
Abstract
We found that blood vitronectin (VTN) leaks into the brain and exacerbates tissue loss after stroke by increasing pro-inflammatory IL-6 expression in female, but not male, mice. VTN signals through integrins and downstream focal adhesion kinase (FAK). Here, a two day systemic treatment with a small molecule FAK inhibitor starting 6 h after middle cerebral artery occlusion reduced ipsilateral brain injury size by ∼40–45% at 7 and 14 d, as well as inflammation and motor dysfunction in wild-type female, but not male, mice. FAK inhibition also reduced IL-6 expression in the injured female striatum at 24 h by 62%. Inducible selective gene deletion of FAK in astrocytes also reduced acute IL-6 expression by 72% only in females, and mitigated infarct size by ∼80% and inflammation at 14 d after stroke. Lastly, VTN−/− females had better outcomes, but FAK inhibitor treatment had no additional protective or anti-inflammatory effects. Altogether, this suggests that VTN is detrimental in females primarily through FAK and that FAK inhibition provides neuroprotection (cerebroprotection) by reducing VTN-induced IL-6 expression in astrocytes. Thus, VTN signaling can be targeted to mitigate harmful inflammation with relevance to treatments for women with ischemic stroke, who often have worse outcomes than men.
Introduction
Ischemic stroke results in worse outcomes in women than men.1,2 Besides female hormones,1–3 few experimental treatments have focused on sex-specific mechanisms even though they might be more efficacious. Vitronectin (VTN) is an abundant blood glycoprotein produced predominantly by the liver. 4 VTN leaks into brain after blood-brain-barrier breakdown caused by injury such as ischemic or hemorrhagic stroke5–8 or multiple sclerosis-like inflammation. 9 We found that, in female mice, but not males, VTN exacerbates brain tissue loss after ischemic stroke by increasing acute IL-6-driven inflammation, with detrimental outcomes correlating with increased VTN blood levels that peak at 24 h. 8 VTN also seems to play a detrimental role in traumatic brain injury 10 and cardiovascular disease. 11 VTN−/− female mice have improved outcomes after stroke, 8 suggesting that inhibiting VTN’s production or its biological effects would be therapeutic. However, little is known about mechanisms that regulate expression of liver VTN and, because of its abundance, neutralizing VTN or blocking binding to its integrin receptors might be difficult. Another avenue might be to interrupt VTN-integrin signaling.
Focal adhesion kinase (FAK or PTK2) is one of the major intracellular mediators of integrin signaling. 12 We have shown in vitro that VTN activates FAK to substantially increase expression of pro-inflammatory cytokines such as IL-66 and also after middle cerebral artery occlusion (MCAO) in female mice. 7 This raises the possibility that pharmacological FAK inhibition might be a therapeutic strategy for stroke in women. FAK inhibitors are in clinical trials for a variety of solid cancers 13 but they have not been tested for neurological disorders yet.
Individual integrins have many ligands and VTN activates several integrins. 14 However, VTN has some selectivity in vitro in inducing cytokine expression and activating FAK compared to other blood proteins. 6 Moreover, VTN does not activate two other major integrin signaling mediators, ILK and PTK2B. 6 Thus, VTN-FAK signaling may be relatively unique, possibly due to its ability to also bind the urokinase-type plasminogen activator receptor, which collaborates with integrins. 6
The cells that produce IL-6 in response to VTN only in females have not been identified, which is complicated by the fact that integrin receptors that bind VTN are present on several cell types in the brain, including astrocytes, 15 and all cells express FAK. Astrocytes are potential candidates because they produce IL-6 after stroke 16 and astrocytic IL-6 deletion reduces inflammation only in female mice in an auto-immune encephalopathy model. 17 Astrocyte end-feet constitute the essential outer layer of the blood-brain-barrier. 18 Thus, astrocytes are in a prime position to be first responders to leaked VTN and to play an important early role in producing the acute peak of detrimental IL-6 after stroke in female mice. Astrocytes have been recognized for their key role in outcomes after ischemic stroke and as a good target for pharmacological intervention for brain tissue protection. 19
Here, we tested whether pharmacological inhibition of FAK would be neuroprotective after ischemic stroke in adult female and male mice. We also determined the contribution of VTN signaling by inhibiting FAK in VTN−/− mice, and the contribution of astroglial FAK by using GFAPcre-FAKfl/fl mice.
Materials and methods
This manuscript and its animal data have followed the ARRIVE 2.0 guidelines. 20 Mice were randomly assigned to different genotype and treatment groups before surgery. All surgeries, post-operative care and behavioral tests were performed blinded to genotype and treatment. All analyses were done blinded to the genotype, sex or treatment. All data generated and analyzed for this study are included. Detailed methods are available from the corresponding author upon reasonable request.
Mice
A total of 230 mice were used. Adult C57BL/6, VTN breeders (originally B6.129S2(D2)-Vtn<tm1Dgi>/J 21 ) and GFAPcre breeders (originally B6.Cg-Tg(GFAP-cre/ERT2)505Fmv/J 22 ) were from The Jackson Laboratory and the FAKfl/fl breeders (originally B6.129X1-Ptk2tm1Lfr/Mmucd 23 ) from the MMRRC. All mice used were essentially on a C57BL/6 background (Table 1). VTN+/+ and VTN−/− littermates were produced from heterozygote breeders. GFAPcre mice were bred with FAKfl/fl mice to generate hemizygous mice who were further backcrossed with FAKfl/fl mice to produce GFAPcre-FAKfl/fl and FAKfl/fl control littermates. Genotyping followed the vendors’ protocols. To induce FAK gene excision in GFAPcre-FAKfl/fl mice, 50 mg/kg tamoxifen (T5648, Sigma) was injected intraperitoneally twice a day for 5 d. Tamoxifen-injected FAKfl/fl mice were used as controls. To exclude potential effects of tamoxifen during and after the stroke, experiments started 14 d after the last injection when concentrations are negligible. 24 Cre recombination was confirmed by PCR23,25 in the contralateral striatal tissue at 24 h post-MCAO. VTN−/− and GFAPcre-FAKfl/fl mice have been characterized as we described previously.7,26 All mouse work was approved by the ETSU University Committee on Animal Care and complied with the NIH Guide on Care and Use of Animals.
Mouse strains used in this study.
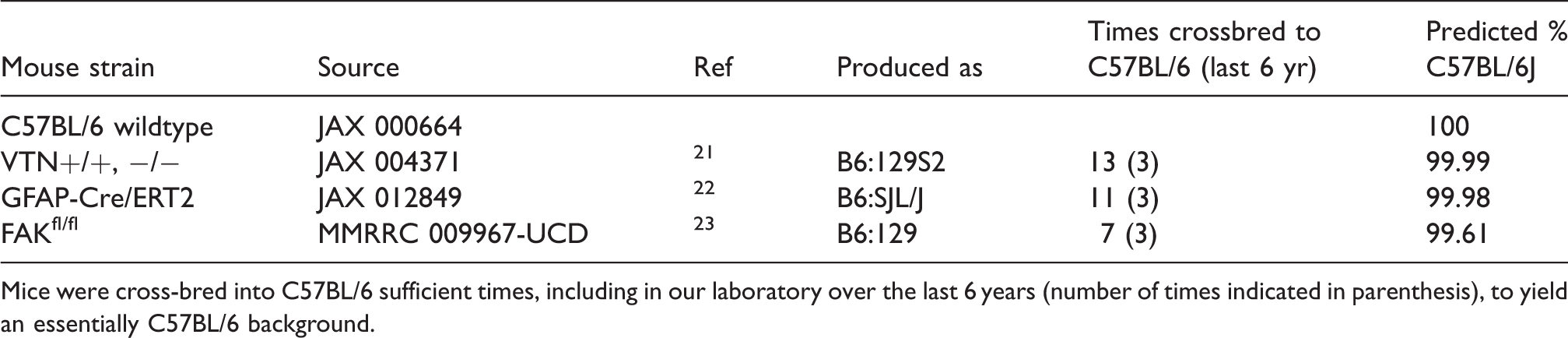
Mice were cross-bred into C57BL/6 sufficient times, including in our laboratory over the last 6 years (number of times indicated in parenthesis), to yield an essentially C57BL/6 background.
