
Abstract
Immunosuppression commonly occurs after a stroke, which is believed to be associated with the increased risk of infectious comorbidities of stroke patients, while the mechanisms underlying post-stroke immunosuppression is yet to be elucidated. In the brains of intracerebral hemorrhage (ICH) patients and murine ICH models, we identified that neuron-derived programmed death-ligand 1 (PD-L1) is reduced in the perihematomal area, associating increased soluble PD-L1 level in the peripheral blood. ICH induced a significant decrease of T and natural killer (NK) cell numbers in the periphery with an upregulation of programed death-1 (PD-1) in these cells. Blocking PD-1 pathway with an anti-PD1 monoclonal antibody prevented the T and NK cell compartment contraction and spleen atrophy post-ICH, with reduced pulmonary bacterial burden and improved neurological outcome. Thus, we here identified that brain-derived PD-L1 as a new mechanism driving post-stroke immunosuppression, and anti-PD1 treatment could be potentially developed to reducing the risk of post-stroke infections.
Keywords
Introduction
Immunosuppression commonly occurs in the aftermath of severe injuries, as seen in stroke, trauma, or burns; post-injury immunosuppression is characterized by the loss of lymphocytes, especially T cells and natural killer (NK) cells, and atrophy of lymphoid organs like the spleen.1–4 Immunosuppression weakens the natural defense to microbes, increasing the risk of post-injury infections. Post-stroke infection is a major cause of death; nevertheless, the mechanisms underlying post-stroke immunosuppression remains elusive. Stress responses, mediated by increased sympathetic innervation and activation of the hypothalamic-pituitary-adrenal axis are major contributors to post-stroke immunosuppression.3,5,6 However, approaches in modulating neurogenic signals to prevent post-stroke infections in clinical practice face translational difficulty due to a variety of factors such as the broad targets of neurotransmitters, especially on the cardiovascular system, might abolishing any benefit of neurogenic pathway blockers.
The alternative strategy of targeting humoral signals that are involved in inducing post-stroke immunosuppression maybe a more feasible approach. Programed death-ligand 1 (PD-L1) is a membrane-bound protein that exerts immune checkpoint roles by binding to programed death-1 (PD-1) receptor; PD-L1 has long been known for its involvement in escaping immune surveillance of tumor cells.7,8 Recent report identified PD-L1 released by tumor cells into the circulation to mediate systemic immunosuppression. 8 We here find that neurons of healthy humans and mice express high basal levels of PD-L1, which are significantly reduced in the perihematomal area upon hemorrhagic stroke. We propose that brain-derived PD-L1 released into the circulation contributes to peripheral immunosuppression. We further investigate the feasibility of anti-PD1 treatment in preventing post-ICH immunosuppression and infections.
Materials and methods
Human brain tissue
Brain tissues were obtained from ICH patients who received hematoma evacuation surgery post-ICH from Zhongda Hospital of Southeast University, China. Four patients had primary ICH syndromes 2–5 h before surgery, their bleeding volume was in the region of 35–50 ml. Informed consent was obtained from all subjects or from a legally acceptable surrogate at the time of enrollment. Control brain tissue slices of 4 natural death adults (2 male, 2 female) without history of neurological or inflammatory diseases were obtained from the brain bank of Beijing Tiantan Hospital. The brain tissues were embedded in paraffin and sectioned into 8 μm-thick slices. Patients study was conducted in accordance with the Declaration of Helsinki. The inclusion of human subjects was approved by the Ethics Committees of Tianjin Medical University General Hospital, Zhongda Hospital of Southeast University, and Beijing Tiantan Hospital. Informed consent was obtained from each patient or their legal surrogate.
Mice
C57BL/6 (B6) male mice, 3–4 months old and weighing 20–25 g, were purchased from Si Pei Fu (SPF, Beijing). We chose male mice to induce experimental ICH due to its high incidence in male population. All mice were assigned randomly in each experiment. Randomization was based on the random number generator function in computer. Tianjin Medical University General Hospital (Tianjin, China) supports a pathogen-free environment for this study. Mice were housed in cage with access to food and water ad libitum, and under a 12–12 h light-dark cycle environ. Mice number were limited to under 5 in each cage. All animal experiments were performed in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health 9 and reported following the ARRIVE (Animal Research: Reporting In Vivo Experiments) guidelines. 10 This study was approved by the Animal Care and Use Committee of Tianjin Medical University and Beijing Tiantan Hospital.
Induction of ICH models
ICH model was induced by collagenase IV (C8160, Solarbio) or autologous blood injection. Mice were anesthetized by intraperitoneal injection of ketamine/xylazine. In brief, following anesthesia, mice were fixed in prone position keeping the head parallel to the bottom of the stereo-locator. Hair on the top of the head was shaved and a 1 cm incision along the central line was made post iodophor disinfection. A 1 mm hole at the coordinates: 2.3 mm lateral and 0.5 mm anterior to the bregma was drilled by a drill bit, then 0.0375 U collagenase IV was injected into the brain parenchyma at a 0.5 μl/min rate by advancing a 10-μl Hamilton microsyringe with a 30-gauge needle 3.5 mm below the skull. The needle was held in the position for 10 min after injection to prevent backflow. For the autologous blood model, 30 µl non-heparinized autologous blood was acquired from the angular vein of the mouse and then injected to the same position in the brain as aforementioned in the collagenase model. With a modified infusion procedure, 5 μL blood was first infused at a rate of 1 μl/min at depth of 3 mm beneath the skull, thereafter, the needle was inserted 0.5 mm deeper and the remaining 25 μL blood was continuously injected at the same rate; the needle was kept in the position for 10 min after injection to prevent any backflow. Sham mice received identical surgery procedures as ICH but injected with equal volume of saline. Mice lacking hematoma or neurological deficit or that died during the experimental time were excluded from data analysis.
