
Abstract
Functional connectivity (FC) is a sensitive metric that provides a readout of whole cortex coordinate neural activity in a mouse model. We examine the impact of experimental SAH modeled through endovascular perforation, and the effectiveness of subsequent treatment on FC, through three key questions: 1) Does the endovascular perforation model of SAH induce deficits in FC; 2) Does exposure to hypoxic conditioning provide protection against these FC deficits and, if so, is this neurovascular protection SIRT1-mediated; and 3) does treatment with the SIRT1 activator resveratrol alone provide protection against these FC deficits? Cranial windows were adhered on skull-intact mice that were then subjected to either sham or SAH surgery and either left untreated or treated with hypoxic post-conditioning (with or without EX527) or resveratrol for 3 days. Mice were imaged 3 days post-SAH/sham surgery, temporally aligned with the onset of major SAH sequela in mice. Here we show that the endovascular perforation model of SAH induces global and network-specific deficits in FC by day 3, corresponding with the time frame of DCI in mice. Hypoxic conditioning provides SIRT1-mediated protection against these network-specific FC deficits post-SAH, as does treatment with resveratrol. Conditioning-based strategies provide multifaceted neurovascular protection in experimental SAH.
Keywords
Introduction
Aneurysmal subarachnoid hemorrhage (SAH) is a devastating condition that affects roughly 30,000 people in the US per year.1 –3 While advances in the mechanistic understanding and management SAH have occurred over the years, morbidity and mortality remain high – 30% of patients die, 4 and 50% of survivors have long term deficits that preclude returning-to-work.5,6 The largest treatable cause of poor SAH outcome is delayed cerebral ischemia (DCI), which leads to death or disability in roughly 30% of all SAH patients. 7 For years, DCI was solely attributed to large artery vasospasm (constriction of large arteries 4–12 days after SAH).8 –11 Recently, several additional pathologies affecting the cerebral microvasculature have been linked to DCI, such as autoregulatory dysfunction,12,13 microvessel thrombosis14 –16 and blood-brain barrier breakdown,17,18 challenging this canonical view of DCI pathophysiology. Past trial failures have likely resulted from targeting only single elements of DCI, which we now know to be a multifactorial process. As a result, future therapies likely need to target both large artery vasospasm and microcirculatory deficits to be effective. This is the primary reason why conditioning-based therapy, which capitalizes on powerful and pleiotropic endogenous protective cascades, is so attractive for SAH patients.
Conditioning harnesses the brain’s inherent resistance to injury by exposure to a sub-lethal injurious stimulus. While hypoxia and ischemia are the most commonly studied conditioning stimuli, many drugs, including some that are already FDA-approved for other conditions, can be used to induce epigenetic-based protective mechanisms.19 –29 In past studies, we have shown that hypoxic conditioning provides profound protection against large artery vasospasm, microvascular thrombi, and neurological deficits in an endovascular perforation mouse model of SAH.30 –32 We have also linked this neurovascular protection to eNOS 32 and most recently Sirtuin 1 (SIRT1).30,31 SIRT1 has been the most extensively studied of all sirtuins and is an established regulator of various molecular pathways that have been implicated in the pathophysiology of DCI. 33 For example, SIRT1 expression results in increased eNOS and decreased MMP-9 expression, which are essential for hypoxic conditioning-induced protection and prevention of large artery vasospasm and microvascular thrombi, respectively.32,34 –37 The protective role of SIRT1 against cerebrovascular injury has also been demonstrated pharmacologically, as treatment with the SIRT1 activator Resveratrol induces protection against ischemic stroke, recurrent stroke, and neonatal hypoxia-ischemia.38 –43