Stroke surgery
For invasive procedures, mice were anesthetized with Avertin that had been prepared fresh within the last 2.5 days and kept at 4 °C (intraperitoneal, 400 mg/kg 2,2,2-tribromoehtanol in 20 ml of 2% 2-methyl-2-butanol in saline). After loss of the righting reflex, mice were placed on a circulating water-based HTP-1500 Heat Therapy Pump system (Kent Scientific) with the controller set at 37 °C. The cages were placed on the same type of heating system during their recovery from surgery and for 7 d afterwards. A unilateral 30 min MCAO or sham operation (without filament insertion) was performed in 12–14 week old mice as we previously described in detail. 27 Occlusion was made consistent by using commercial filaments (602156PK10Re, Doccol) and clamping of the ipsilateral common carotid during the MCAO. This results in infarcts 27 and neurological deficits seen by others using longer occlusion times. Our current set of mice had injuries at 7 d involving almost 50% of the hemisphere area. Others have also described large injuries with 30 min MCAO in mice, close in size to 60 min. 28 Previously, increasing the MCAO to 45 min in males caused more and earlier mortality than the 17% we had seen with 30 min. 27 Here, MCAO-related mortality was 6% for males and 11% for females (Supplemental Table 1). The mice were monitored for vascular perfusion of the cortex before (baseline) and during the MCAO using transcranial laser-doppler flowmetry. Mice with more than 20% of baseline were excluded from the experiments (1% females, 8% males, Supplemental Table 1). Cerebral blood flow during MCAO was not significantly different between female and male C57BL/6 (15.1 ± 3.1, 16.7 ± 3.3, percent of baseline ± standard deviation), between VTN+/+ and VTN−/− female (15.7 ± 5.0, 17.1 ± 4.6) and male (18.6 ± 5.6, 18.5 ± 4.8), or between FAKfl/fl and GFAPcre-FAKfl/fl female (13.5 ± 3.2, 11.8 ± 4.3) and male (15.5 ± 4.9, 15.5 ± 4.0) mice. In a recent separate study, we found that 10 out of 10 C57BL/6 female mice recovered baseline flow values by 3–5 min after quickly removing the filament (unpublished results). Core body temperatures, measured in a recent separate study by fully inserting a 19 mm long probe of a digital rectal thermometer (Microtherma 2, Thermoworks), were not different between female and male C57BL/6 mice before, during or over a 4 h period after the MCAO (unpublished results). However, cerebral blood flow, temperature and other physiological changes during MCAO are known to affect outcomes and the lack of monitoring is a limitation of our study.
FAK inhibitor treatment
FAK14 (also named FAK inhibitor 14 or Y15: 1,2,4,5-Benzenetetramine tetrahydrochloride, MW = 284, PubChem # 78260) docks directly into the FAK Y397 site and inhibits its autophosphorylation, 29 which is essential for subsequent phosphorylation of other residues and activation of FAK. FAK14 is very specific for FAK and did not inhibit nine other recombinant kinases, importantly, the related Pyk2, which is also downstream of integrins. 29 We have shown that FAK14 and siRNA against FAK have similar reducing effects on mitochondrial respiration. 30 FAK14 injected i.p. at 30 mg/kg daily for 28 days did not cause mortality or produce toxic, hematological, or histopathological effects in different mouse organs. 31 Here, similar numbers of mice died after MCAO whether treated with saline or FAK14 (Supplemental Table 2). FAK14 is water-soluble with an IC50 of 1 μM and FAK phosphorylation is much decreased at 1 μM in vitro. 32 In vivo pharmacokinetics study in mice shows that FAK14 at 30 mg/kg (i.p.) is rapidly absorbed to reach maximum plasma concentration in less than 5 min and, after an initial drop, FAK14 plasma levels remain stable over 24 h. 31 I.p. injections provide a reservoir which can be replenished by daily injections. A preliminary study showed that i.p. injections of 3 mg/kg/d caused a maximal increase in CNTF expression in the naïve mouse brain, as confirmed by finding similar induction levels with 325 and 30 mg/kg/d. 33 We chose to test a 6 h delay after the stroke, in addition to the immediate treatment, because it would increase the clinical relevance. The treatment was given over a two-day period to overlap with the 24 h peak increase of plasma VTN 8 and IL-627 levels. Thus, adult C57BL/6, or VTN+/+ and their VTN−/− littermates were injected i.p. with saline or FAK14 (3 mg/kg, Tocris Bioscience, #3414) at zero, 3, 6 or 12 h after cessation of the MCAO and again at 24 and 48 h.
Grid walk test
Forelimb motor deficit was measured by impaired forelimb placement in a grid walk as we described. 8 Mice were placed on a suspended and unsteady metal wire grid (45 cm × 33 cm with 2.1 × 2.1 cm holes) one day before, and 7 and/or 14 days after MCAO. Mice were allowed to explore for 2 min and the last 1.5 min was video-recorded from underneath and used for data analysis. Errors made over the first 50 steps of the 1.5 min were identified as the paw protruding through the hole, improperly gripping the rung or slipping off when the mouse neared full body weight support. The impaired forelimb errors were calculated as a percentage of the total 50 steps. This test is very sensitive in that 10–15% of the steps taken by naïve mice are faulty and mice with an MCAO misstep a third to half of the time (8, see results below).
Histological and cytokine analyses
Coronal 30 µm thick microtome sections through the injury site were collected between the genu of the corpus callosum to the anterior commissure decussation. As we described, 8 a series of every sixth 30 µm sections per mouse was stained for immunofluorescence with antibodies against GFAP (1:1000, MAB3402, Millipore, RRID: AB_94844), NeuN (1:200, MAB377, Millipore, RRID: AB_2298772), or CD68 (1:500. MCA1957, Bio-rad, RRID: AB_322219). For any antibody, all sections of sets of mice containing all groups of an experiment were processed at the same time. As described, 8 whole sections were imaged with a 10x objective with the same exposure settings within a study, and the injury size was measured by GFAP-negative areas surrounded by reactive GFAP-positive astrocytes or NeuN-negative areas surrounded by NeuN-positive nuclei in the striatum and cortex, i.e. the epicenter without penumbra (Image J). The values were normalized to the area of the contralateral hemisphere in each section. Inflammation was quantified by the area and integrated density (the product of area and mean gray value) of CD68. This type of measurement is the least variable and most time-efficient method across different types of CNS injuries and between users for quantifying activated microglia/macrophages. 34 We have used it with good success in mouse stroke8,27 and spinal cord injury models. 35
mRNA analysis and Western blotting
The ipsilateral striatum was dissected following a transcardial perfusion with ice-cold phosphate buffered saline. After RNA extraction (RNeasy Mini Kit, #74104, Qiagen), mRNA expression levels of mouse inflammation-related genes were quantified using Taqman RT-qPCR as described before, 27 with primer-probe sets for IL-6 (Assay ID Mm00446190_m1), LIF (Mm00434762_g1), CNTF (mM00446373_m1), CD68 (Mm03047343_m1), CD45 (Mm01293575_ m1), TNF (Mm00443258_ m1), IL-1β (Mm00434228_m1), TGFβ1 (Mm01178820_m1), INFγ (Mm01168134_m1), and IL-13 (Mm00434204_m1), with GAPDH as internal normalizing control (Mm99999915_g1). Western blotting was performed as described, 33 using antibodies against pFAK (1:1000, #3283, RRID:AB_2173659), FAK (1:1000, #3285, RRID:AB_2269034), and β-actin as loading control (1:5000, #4967, RRID:AB_330288).