Evaluation of hematoma volume
Mice brains were collected at day 3 after ICH. The whole brain was sectioned into 1 mm slices serially. Image J software was used to evaluate the hematoma area of each brain slice. The total volume of hematoma for each brain were calculated by following formula: Hematoma volume (mm3) = hematoma area of each slice × the thickness of the brain slices with hematoma.
Brain water content
Brain water content were evaluated at day 3 after ICH. Mice brain tissue were dissected without perfusion and divided into contralateral hemisphere, ipsilateral hemisphere, and cerebellum parts. The wet weight of different brain part was recorded immediately after dissected and the dry weight was documented after dried at 100°C for 24 hours. Brain water content was calculated by using the following formula: (wet weight−dry weight)/wet weight × 100%.
Drug administration
Blockade of PD-1 pathway was achieved by injecting 200 μg of anti-PD-1 monoclonal antibody (29 F.1A12; Biolegend) intraperitoneally into mice immediately after ICH induction or sham operation. An isotype control IgG (RTK2758; Biolegend) was given to sham or ICH mice in a same regimen.
Immunofluorescence staining
Mice brains were separated at day 3 after ICH and fixed in 4% paraformaldehyde at 4°C overnight. The brain was then dehydrated in 15% and 30% sucrose sequentially until the tissue was saturated and sank to the bottom. After embedding in OCT (Sakura Finetek, Torrance, CA), brain tissues were cut into 7 μm-thick slices by a cryostat. Staining was conducted as previously described; 11 primary antibodies used is as follows: rabbit anti-mouse/human PD-L1(1:500; Invitrogen), guinea pig anti-mouse NeuN (1:1000; Merck), mouse-anti human NeuN (1:800; Abcam), goat anti-mouse GFAP (1:500; Wako), goat anti-mouse Iba1(1:200; Abcam). Secondary antibodies: donkey anti-rabbit 488 (1:1,000; Invitrogen), donkey anti-mouse 546 (1:1,000; Invitrogen), donkey anti-goat 546 (1:1,000; Invitrogen), goat anti-guinea pig 546 (1:1,000; Invitrogen). Images were acquired with a fluorescence microscope (Nikon, Japan) and analyzed by Image J.
Cell culture and treatment
A mouse hippocampal neuronal cell line HT22 was purchased from American type culture collection (ATCC). HT22 cells were seeded in a 6-well culture plate and grown in Dulbecco’s modified Eagle’s medium (GIBCO, USA) with 10% fetal bovine serum (GIBCO, USA). Cells were cultured in a 37°C incubator maintained at 5% CO2 and 5% O2 till they reached 80% confluency, then treated with culture medium contains either hemin (100 μM; Sigma, USA) or equal volume of saline for 24 hours. Hemin- and saline-induced neuron supernatant were collected separately for followed experiments.
In vitro detection of splenocyte apoptosis
Spleen cells isolated from healthy mice were resuspended in medium with 10% fetal bovine serum and then seeded in a 12-well culture plate (1 × 106/well). The collected HT22 cell supernatant either exposed to hemin or saline were added into the culture medium of spleen cells at a ratio of 1:1 with the original medium volume, cells were then cultured for 24 hours in a 37°C incubator maintained at 5% CO2 and 5% O2. After which, spleen cells were collected and counted, a part of cells were resuspended to a concentration of 105 cells/mL for apoptosis detection by FITC AnnexinV Apoptosis Detection kit (5565447, Biolegend) following manufacturer’s protocol. After staining, cells were analyzed by a FACSAria™ III flow cytometer (BD,Biosciences) and analyzed with FlowJo software.
Enzyme-linked immunosorbent assay
Enzyme-linked immunosorbent assay (ELISA) was performed to determine the concentration of soluble PD-L1 in mice circulation and HT22 cell supernatant. Mice blood samples were acquired at 12 h, 24 h and 72 h after ICH or sham surgery. Plasma was collected by centrifuging the blood at a speed of 3000 rpm for 10 min. After hemin or saline induced HT22 cell for 24 hours, the cell supernatant was collected. Measurement of PD-L1 was conducted by an ELISA kit (DY1019-05; R&D Systems) following manufacturer’s protocol.
Flow cytometry
Single cell suspensions of spleen and peripheral blood mono-nuclear cells and flow cytometry were conducted as described.11,12 Cells were suspended in 100 μL 1% bovine serum albumin solution for staining. For surface staining, cells were incubated with conjugated antibodies for 30 min at room temperature. For intracellular staining, cells were fixed with fixation buffer (eBioscience) at 4°C for 20 min after surface staining. Then the fixed cells were washed in permeabilization buffer (eBioscience) twice and incubated with corresponding antibodies for 30 min. The following antibodies were used: CD3 (100236, Biolegend), CD4 (100432, Biolegend), CD8 (100753, Bio-legend), NK1.1 (108708, Biolegend), PD-1 (109112, Biolegend). The data and images were acquired by a FACSAria™ III flow cytometer (BD,Biosciences) and analyzed with FlowJo software.
Microbiologic analyses (colony forming unit assay)
After euthanizing, the left lungs of mice were dissected with sterilized instruments and homogenized with 1 ml PBS using a glass dounce. The homogenized medium was centrifuged at a speed of 3000 r/min for 10 min at 4°C, the clear supernatant was collected as the original concentrate. PBS was added to process a dilution range from 1:1 to 1:100. Then the media at different dilution were incubated on an agar plate (L2020, Solarbio) for 48 h at 37°C. The colony forming units (CFU) were calculated by following formula: CFU/mL = (number of colonies × dilution ratio)/volume of cultured plate. All operation steps were performed in a sterile manner.