In the present study, we examine SAH in mice using a functional neuroimaging readout. While many functional neuroimaging techniques, such as functional magnetic resonance imaging (fMRI), have significantly improved the field of human cognitive neuroscience, 44 the small brain size makes it difficult to use these technologies to yield the same level of detail in a mouse model. Fortunately, there are optical methods for analyzing and mapping the mouse cerebral cortex that provide comparable resolution to fMRI. 45 –48 Functionally related brain networks exhibit patterns of spontaneous activity that are temporally coherent and can be quantified via functional connectivity (FC) analysis, referring to the zero-lag Pearson correlation analysis performed across cortical regions. This phenomenon has been observed in the mouse cerebral cortex with optical intrinsic signal (OIS) imaging, where changes in reflected light intensity off the surface of the brain are converted to changes in local hemoglobin concentrations.49 –53 Due to the close coupling of neurovascular activity, this hemoglobin-based imaging strategy gives a population-based surrogate readout of neural, as well as vascular reactivity.45,54,55 Mouse fcOIS has proven to be a sensitive and effective assay for many neurological diseases, including Alzheimer’s disease, 56,57 ischemic stroke, 58 and glioma growth. 59 Recently, fcOIS was used to map deficits post-SAH using a prechiasmatic injection model of SAH induction in both the acute (4 days) and chronic (3 months) phases of this disease process. 60 Here, we use the endovascular perforation model of SAH induction, which is widely considered to be the closest representation of human SAH, replicating the trauma experienced by the cerebrovascular system following aneurysm rupture.61 –64 It is not clear if global metrics, such as cortical functional architecture, are altered by multiple models of SAH or its sequela (e.g. DCI) and if treatments are effective at resolving any alterations. Given that DCI is the largest modifiable risk factor for patient outcome, mapping the differences in FC post-SAH during DCI allows for further understanding of the neurovascular deficits caused by SAH and its sequela. By applying a translational therapeutic framework, we can investigate any potential alterations in functional architecture post-SAH as well as determine if previously proven efficacious treatment strategies attenuate these alterations in the mouse. This will allow for further exploration of the protective effects afforded by hypoxia and other SIRT1 based treatments against SAH.
Materials and methods
Ethics statement
All animal studies were approved by the Washington University School of Medicine Animal Studies Committee under guidelines and regulations consistent with the Guide for the Care and Use of Laboratory Animals, Public Health Service Policy on Humane Care and Use of Laboratory Animals, and the Animal Welfare Act and Animal Welfare Regulations. Animal reporting is according to ARRIVE guidelines. 65
Animals
Three to four-month-old male wild-type (WT) mice (C57BL/6J) mice from Jackson Laboratories (Bar Harbor, Maine, stock#000664) were used. Experimental animals were housed in an AAALAC-accredited facility in temperature- and humidity-controlled rooms with a 12 h light-dark cycle. Mice were housed five to a cage and had ad libitum access to laboratory chow and tap water. Allocation of animals into experimental groups was performed randomly.
Cranial window technique
Cranial window placement occurred as described previously, 66 and briefly summarized here (Figure 1(a)). Each mouse head was shaved, and a midline sagittal incision was made across the dorsal cortical surface. The scalp was retracted, and periosteal membranes were removed. A Plexiglass plate was adhered to an intact skull using dental cement (C&B MetaBond), which allowed for consecutive, repeatable imaging experiments. The mice were placed in an incubator post-surgery until they sufficiently recovered to return to a clean cage with laboratory chow and tap-water.