Statistical analyses
The sample size was estimated according to our prior studies in mice MCAO7,8 and power analyses with 80% power and α = 0.05 (GPower 3.0). The data were collected from 2–3 independent experiments (replications). Statistical outliers were identified by ROUT outlier test (Q = 1.0%) and normality was tested by skewness and kurtosis. Significance was set at P < 0.05 (GraphPad Prism 7) and determined by one-way or two-way ANOVA with Bonferroni post hoc multiple comparisons, as well as two-tailed t-test where stated. Repeated measures two-way ANOVA with post hoc Bonferroni multiple comparisons was used for behavioral data analysis. Data are presented as mean ± standard deviation.
Results
Systemic FAK inhibitor treatment reduces brain injury and inflammation after ischemic stroke
To determine whether FAK inhibition would be neuroprotective, adult female C57BL/6 mice received an i.p. injection of the small molecule FAK inhibitor, FAK14, at either 0, 3, 6 or 12 h following termination of a unilateral 30 min MCAO. Injections were repeated at 24 and 48 h after MCAO. At 7 d, only the 6 h delayed treatment resulted in a smaller injury size on the ipsilateral side of the brains of these female mice, predominantly the striatum and cortex (Figure 1(a) and (c)). The inflammatory response was also diminished by the 6 h delayed treatment, with less microglial/macrophage activation, as measured by the area and intensity of CD68 immunostaining (Figure 1(b), (d) and (e)). At 7 d, the core of the injury in the saline-treated females involved a large ∼50% area of the affected hemisphere. Surprisingly, the treatment that started immediately or at 3 h did not reduce the injury size or microglial activation. The 6 h, but not the 0, 3 h, delayed FAK14 treatment also reduced MCAO-induced neurological deficits as measured by the number of errors made in a sensitive grid walking test (Figure 1(f)). While the 12 h delayed treatment had no significant effect on injury size (Figure 1(c)), it reduced MCAO-induced grid walking deficits at 7 d (Figure 1(f)).
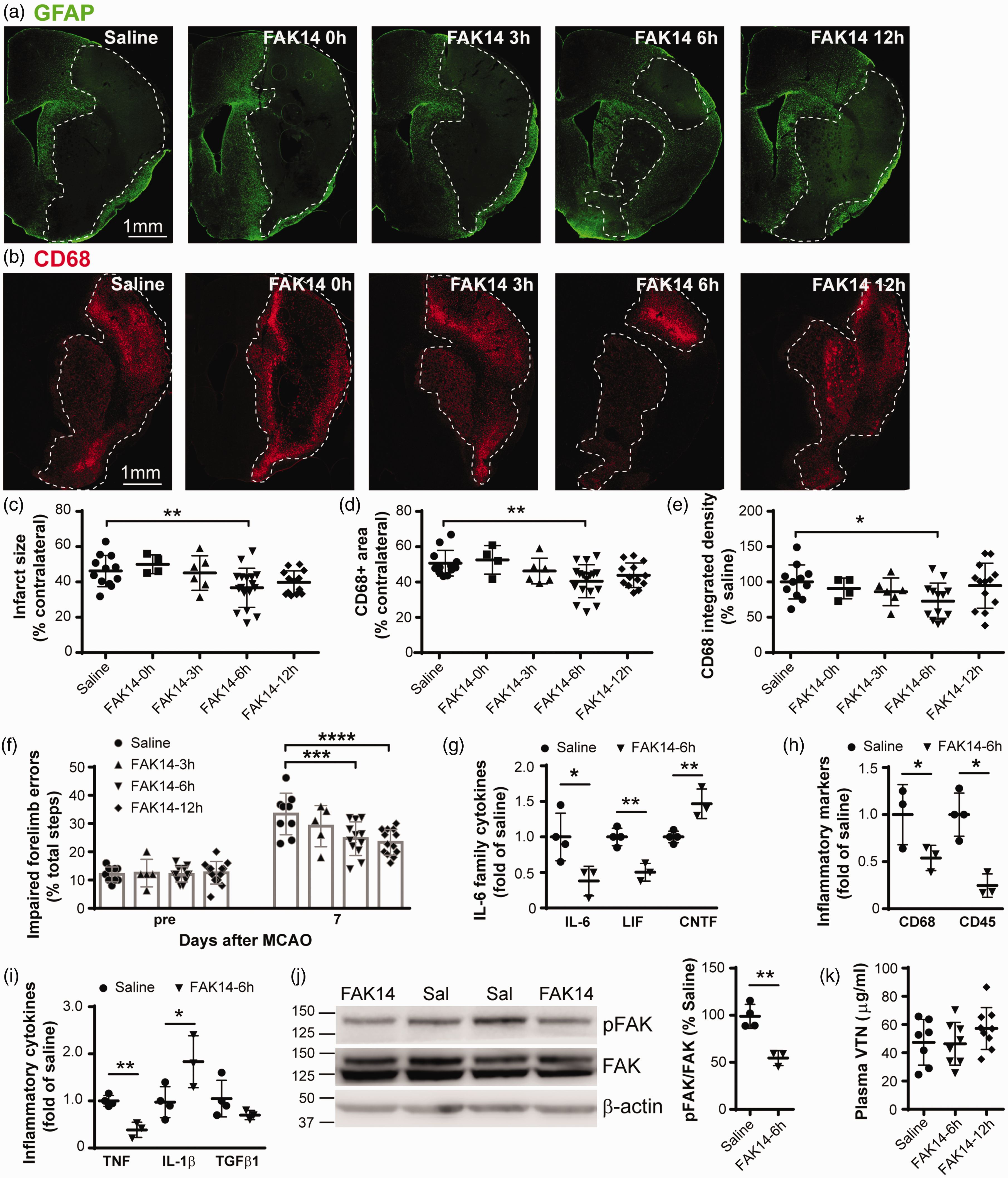
Delayed systemic FAK inhibitor treatment reduces infarct size and inflammation in female mice after ischemic stroke. (a) At 7 d, adult C57BL/6 female mice injected intraperitoneal with FAK inhibitor FAK14 three times over two days starting 6 h after termination of a 30 min MCAO had smaller injuries in the brain as shown in GFAP or (b) CD68 immunostained sections. Scale bar is 1 mm for all micrographs. (c) Measurements of the infarct as defined by the GFAP-negative area surrounded by the GFAP-positive reactive astrocytes showed that the treatment was neuroprotective if delayed by 6 h, but not 3 or 12 h or when given immediately (0 h) after the unilateral MCAO. Values are expressed as a percentage of the area of the contralateral hemisphere. N = 12,4,6,17,13 mice (three statistical outliers in saline, FAK14-0 h and FAK14-3h groups with infarct size = 2.1, 0.8 and 5.8%, respectively, were not included). Microglial and macrophage activation (CD68) was reduced by the 6 h delayed FAK14 treatment, as shown by (d) the CD68-positive area (N = 12,4,6,17,13 mice) and (e) integrated density (percent of saline, N = 11,4,6,13,13 mice). (f) Female C57BL/6 mice treated with FAK14 starting at 6 or 12 h after MCAO made fewer placement errors with the affected forelimb over 50 steps in a 1.5 min period of walking on a suspended grid at 7 d compared to those treated with saline. N = 9,5,12,13 mice. (g and i) At 24 h after MCAO, mRNA expression of pro-inflammatory IL-6 was much reduced in the striatum by FAK14 treatment at 6 h, as measured by qPCR. The related LIF, inflammatory markers CD68 and CD45, and TNF were also reduced by FAK14, whereas CNTF and IL-1β were increased. Data are calculated as a fold of saline controls. N = 4,3 mice. (j) FAK14 treatment at 6 h reduced phosphorylated FAK in striatum tissue at 24 h after MCAO compared to saline injections, as shown in western blots and quantified by densitometry. N = 4,3 mice and (k) Compared to saline, FAK14 treatment at 6 or 12 h did not affect plasma levels of VTN at 24 h after MCAO. N = 7,8,9 mice. Data are presented as individual values with horizontal lines indicating mean ± standard deviation. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
We had previously shown that acute pro-inflammatory IL-6 expression in the stroke injury is detrimental in females 8 and that FAK14 reduces pro-inflammatory cytokine expression in vitro. 6 Here, the mice with 6 h delayed FAK14 treatment had lower levels of IL-6 mRNA expression (172 vs. 452 fold of sham) in the injured striatum 24 h after MCAO (Figure 1(g)). Expression of the related LIF was also decreased, but the related CNTF was increased, as expected. 33 The systemic FAK14 treatment decreased microglial activation (CD68) and influx of leukocytes (CD45)(Figure 1(h)). The pro-inflammatory cytokines TNF, IL-1β and TGFβ1 were variably affected by FAK14 (Figure 1(i)), whereas expression of late immune response genes IFNγ and IL-13 was negligible (data not shown). The 6 h delayed systemic FAK14 treatment reduced FAK activation in the striatum as shown in western blots probed for phosphorylated FAK (Figure 1(j)). This suggests that FAK14 injected i.p. at 6 h is still at biologically active concentrations in the bloodstream at 24 h, given its short-half life when taken up by cells as indicated by its metabolic activity in liver microsomes. 31 FAK14 treatment started at 6 or 12 h after MCAO did not affect VTN plasma levels at 24 h after MCAO (Figure 1(k)). This indicates that FAK14 entered the brain and inhibited its intended target, rather than having an indirect effect by reducing VTN integrin ligand.