Lung immunohistochemical staining and assessment
Lung tissues were fixed in 4% paraformaldehyde at 4°C overnight. The tissue was then embedded with paraffin after dehydration in different concentrations of ethanol and cut into 7 μm section. Hematoxylin-Eosin (HE) staining was performed by a kit (G1120, Solarbio). Images were acquired by a microscope (Nikon, Japan) and analyzed by Image J. Lung immunohistochemical assessment was graded as follows: (1) normal; (2) focal (<50% lung section) interstitial congestion and inflammatory cell infiltration; (3) diffuse (>50% lung section) interstitial congestion and inflammatory cell infiltration; (4) focal (<50% lung section) consolidation and inflammatory cell infiltration; (5) diffuse (>50% lung section) consolidation and inflammatory cell infiltration, as previously described. 13
Neurological assessment
The neurological assessment was performed independently on day 1 and day 3 post ICH or sham operation by 2 investigators who were blinded to the treatment of mice. The motor function, sensory function, reflex, and balance of mice were assessed by the modified Neurological Severity Score (mNSS).11,14 Corner turning test and Rota- rod test was performed to assess fine sensorimotor function and motor coordination function of mice as previous described.15,16
Statistical analysis
Data analyses were conducted independently by investigators who were blinded to experiment alignment. The sample size was determined by power calculation to detect difference by calculating at α = 0.05 and 80% power. Each experiment was repeated at least 3 times. The Kolmogorov-Smirnov normality test and Shapiro-Wilk test were performed on datasets firstly and all data are normally distributed. Data are expressed as mean ± SD and P < 0.05 was considered significance. Two-tailed unpaired Student’s t test was used for comparisons between two group. One-way and two-way Analysis of variance (ANOVA) followed by Tukey post hoc test were used to determine the significance among three or more groups. All analysis was performed using GraphPad Prism 8 software.
Results
PD-L1 is released from perihematomal area into circulation post-ICH
To investigate the role of PD-L1 in post-stroke pathophysiologic changes, we first determined the expression of PD-L1 in healthy and hemorrhagic brains of mice. Immunostaining of mice brain slices shows that PD-L1 is broadly expressed in the control brain (Figure 1(a)). In mice model of ICH induced by either autologous blood or collagenase injection, PD-L1+ cells are significantly reduced in the perihematomal areas of the brain (sham versus autologous blood ICH: 314 ± 60.84 versus 187.3 ± 38.38/mm2 brain tissue, p = 0.0002; sham versus collagenase ICH: 314 ± 60.84 versus 170.9 ± 27.8/mm2 brain tissue, p < 0.0001). In addition, the diminished expression of PD-L1 in hemorrhagic brain is increased over time (Figure 1(b)). We next determined the cellular source of PD-L1 in the brain. Co-staining of PD-L1 with neuronal and glial cell markers indicated that PD-L1 is mainly expressed by neurons (Sham versus ICH, 312.5 ± 32.39 versus 173.4 ± 34.95/mm2 brain tissue, p < 0.0001), but not glial cells like astrocytes and microglia cells (Figure 1(c)). PD-L1+ cell counts are positively correlated with NeuN+ cell counts in the perihematomal area of ICH mice (Supplemental Figure 3), suggesting that PD-L1 and NeuN are simultaneously reduced in the perihematomal area of ICH mice.
