are also grateful to Korea Ginseng Corporation for the generous provision of Panax Ginseng CA MEYER. The secretarial assistance of Ms K Hiraoka is acknowledged.