FAK inhibitor treatment reduces brain stroke injury by reducing the effect of VTN in females
We next tested whether FAK14 treatment started 6 h after MCAO would reduce the detrimental effect of VTN, using VTN+/+ and VTN−/− littermates (almost pure C57BL/6 background), and compared its efficacy in females and males. In WT VTN+/+ females treated with FAK14, the injury size in the ipsilateral hemisphere at 14 d was only about half compared to saline control treatment (Figure 2(a) and (b), 22.4% vs. 11.9%). As reported before, 8 VTN−/− females had smaller injuries compared to VTN+/+ littermates in the saline-treated groups (Figure 2(a) and (b)). However, FAK14 had no additional beneficial effect on the injury size of VTN−/− females (Figure 2(a) and (b)). FAK14 had no effect in either VTN+/+ or VTN−/− males (Figure 2(c) and (d)), and there was no difference in the injury size between the two genotypes in males, as we reported before. 8 The injury size of VTN−/− or FAK14 treated WT females was similar to that of males (12% vs. ∼14–15%), suggesting that VTN-FAK signaling makes WT females more sensitive to this type of stroke injury. The finding that at 14 d, our females had larger injuries than the males is in apparent contrast to findings that females have smaller lesions early after MCAO due to the neuroprotective effect of estrogen and progesterone.3,36,37 It is possible that our females had smaller initial injuries than males soon after the MCAO, but that the injuries worsened more because of their VTN-induced IL-6 peak expression at 24 h, which exacerbates cytotoxic inflammation. The injury size in these 14 d MCAO WT female mice was smaller (22%) than seen in females at 7 d (Figure 1(c), 46%), consistent with the shrinkage of the injury over time.
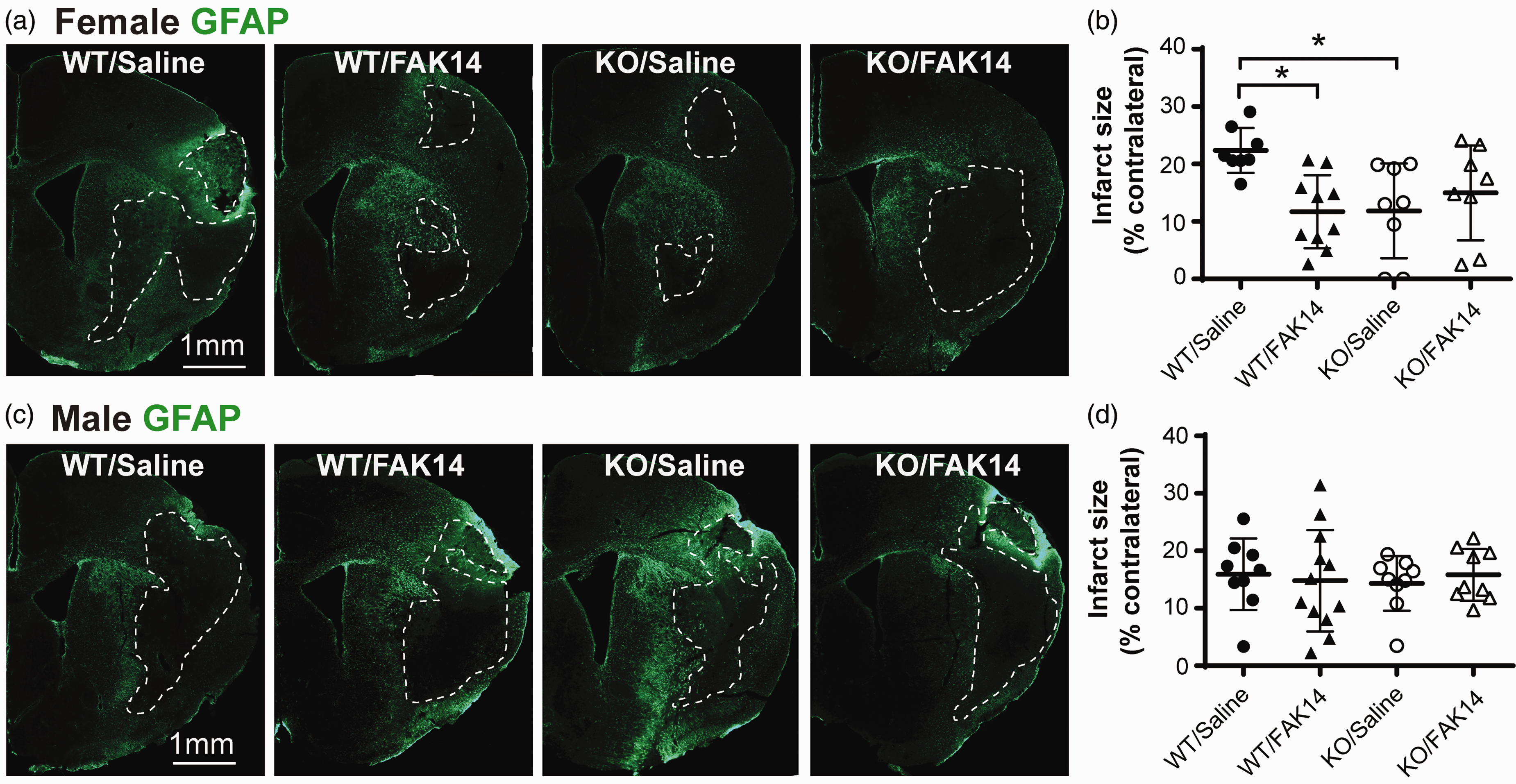
VTN worsens stroke injury in female mice by activating FAK. (a) VTN+/+ female mice (C57BL/6 background) treated with FAK14 started 6 h after an MCAO had smaller brain injuries at 14 d compared to the saline-injected group as shown in GFAP-stained sections. VTN−/− females also had smaller injuries, which was not affected by the FAK14 treatment. Scale bar is 1 mm for all micrographs. (b) Injury area measurements expressed as a percentage of the contralateral hemisphere confirmed the effects of the gene deletion and FAK14 in VTN+/+ females. N = 8,10,8,8 females. *p < 0.05 and (c) Males treated with FAK14 or male VTN−/− mice had comparable injuries with VTN+/+ males treated with saline as confirmed by measurements of the injured area (d). N = 9,12,9,9 males.