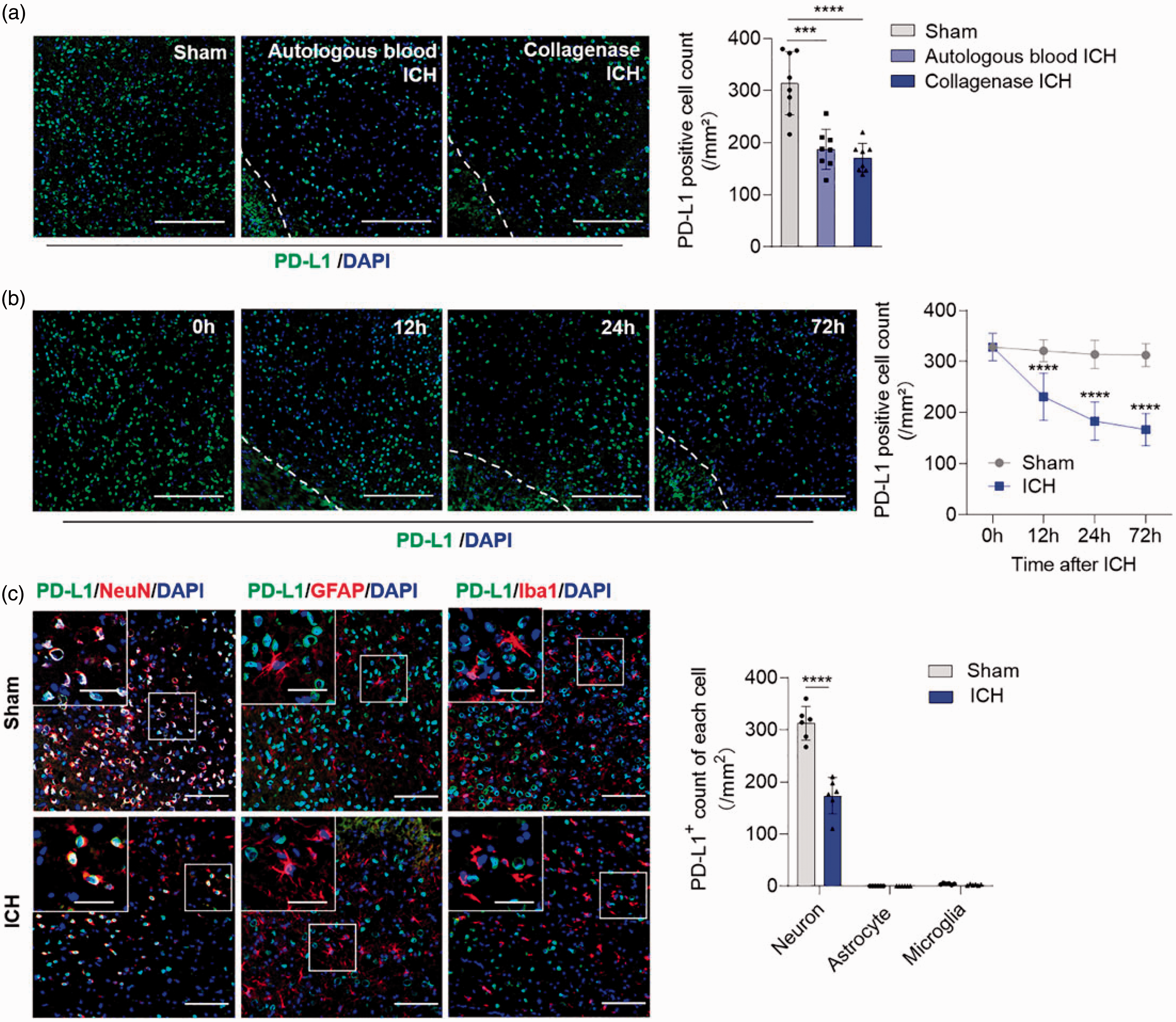
PD-L1 is progressively reduced in the perihematomal area of ICH mice. (a) Immunofluorescence images show the expression of PD-L1 in perihematomal area of mice brain section between sham and ICH model. ICH model was induced by injecting autologous blood or collagenase IV separately. Dashed lines indicate the borders of hematoma. Scale bar = 200 µm. PD-L1 positive cells were quantified among the 3 groups. n = 8 per group; (b) Mice brain sections were collected in different time points post ICH and immunostained for PD-L1. Scale bar = 200 µm. n = 8 per group; (c) Co-staining of PD-L1 with neuron (NeuN+), astrocyte (GFAP+), microglia (Iba1+) post ICH and quantification of PD-L1+ cells of each cell type in the perihematomal area. Scale bar = 100 µm, inset scale bar = 40 µm, n = 6 per group. Data are shown as mean ± SD, ***P < 0.001, ****P < 0.0001.
To confirm the discovery in ICH patients, we collected perihematomal tissues of ICH patients who underwent craniotomy to clear hemorrhage. Compared to healthy controls, total number of neurons and PD-L1+ cells were reduced simultaneously in the perihematomal area of ICH patients (PD-L1+ cells: control versus ICH, 128.3 ± 10.53 versus 41.75 ± 23.22/mm2 brain tissue, p = 0.0005), while the % of PD-L1+ neurons was similar between two groups. It suggests that the reduction of PD-L1 in the perihematomal area of ICH patients was potentially due to the loss of neurons (Figure 2). To examine if neuronal PD-L1 is released from the injured brain as a “danger signal”, we measured the level of soluble PD-L1 in the plasma of mice after ICH. Corresponding to the reduction of PD-L1 in the brain, plasma PD-L1 levels were found to be upregulated as early as 12 h after ICH induction (Sham versus ICH, 39.14 ± 17.92 versus 61.61 ± 21.71 pg/ml, p = 0.0514), and continuously increased to day 3 after ICH (Sham versus ICH, 42.88 ± 15.43 versus 68.34 ± 15.73 pg/ml, p = 0.007) (Figure 3(a)), corresponding to the reduction of PD-L1 in the perihematomal area. These data suggest that neuron-derived PD-L1 is progressively released from died neurons into the peripheral blood after ICH.
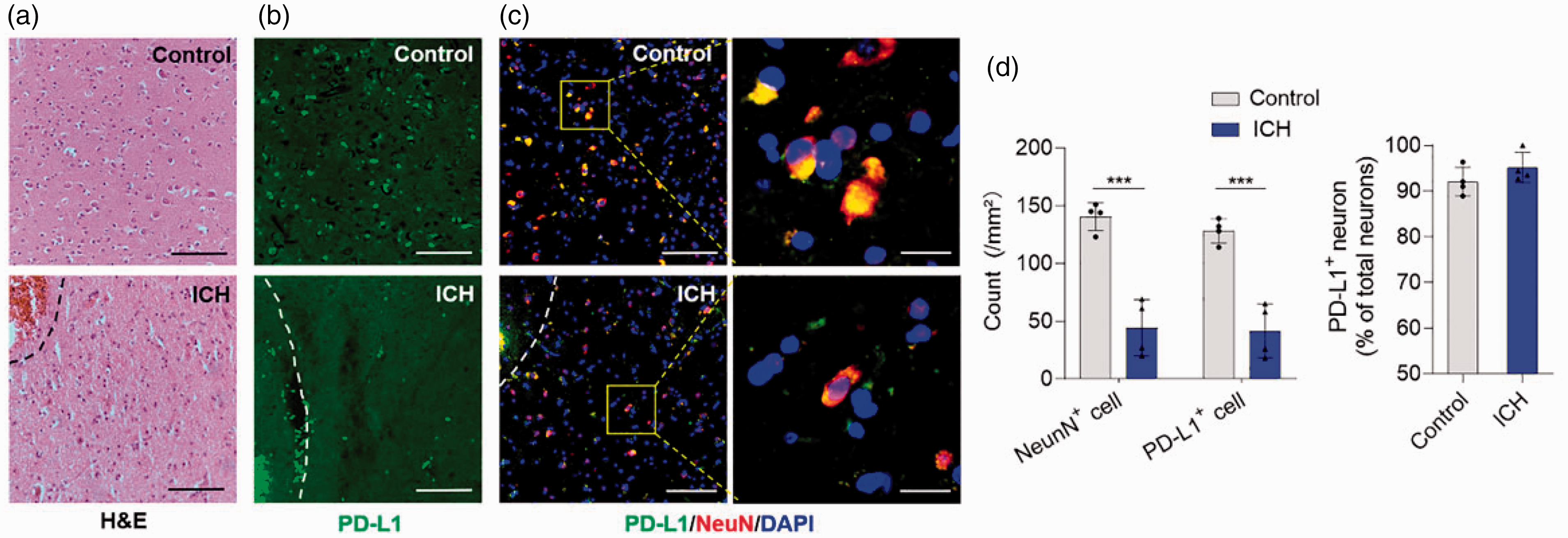
PD-L1+ Neurons are decreased in the perihematomal area of ICH patients. Perihematomal tissues of ICH patients undergoing craniotomy hematoma clearance were harvested for H-E staining and immunostaining, brain slices of subjects without neurological disorders were used as control. (a) HE staining images show the hematoma and perihematomal area of human brain section between control and ICH patients. Dashed lines indicate the borders of hematoma. Scale bar = 100 µm; (b) Immunofluorescence images show the expression of PD-L1 in perihematomal area of human brain section of control and ICH patients. Dashed lines indicate the borders of hematoma. Scale bar = 100 µm. (c) Images showing the expression of PD-L1 in neurons. Left scale bar = 100 µm, right scale bar = 20 µm; (d) PD-L1+ neurons and percentage of PD-L1+ neurons among total neurons are quantified between two groups, n = 4 per group. Data are shown as mean ± SD, ***P < 0.001.