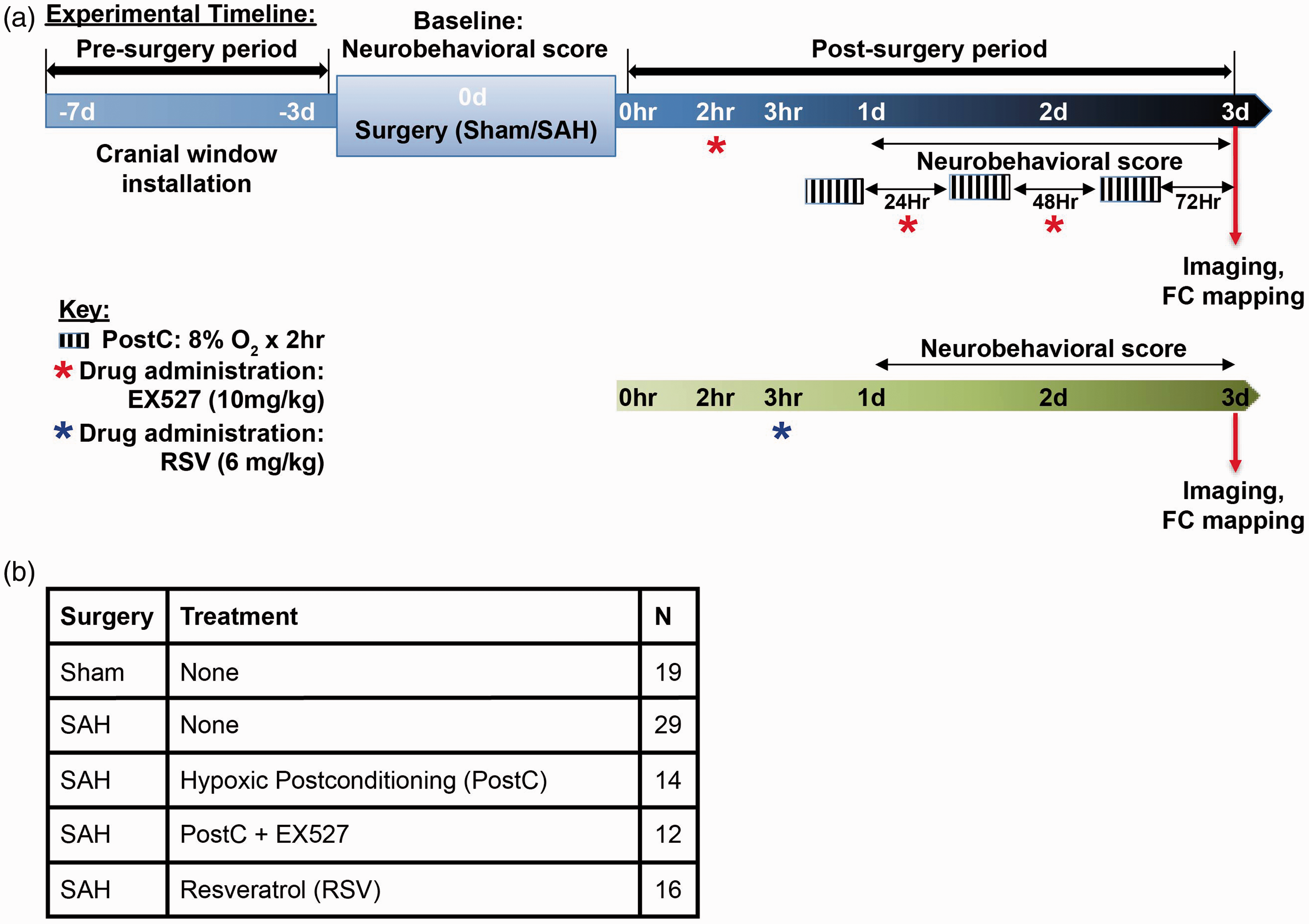
Optical Intrinsic Signal (OIS) Imaging of Subarachnoid Hemorrhage (SAH). (a) Timeline outlining the surgical, treatment, behavior recording, and imaging schedule for the present study. (b) Final number of mice used in the analysis for each condition.
Experimental SAH
Endovascular perforation SAH was performed as described (Figure 1(a) and (b)). 67 Briefly, mice were anesthetized with isoflurane (4% induction, 1.5% maintenance) in room air, with their core body temperature maintained at 37 °C by a thermoregulated heating pad. A midline incision was made in the neck to expose the external carotid artery (ECA). Next, a 5-0 nylon suture was introduced into the ECA and advanced through the internal carotid artery (ICA) until the ICA bifurcation. The suture was then advanced to induce SAH, then removed and the ECA ligated. Mice in the sham surgery groups underwent the above procedures except for suture-perforation. 67
Hypoxic postconditioning
Mice were placed in a hypoxia chamber and exposed to air containing 8% O2/92% N2 for 2 h with access to food and water ad libitum as indicated (Figure 1(a) and (b)). Mice were then returned to their vivarium containing room air. This was performed 3 h post-operatively and repeated every 24 hours until mouse sacrifice on day 3, for a total of three 2-hour hypoxia treatments.