Excessive inflammation in the injured brain is detrimental after stroke. The total area occupied by CD68-positive cells, i.e., the region showing microglial/macrophage activation, was lower but not significantly different at 14 d after MCAO with the FAK14 treatment or the VTN gene deletion (Figure 3(a) to (c), C57BL/6 background). It was also not different between females and males (29 ± 8 vs. 27 ± 6% of the contralateral hemisphere area in saline-treated WT VTN+/+ mice, P = 0.61, two-tailed t test). However, the FAK14 treatment reduced the intensity of inflammation in those affected areas in female VTN+/+ mice, as measured by CD68 integrated density (area × gray level) in the injured hemisphere (Figure 3(c)). The FAK14 treatment had no effect in VTN−/− females (Figure 3(c)). The genetic deletion of VTN, as shown in VTN−/− females, had a similar effect as FAK14 in VTN+/+ females on the CD68 integrated density (Figure 3(c)). In males, neither FAK14, nor VTN gene deletion, had an effect on the CD68 integrated density (Figure 3(f)). The raw values of CD68 integrated density of WT mice treated with saline were not different between females and males (4.6 ± 3.1 vs. 4.1 ± 3.1, p = 0.72, 2-tailed t test, not shown). Together, these results suggest that both FAK14 and VTN deletion reduce the intensity of microglia/macrophage activation in injured area in female mice as seen at 14 d after MCAO. The total CD68 area in VTN+/+ females was about half as much as seen at 7 d after MCAO (Figure 3(b), 28% vs. Figure 1(d), 57%). The raw values for the CD68 integrated density were also much reduced at 14 d compared to 7 d (4.6 vs. 29.0, n = 8 and 3 mice, 2-tailed t test, t(9)=11.84, p < 0.0001, data not shown). This is consistent with the resolution of the early inflammatory response to injury.
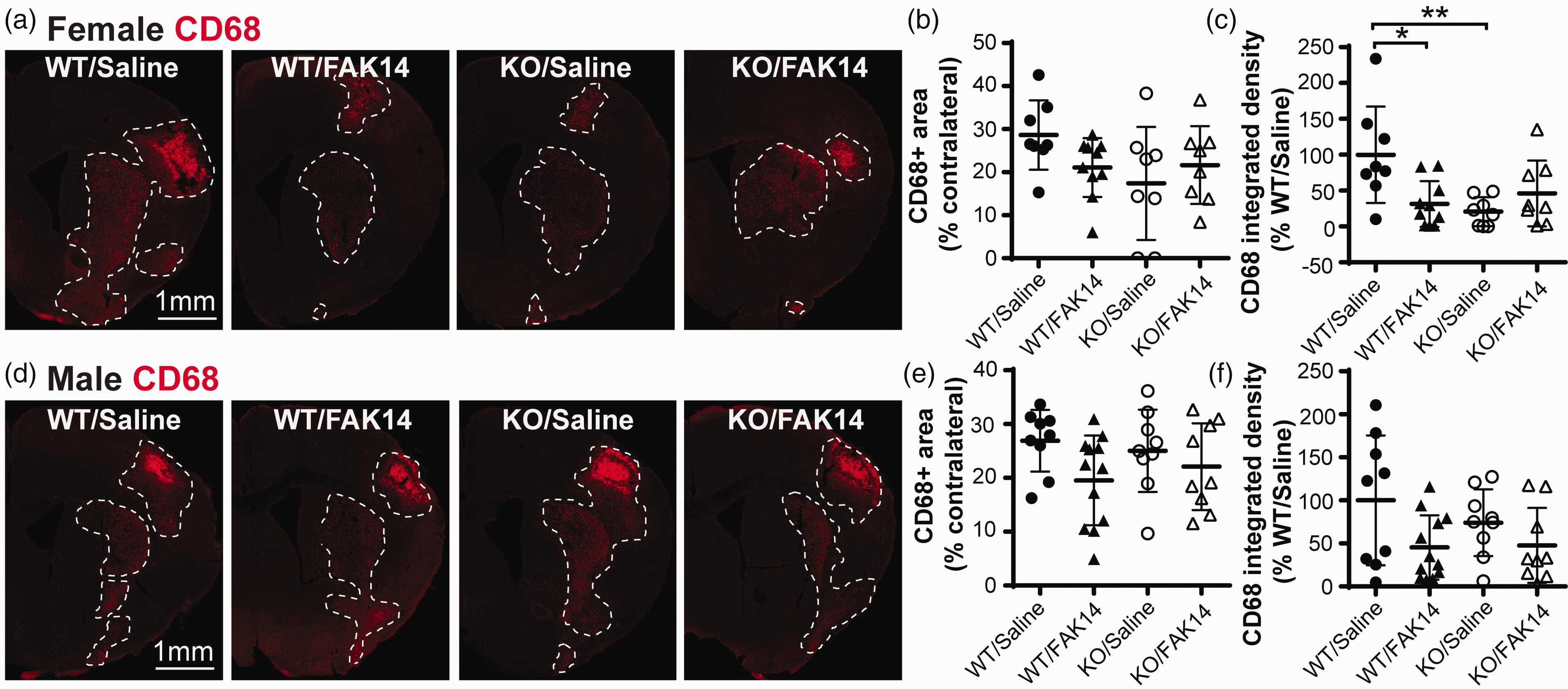
VTN exacerbates microglia/macrophage activation after stroke in female mice by activating FAK. The mice in Figure 2 were analyzed by CD68 immunostaining. (a) FAK14 treatment reduced MCAO-induced microglia/macrophage activation in VTN+/+ female mice (C57BL/6 background) at 14 d. This effect was the same as seen in VTN−/− females. The lesser response of VTN−/− females did not seem to be further reduced by FAK14. Scale bar is 1 mm for all micrographs. (b) Quantification showed that the brain area with CD68 staining was not significantly affected by genotype or FAK14 treatment, whereas (c) the CD68 integrated density, i.e., the product of area and mean gray value, was much reduced in FAK14 treated VTN+/+ and saline-treated VTN−/− females. FAK14 did not have additional effects in VTN−/− females. N = 8,10,8,8 mice. *p < 0.05, **p < 0.01. (d) In males, genotype or FAK14 treatment did not have any obvious effect, as confirmed by the measurements of (e) CD68 area and (f) CD68 integrated density (one outlier in WT/FAK14 group with CD68 integrated density = 257 was not included). N = 9,12,9,9 males.
A reduction in injury severity after MCAO stroke is often associated with reduced neurological motor dysfunction. Here, FAK14 treatment started 6 h after MCAO reduced the numbers of errors or footfalls by the impaired forelimb in a grid walking test at 7 d in VTN+/+ females (Figure 4(a), C57BL/6 background). This suspended grid test is very sensitive as shown by the ∼10–15% error rate over the first 50 steps measured in uninjured mice prior to the MCAO, and ∼25% and 53% (p = 0.0006, n = 9 male and 8 female mice. 2-tailed t test) at 7 d in saline-treated VTN+/+ males and females, respectively. The effect of FAK14 did not last until 14 d, suggesting that longer treatments might be necessary. At 7 d, the female FAK14-treated VTN+/+ mice and saline-treated VTN−/− mice had a similar extent of improvement compared to saline-treated VTN+/+ mice, suggesting that WT females make twice more grid errors than VTN−/− females (and WT males) because of VTN signaling through FAK. In VTN−/− female mice, FAK14 did not have a significant effect compared to saline treatment (Figure 4(a)). In males, FAK14 or VTN gene deletion did not have any effect on the grid walking performance (Figure 4(b)). Collectively, the data so far suggest that VTN exacerbates brain injury after ischemic stroke by signaling through FAK only in females and that this pathway can be inhibited as a female-specific treatment to improve outcomes.