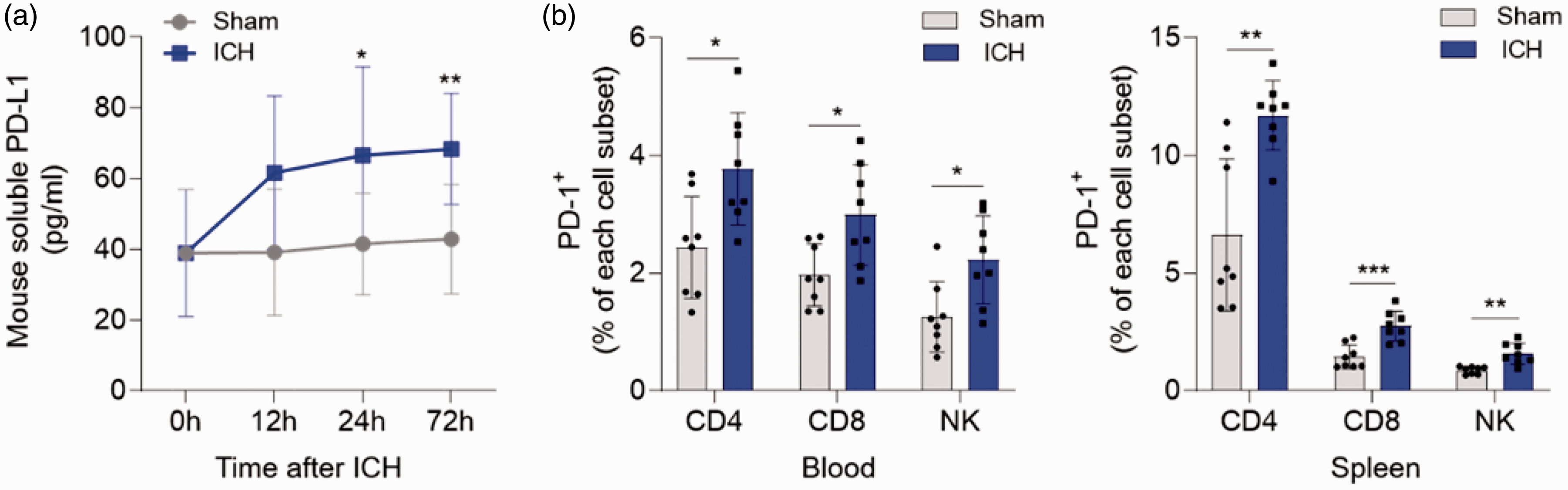
The expression of PD-L1 and PD-1 in peripheral system is enhanced after ICH. ICH model was induced by collagenase IV injection in C57BL/6 mice. (a) Soluble PD-L1 level was evaluated by ELISA in the plasma of ICH mice at indicated time points, with sham mice used as control. n = 10 in ICH group and n = 6 in sham group at each time point sequentially from 0h to 72h post ICH. (b) Expression of PD-1 was evaluated by flow cytometry in CD4+ T, CD8+ T, and NK cells from blood (left) and spleen (right) of mice at day 3 post ICH. Gating strategies of immune cells are shown in Supplemental Figure 1. n = 8 per group. Data are shown as mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.001.
Blocking PD1 reversed lymphocyte deficiency post-ICH
We have previously reported that ICH patients experienced a clear deficiency of T cells and NK cells in circulation, which was associated with increased risk of post-stroke infections like pneumonia or urinary tract infection. 17 PD-1 is mainly expressed on T cells and plays key roles in T cell priming and memory formation, and to a lower extent on NK cells. 18 Given the prior observations, we wanted to examine whether PD-1 contributes to the deficiency of T cells and NK cells after ICH. ICH induced a severe contraction of T and NK cell in both the blood and spleen compartments at day 3 post ictus (Figure 4(a) and (b)), which was similar to our prior reporting in ICH patients. 17 Interestingly, the ICH group exhibited a greater number of PD-1 expressing T cells and NK cells relative to sham control (Figure 3(b)). We then tested whether blocking of PD-1 signaling could reverse the deficiency of T cells and NK cells in the periphery. An anti-PD-1 monoclonal antibody (mAb) was given to mice immediately after ICH induction. Anti-PD-1 treatment didn’t impact the hematoma volume and brain water content of ICH mice (Supplemental Figure 2). Blocking of PD-1 nearly completely reversed the contraction of T cell and NK cell in the circulation and spleen reservoir, and further reversed spleen atrophy (ICH + IgG versus ICH + Anti-PD-1 mAb, 53.27 ± 7.35 versus 63.54 ± 11.81 mg, p = 0.0314) at day 3 after ICH (Figure 4). These data indicated that the elevated PD-L1 contributes periphery immunosuppression after ICH.
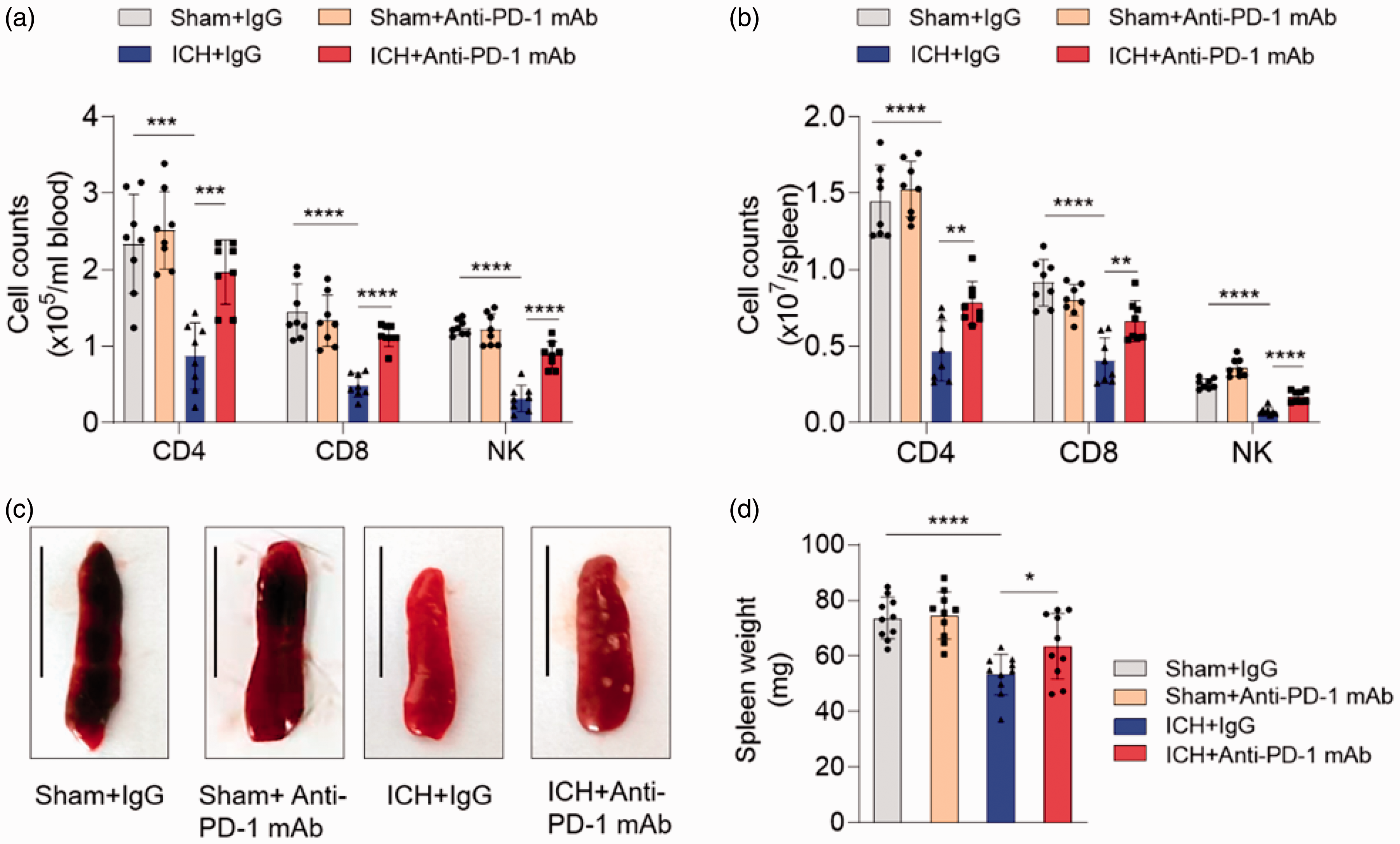
Blockade of PD-1 signaling reversed peripheral immunosuppression post-ICH. ICH model was induced by collagenase IV injection in C57BL/6 mice, sham mice were used as control. An anti-PD1 monoclonal antibody (mAb) was given to ICH mice immediately after ICH to block PD-1 signaling, an isotype control IgG was given to control group. Flow cytometry of blood and spleen sample was performed at day 3 after ICH. (a, b) Quantification of CD4+ T cell, CD8+ T cell and NK cell in the blood (a) and spleen (b) of mice by flow cytometry in indicated groups. n = 8 per group. (c) Representative Spleen images of indicated groups, scale bar = 1 cm. (d) Quantification of spleen weights of indicated groups. n = 10 per group. Data are shown as mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
It has been reported that PD-L1 expressed in some peripheral immune cells such as macrophage can also be up-regulated after inflammatory stimulation. 19 In order to test whether neuron-derived PD-L1 was sufficient to induce peripheral lymphocyte deficiency, we established an in vitro hemorrhage model, 20 exposure of neurons to hemin for 24 hours significantly increased the PD-L1 level in the culture medium (Supplemental Figure 4 A). Hemin-induced neuron supernatant was then adopted to stimulate spleen cells in vitro, which promoted the apoptosis of lymphocytes as well as reduction of the counts of T and NK cells (Supplemental Figure 4B–C).