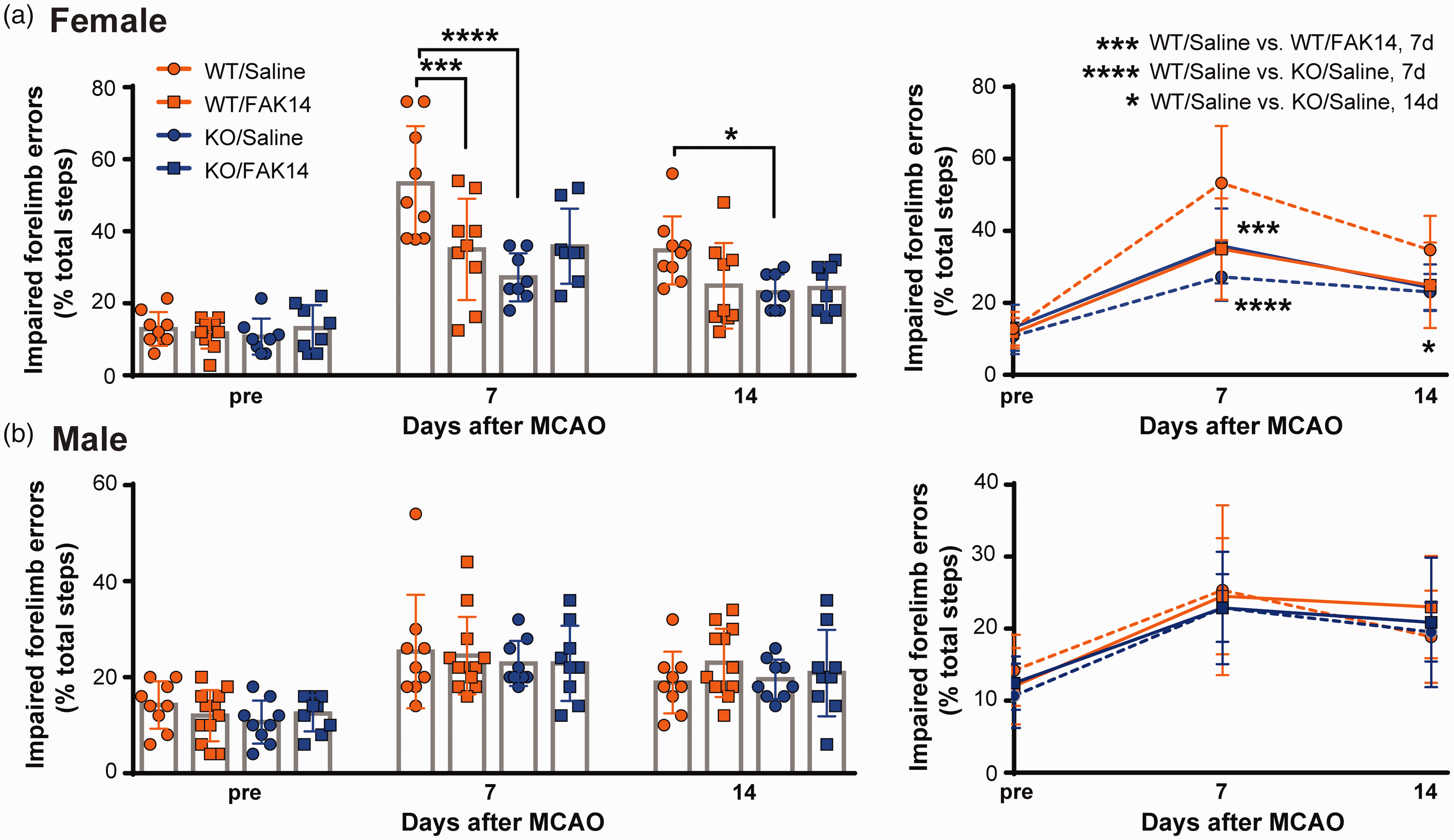
VTN contributes to motor deficits in female mice through FAK activation. (a) WT VTN+/+ female mice (C57BL/6 background) treated with FAK14 after MCAO made fewer placement errors with the affected forelimb in a grid test compared to those treated with saline. VTN−/− females treated with saline also made fewer errors than VTN+/+ females, but FAK14 did not have any additional benefit. The same data were presented as both individual values (column indicates group average) and as lines (right). N = 8,10,8,8 females and (b) In males, forelimb placement errors were not affected by genotype or FAK14 treatment. N = 9,12,9,9 males. *p < 0.05, **p < 0.01. Data are presented as mean ± standard deviation.
Astrocytic FAK deletion reduces acute IL-6 expression in the injured striatum only in females
We had previously shown that VTN exacerbates the peak expression of IL-6 and worsens the injury outcomes only in females when it leaks into the injured brain. 8 This VTN-FAK regulated IL-6 could be produced by astrocytes. Here, inducible conditional GFAPcre-FAKfl/fl mice were treated with tamoxifen to delete FAK in astrocytes two weeks before MCAO. FAKfl/fl mice injected with tamoxifen served as no gene deletion controls. These mice are at least 99.61% C57BL/6 (Table 1). In control female FAKfl/fl mice, MCAO caused a substantial increase in acute IL-6 expression at 24 h in the injured striatum compared to their sham injury controls (Figure 5(a)). In astrocyte FAK deleted females, the IL-6 expression was only about one third as high (Figure 5(a)). The FAK gene deletion did not affect the stroke-induced expression of related LIF but further increased CNTF expression, as expected. 33 Local microglial activation was reduced at 24 h as shown by CD68 mRNA expression levels but not CD45 of infiltrating leukocytes (Figure 5(b)). The FAK deletion in astrocytes did not affect MCAO-induced expression of other pro-inflammatory cytokines TNF, IL1β or TGF1β (Figure 5(c)), with expression of late immune response genes IFNγ and IL-13 being negligible (not shown). In males, astrocyte-specific FAK deletion had no effects on any of the inflammatory cytokines or markers (CD68 and CD45) in the injured striatum 24 h after MCAO (Figure 5(d) to (f)). CNTF expression was further increased, as expected. 33 The FAK deletion did not affect gene expression in female and male sham operated mice (data not shown).
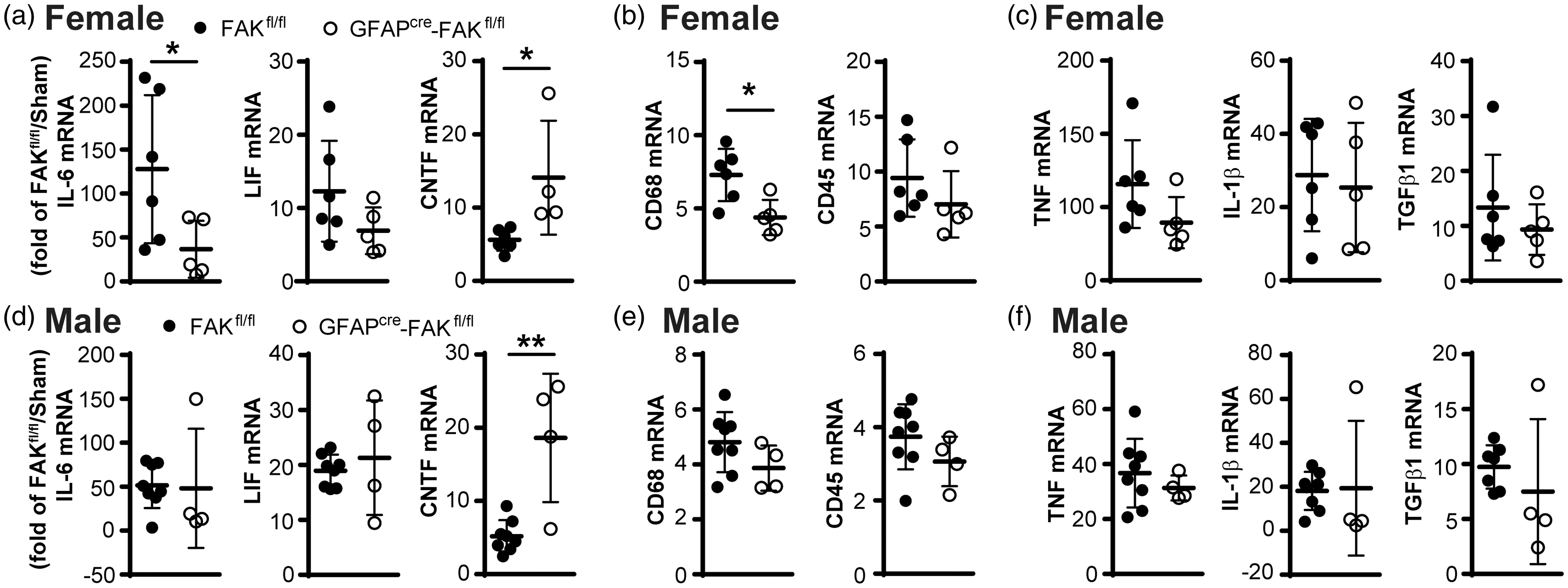
Astrocytic FAK deletion reduces pro-inflammatory IL-6 and CD68 expression only in female mice. Female and male GFAPcre-FAKfl/fl mice with an FAK gene deletion in astrocytes induced by tamoxifen two weeks earlier received a 30 min MCAO. At 24 h after MCAO, (a) GFAPcre-FAKfl/fl females had much lower expression levels of IL-6 mRNA in the injured striatum than their tamoxifen-injected FAKfl/fl controls (one outlier in GFAPcre-FAKfl/fl group with IL-6 = 320 fold was not included). Expression of the related LIF was not affected but CNTF was increased. (b) Microglial and macrophage activation was reduced as shown by CD68 mRNA levels, while leukocyte infiltration was no altered as shown by CD45 mRNA. (c) Pro-inflammatory TNF, IL-1β and TGFβ1 were not affected. (d–f) In males, the astrocyte FAK deletion did not affect pro-inflammatory cytokine expression or the inflammatory markers, but increased CNTF. N = 6,5 MCAO and 5,5 sham females, and 8,4 MCAO and 5,5 sham males. Values are expressed as fold change compared to sham operated FAKfl/fl control mice. *p < 0.05, **p < 0.01.