PD-1 blockade reduced pulmonary bacterial burden and improved ICH outcome
T and NK cell deficiency have been associated with infections of ICH patients. 17 In confirming that anti-PD-1 treatment reversed the deficiency of T cells and NK cells after ICH, we were then keen to see whether PD-1 blockade could effectively reduce post-ICH infections and improve the clinical outcome. Pneumonia is the most common infectious complication in stroke patients. Therefore, we tested whether anti-PD-1 treatment could reduce the pulmonary bacterial burden of ICH mice, an established method in evaluating lung infections of mice. 21 In our experiment, ∼90% ICH mice developed pneumonia-like symptoms with positive lung tissue bacteria culture in day 3 after ICH. ICH significantly increased pulmonary bacterial burden of mouse lungs compared to sham controls. Treatment of anti-PD-1 mAb reduced pulmonary bacterial burden (ICH + IgG versus ICH + Anti-PD-1 mAb, 3.286 ± 1.27 versus 2.17 ± 1.21 units/μl lung supernatant, p = 0.0288) significantly (Figure 5(a)) and decreased the incidence of pneumonia to ∼50%. Histological staining of lung tissue identified characteristic pneumatic changes of ICH mice compared to sham control, i.e. thickening of alveolar wall, increased neutrophilic inflammatory infiltration, as well as alveolar structure destruction. ICH mice treated with anti-PD-1 mAb exhibited improved pathological lung alterations with reduced inflammatory infiltration and preservation of alveolar structures (ICH + IgG versus ICH + Anti-PD-1 mAb, 3.667 ± 0.52 versus 2.667 ± 0.82 scores, p = 0.0296). In addition, evaluation of the clinical outcomes showed that anti-PD-1 treatment improved the neurological deficits of ICH mice. These results indicate that anti-PD-1 treatment could reduce infection risk and improve ICH outcomes in mice.
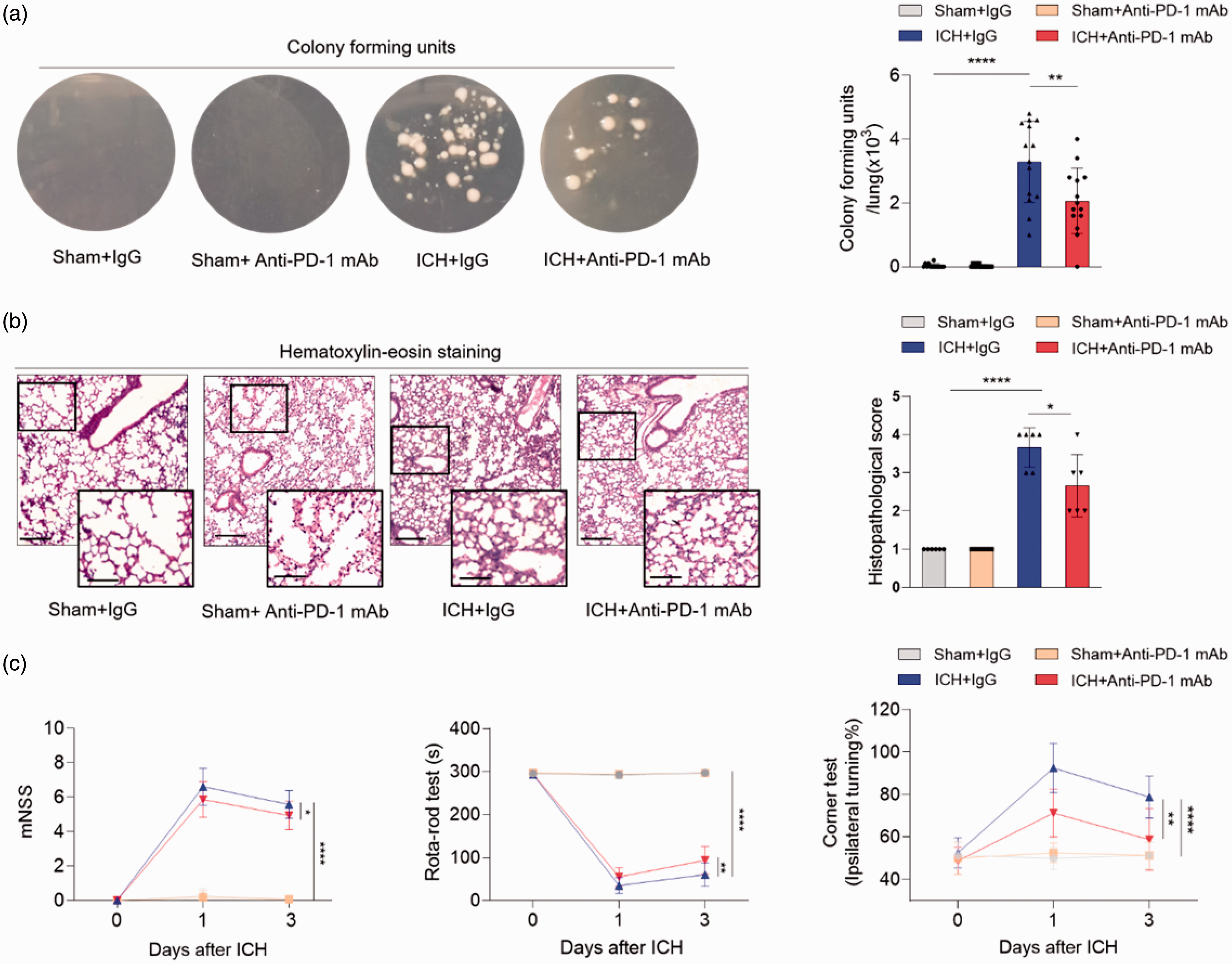
Blocking PD-1 ameliorated pulmonary bacterial burden and improved the neurological deficits of ICH mice. ICH model was induced by collagenase IV injection in C57BL/6 mice, sham mice were used as control. Anti-PD1 monoclonal antibody (mAb) was given to ICH mice immediately after ICH. Lung tissue was obtained from sham, ICH, and ICH+anti-PD-1 mAb groups for bacterial analysis. (a) Images show the bacterial colony forming of lung tissue homogenates been cultured on the agar plate for 24 h. Bacterial burden was quantified as colony forming units (CFU) per lung tissue, n = 14 per group. (b) Left panel: Hematoxylin-Eosin (HE) stained of lung tissue. Characteristic pneumatic changes such as thickening of alveolar walls and infiltration of neutrophils were proven in the lung tissue of ICH mice. Scale bar = 200 µm, inset scale bar = 100 µm. Right panel: Quantification of lung histopathological scores. n = 6 per group. (c) Neurological deficits evaluated by mNSS, rota-rod test and corner test between ICH and anti-PD-1 group were evaluated in 1 day and 3 day post ICH. mNSS test n = 14 per group. Rota-rod test and corner test n = 8 per group. Data are shown as mean ± SD, *P<0.05, **P < 0.01, ****P < 0.0001.