Astrocytic FAK deletion reduces stroke injuries in females
Lastly, at 14 d after MCAO, GFAPcre-FAKfl/fl females had substantially smaller brain injuries as measured by the GFAP-negative injured area (Figure 6(a) and (b)). The control “WT” FAKfl/fl females (99.61% C57BL/6) had ∼15% injuries, within the range of the WT VTN+/+ (C57BL/6) females also analyzed 14 d after MCAO (Figure 2(b)). Because the gene deletion was in astrocytes we also measured the area of loss of neuronal nuclear NeuN staining (Figure 6(c)), which showed a similar percentage of injury (Figure 6(d)). The FAK gene deletion in astrocytes also greatly reduced microglial and macrophage activation as shown by CD68-positive area (Figure 6(e) and (f)) and CD68 integrated density (Figure 6(g)) compared to their FAKfl/fl controls. At this 14 d time point, GFAP, NeuN and CD68 in the cre-lox mice all showed injury sizes in the range of 5-27% and NeuN values correlated with GFAP and CD68 values within the same mice (r = 0.705, p = 0.023 and r = 0.816, p = 0.004, respectively). This validates the use of GFAP staining, as we found previously. 8 Altogether, these data are consistent with the idea that IL-6 produced by astrocytes mediates the detrimental effects of VTN-FAK signaling in females after stroke.
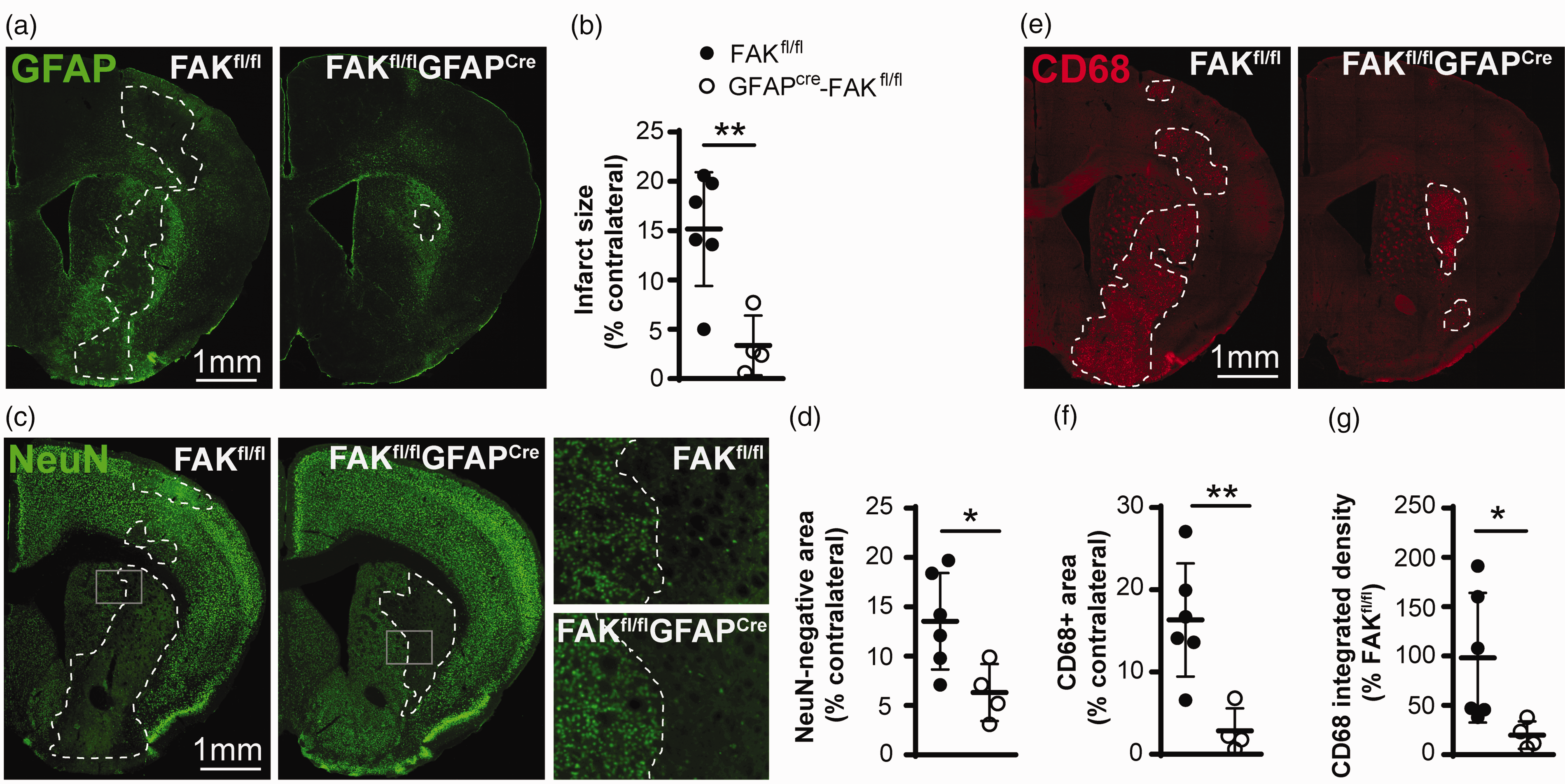
Astrocytic FAK deletion reduces injury size and microglia/macrophage activation in female mice. (a) GFAPcre-FAKfl/fl female mice had smaller brain injuries 14 d after a 30 min MCAO than their FAKfl/fl controls as shown in GFAP-stained sections and area measurements (b). (c) These results were confirmed by NeuN staining and (d) area measurement. CD68-stained sections (e) from these GFAPcre-FAKfl/fl mice show a reduced area and less intensity of microglial and macrophage activation, as confirmed by measurements (f, g). Scale bar is 1 mm for all micrographs N = 6,4 mice. *p < 0.05, **p < 0.01.