Discussion
In this study, we identified a previously unrecognized mechanism underlying post-stroke immunosuppression. We show that PD-L1 is constitutively expressed by neurons in the normal brain and is released as an “alarmin” upon ICH injury to modulate peripheral immune responses. Loss of PD-L1 mainly occurs in neurons located proximally to the hematoma, the perihematomal region, and is associated with the increased soluble level of PD-L1 in the peripheral blood. Increased circulating PD-L1 leads to the contraction of T and NK cell compartments in the blood and spleen. Additionally, blockade of PD-1 pathway via anti-PD1 monoclonal antibody nearly completely reversed post-ICH immunosuppression and reduced the pulmonary bacterial burden of ICH mice.
Post-stroke infections occur in about a third of ICH patients, and is associated with deteriorating neurological deficit and worsened clinical outcomes.3,21 We have previously reported that ICH patients developed severe deficiencies in T and NK cells, which was associated with post-ICH infections, mainly pneumonia or urinary tract infections. 17 Post-stroke immunosuppression is a main contributor to the increased infection risk following ictus. Stress pathways, such as increased sympathetic nervous input and activation of the hypothalamus-pituitary-adrenal gland axis have been commonly postulated as the causes of post-stroke immunosuppression.3,5,6,21 Blockade of adrenergic innervation via β-adrenergic receptor blockers is associated with rescued immune function and reduced infection risk.5,6 However, currently there are still no clinical trials of β-blockers showing benefit in reversing lymphopenia or prevention of pneumonia in stroke patients.3,22 The broad targets and impacts of β-blockers, especially on the cardiovascular system, maybe a major reason halting its benefit in clinical practice. Here, we identified that ICH induced the release of neuron-expressed PD-L1 into the peripheral blood, resulting in peripheral immunosuppression. This demonstrates the contribution of soluble molecules in the induction of post-stroke immunosuppression, and more importantly, identified a treatable target in preventing this phenomenon. Interestingly, another group recently reported that severe injury induced FasL-expressing monocyte drive bystander T cell death after ischemic stroke and burn injury. 2 Together, these discoveries suggests that the mechanisms underlying post-stroke immunosuppression are manifold, and thus a full illustration of a mechanism map is of paramount importance in identifying the most suitable targets to accelerate the development of treatments in preventing post-stroke infections.
The reported roles of PD-L1 and PD-1 in post-stroke immunopathology has been mixed. For example, PD-1 signaling is reported to be associated with attenuated neuroinflammation after ischemic brain injury in mice; 23 moreover, PD-L1−/− mice showed reduced infarct volume and pharmacological blocking of PD-L1 is beneficial in mice upon brain ischemia. 24 Correspondingly, administration of recombinant PD-L1 to a mice ICH model induced by autologous blood injection showed attenuated neuroinflammation and improved outcome. 25 Nevertheless, how PD-L1 and PD-1 execute their roles in the setting of stroke is not fully elucidated. Here, we show that PD-L1 is reduced from the perihematomal area of ICH patients and mouse models and its soluble form is increased in the peripheral blood, contributing to the peripheral immunosuppression after ICH. Blocking of PD-1 partially reversed post-ICH immunosuppression and reduced pulmonary bacterial infection. However, it has also been reported that anti-PD-L1 treatment is potentially associated with increased risk of hemorrhagic transformation of mice with brain ischemia; 24 in the context of ICH, we did not observe any enlargement of the hemorrhage in the ICH mice treated with anti-PD-1, nor increasing of brain edema. Conversely, anti-PD-1 treatment improved the neurological deficits in these ICH mice. Nevertheless, we cannot fully exclude the possibility that peripheral-derived PD-L1 signaling also play a role in inducing post-ICH immunosuppression, which limited the interpretation of our results, adopting conditional mutant mice will assistant us to answer this question in future pursuing.
In summary, our study identified neuron-derived PD-L1 as a potential brain alarmin molecule that is released to the circulation after injury and modulate peripheral immune responses. Targeting PD-1 pathway could be a potential treatment to reverse post-stroke immunosuppression and prevention of infectious complications.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X221116048 - Supplemental material for Brain-derived programmed death-ligand 1 mediates immunosuppression post intracerebral hemorrhage
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X221116048 for Brain-derived programmed death-ligand 1 mediates immunosuppression post intracerebral hemorrhage by Nuo Cheng, Hong Wang, Ming Zou, Wei-Na Jin, Fu-Dong Shi and Kaibin Shi in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported partially by National Science Foundation of China (91949208, 81830038, and 82101364), Tianjin Natural Science Foundation (19JCYBJC27500, 18JCYBJC43800), and Young Talents Supporting Program of Tianjin.
Acknowledgements
We thank our patients and their family members for participating this study and Samuel X Shi (Tulane University) for editorial assistance.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
KS and HW formulated the concept and designed the study. HW and WNJ obtained human brain slices. NC, HW, and MZ performed the experiments and data collection. NC, HW and KS analyzed the data, NC, HW, WNJ, FDS and KS drafted the manuscript.
Supplementary material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.