Discussion
The results of this study show that systemic treatment with an FAK inhibitor is neuroprotective (cerebroprotective) after ischemic stroke in female mice, but not in males, by interrupting VTN-FAK-IL-6 driven inflammation in the injured brain tissue. Thus, FAK inhibition has pleiotropic effects by reducing excessive detrimental inflammation to rescue brain tissue and function, which is a desirable feature of treatments for ischemic stroke. 38
The need to study sex-specific mechanisms after stroke and the opportunities they may provide to improve treatments is increasingly recognized. 1 Women generally have worse outcomes after stroke, especially after menopause when production of gonadal hormones greatly diminishes.1–3 In both female and male rodents, estrogen treatment during the acute stroke phase is neuroprotective, 39 increases neurogenesis, 40 is associated with less reactive astrocytes, 41 and partially mediated by protective IL-4. 42 Progesterone is also neuroprotective in rodent models of stroke 43 but combinations do not seem to be more effective. 44 Chronic hormone replacement therapy, which contains estrogen and progesterone, has failed to reduce the risk of stroke and its deficits in post-menopausal women in large clinical trials.1,39 Treatment with sex hormones during the acute stroke phase has not been tested in humans yet. Inhibition of protective X-link inhibitor of apoptosis (XIAP), which causes caspase activation, increases stroke injuries only in females. 45 Conversely, improved outcomes after stroke with a miR-181 antagomir was associated with an increase in XIAP, but this was tested in males. 46 Thus, we seem to have discovered the first female-specific detrimental mechanism whose inhibition leads to a female-specific treatment. Male-specific treatments include injection of antibodies against programmed cell death 1 ligand 2, which reduce numbers of certain inflammatory cells in the spleen only in male mice after stroke, 47 consistent with the finding that splenectomy is neuroprotective only in males. 48
A clear advantage of FAK14 inhibitor is its efficacy when given over a relatively brief period of two days after the ischemic stroke, consistent with the brief peak of increased plasma VTN 8 and IL-6 27 expression. The ability to start FAK14 treatment at 6 h after cessation of the primary ischemic injury to reduce injury and at 6 or 12 h to reduce neurological deficits makes this clinically relevant, suggesting that such adjuvant treatments could be beneficial well after the therapeutic 3 h window of tissue plasminogen activator. A few other neuroprotective studies have shown efficacy when the treatment was started with a delay of 3–6 h after initiation of ischemic stroke. These include progesterone 43 and an αvβ3 integrin peptide antagonist 49 in male rats, a 12/15-lipoxygenase inhibitor 50 or E-selectin antibody 51 in male mice. Another advantage of FAK inhibitors is that they lack obvious toxicity in mice 31 and seem to be tolerated well in clinical trials for cancer. 13 Here, the FAK inhibitor treatment started within the first 3 h after the stroke did not reduce the brain tissue damage in females. This suggests that FAK14 did not affect the initial ischemic injury. It remains to be determined why the early treatments are not neuroprotective. The improvement in motor function with the two days of FAK inhibitor treatment was seen at one but not two weeks after the stroke. It remains to be determined whether more frequent and longer treatments would provide more lasting benefits. The inflammatory response in the injured brain tissue peaked at 7 d and was much less at 14 d. This is consistent with the spontaneous recovery in neurological function which is typical for rodents after different neural injuries including stroke, 52 despite injury sizes that would cause devastating and permanent neurological deficits in human patients. Thus, the better tissue sparing achieved with the FAK inhibitor would be expected to result in marked improvements in humans.
Remarkably, FAK inhibitor treatment had no additional neuroprotective effects in VTN−/− female mice after stroke and the extent of the injury was similarly reduced from ∼22% to ∼12% of the contralateral hemisphere. This suggests that most or all of the neuroprotective effects of the FAK inhibitor were due to the inhibition of VTN signaling through integrin receptors and that VTN acts entirely through FAK. FAK14-treated WT VTN+/+ and VTN−/− females (C57BL/6 background) had similar functional deficits at 7 d after stroke as males (∼23% grid errors), suggesting that WT VTN+/+ females performed worse (∼53%) because of deficits induced by VTN-FAK signaling. In vitro, inhibition of FAK, but not the related PTK2B or ILK, blocks VTN-induced IL-6 expression. 6 The specificity of VTN-FAK signaling is surprising because fibrinogen 53 and fibronectin 54 also leak into the brain after stroke, and can also bind to αvβ3 integrin. 14 Moreover, fibronectin seems to inhibit FAK activation in vitro, 6 which would be consistent with the finding that it is neuroprotective after stroke in male mice. 54 One potential mechanism by which VTN, and not these other ligands, activate FAK could be its unique ability to also bind to the urokinase-type plasminogen activator receptor.6,55,56
The female-specific neuroprotective effects of inhibiting VTN-activated FAK is most likely due to a female-specific reduction of IL-6-driven detrimental inflammation. Our data show that the brain area and the intensity of microglial activation after stroke is similar between WT females and males. FAK inhibition and VTN knockout only reduce the intensity of the response in females but not males, suggesting a specific difference in the source and nature of the inflammation. In female astrocyte FAK knockout mice, IL-6 and CD68 expression in the injured striatum was reduced at 24 h after MCAO, but levels of several other pro-inflammatory cytokines and CD45 (leukocytes) were not affected. This suggests that IL-6-driven microglial activation is very specific in exacerbating brain injury in females. Moreover, IL-6 levels at 24 h correlate with injury size only in females and injection of IL-6 after MCAO reverse the female-specific therapeutic benefits of VTN deletion. 8 CNTF expression was increased, as expected by FAK inhibition in astrocytes, 33 whereas smaller injuries per se would be expected to have much less induction of CNTF. 27 Also, the neurotrophic CNTF was increased in both females and males but only the females benefitted from the FAK deletion. Thus, the reduction in IL-6 was not caused by smaller injuries in these astrocyte FAK deleted females but most likely resulted in lesser penumbral injury during the sub-acute injury phase.
IL-6 expression increases hundreds-fold from very low levels in the injured striatum after stroke in both female and male mice but is only reduced in VTN−/− females. 8 This sex dimorphism in VTN activity is most likely not caused by female gonadal hormones as it is not affected by ovariectomy. 8 Thus, there might be sex differences in FAK signaling to regulate IL-6 that remain to be investigated. FAK or downstream signaling effectors underlie sex differences in migration of human endothelial cells. 57 The EGF receptor can promote integrin-FAK signaling 58 and female mice reportedly have higher EGFR and TGFα ligand expression in some brain regions, although the cortex and striatum were not analyzed. 59 It also remains to be determined whether VTN-binding integrins are differentially expressed in females compared to males under physiological or stroke conditions.
There are several potential cellular sources of IL-6 after brain injuries such as stroke, including resident astrocytes and microglia.16,60,61 Both astrocytes and microglia62–64 have known sex differences but it remains to be determined which dimorphically expressed genes could interact with integrins and FAK. Our data with the astrocyte-specific deletion of FAK suggest that approximately 2/3rd of IL-6 expression in the injured striatum of females after stroke is regulated by FAK in astrocytes, but not in males. Compared to astrocyte FAK deletion, systemic FAK inhibition had a broader effect on inflammatory cytokines at 24 h after MCAO, suggesting that FAK14 affects infiltrating leukocytes. The effects of the astrocyte FAK deletion in the injured striatum seem to be opposite to that in the neighboring subventricular zone, where IL-6 was increased. 7 This difference could be related to regional differences between astrocytes 65 or between reactive astrocytes and quiescent astrocytes, 66 as are present in the injured striatum and neighboring subventricular zone, respectively. The current findings in the striatum are consistent with our in vitro studies with proliferating astroglioma cells showing that pharmacological- or siRNA-mediated inhibition of FAK reduced expression of IL-6, also after traumatic cell injury. 6
In conclusion, VTN-FAK signaling in astrocytes is detrimental in female mice by enhancing IL-6 expression and its promotion of harmful inflammation. Importantly, this female-specific pathway can be pharmacologically inhibited to improve outcomes in female, but not male, mice, using a clinically relevant treatment protocol of a systemic injection initiated 6 h after stroke. This may be relevant to developing treatments for women who generally have worse outcomes after ischemic stroke than men.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X221107871 - Supplemental material for Female-specific neuroprotection after ischemic stroke by vitronectin-focal adhesion kinase inhibition
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X221107871 for Female-specific neuroprotection after ischemic stroke by vitronectin-focal adhesion kinase inhibition by Cuihong Jia, Chiharu Lovins, Hannah M Malone, Matthew P Keasey and Theo Hagg in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: NIH grant NS102745 (TH).
Acknowledgements
We are grateful to DEL Lovins for technical support, and thank the Molecular Biology and the Microscopy Core Facilities for equipment support.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
CJ, MPK and TH designed research. CJ, CL, HM and MPK performed experiments and analyzed the raw data. CJ and TH performed final analyses and wrote the paper.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.