Drug administration
The Sirt1 inhibitor EX527 (10 mg/kg i.p., QD, Tocris Bioscience, Bristol, UK, 2780) was first dissolved in dimethyl sulfoxide (DMSO) and then diluted to the final concentration with 1.2% beta-cyclodextrin in PBS. WT mice underwent sham or SAH surgery and were then administered EX527 beginning 2 h after surgery. One hour later (3 h after sham or SAH surgery) hypoxic postconditioning was administered for 2 h and continued once daily thereafter (Figure 1(a) and (b)). Resveratrol (RSV, 6 mg/kg i.p., BID Sigma, R5010) was prepared in DMSO. Mice were administered RSV starting 3 h after SAH or sham surgery and again every 24 h until mouse sacrifice. (Figure 1(a) and (b)). In pilot studies, EX527 or resveratrol administration to Sham‐operated mice did not induce any changes in body weight.
Neurobehavioral testing
Neurobehavioral outcome was examined daily using Neuroscore tests, as described. 67 Briefly, neurological function was graded based on a motor score (0–12) that evaluated spontaneous activity, symmetry of limb movements, climbing, balance and coordination, and a sensory score (4–12) that evaluated body proprioception and vibrissae, visual, and tactile responses (Figure 1(a)). Blinded observers performed all assessments.
Imaging
Mice were head-fixed in a stereotaxic frame under a high-powered, cooled, frame-transfer EMCCD camera (iXon 897, Andor Technologies, Belfast, Northern Ireland, United Kingdom,) . Sequential illumination was provided by a ring (7 cm diameter) of LEDs: 478 nm, 588 nm, 610 nm, and 625 nm, Roithner Lasertechnik. External triggering of the camera and LEDs was provided by custom MATLAB software (MathWorks) at a frame rate of 30 Hz per LED. The field-of-view (∼1cm2) covered the anterior-posterior axis from the olfactory bulbs to the superior colliculus, and the majority of the cerebral cortex convexity. Time series were binned in 128x128 pixel images with a resolution of about 80 μm per pixel. Mice were imaged on day 3 under each condition, corresponding with the timeframe of DCI in mice.65,68 Up to 45 minutes of resting state data was collected for each mouse in 5-minute data sets. Blinded observers performed all imaging.
Image processing
Imaging data was processed as described 45 and briefly summarized here. Reflected light intensities were filtered to 0.009–0.08 Hz and translated to concentrations of oxy- and deoxy-hemoglobin by solving the modified Beer Lambert law. For each mouse, the outline of brain region was traced manually to create a binary mask (using the roipoly.m MATLAB function) indicating brain regions for processing within the field-of-view. The global signal within this brain region was averaged and regressed from the time series data. Zero-lag functional connectivity (FC) analysis was performed using pre-specified seed regions corresponding to various cortical networks. Hemoglobin time traces within these seed regions were averaged and the resultant Pearson correlation with remaining brain pixels was calculated and plotted. Bilateral FC analysis was plotted by computing the Pearson correlation coefficient between each left hemisphere pixel and its corresponding symmetrical right hemisphere pixel.
Data exclusion
Each imaging run was inspected manually. Runs with light level variance >1% across the 5 minutes recorded were discarded. Additionally, the average bilateral FC Pearson correlation coefficient was calculated over the entire field-of-view for each mouse and calculated outliers within each experimental group were discarded (N_discarded = 5, 3 from SAH, 1 from Sham, and 1 from SAH Hypoxia + EX527).
Statistical analysis
D’Agostino-Pearson and Jarque-Bera tests of normality were performed in order to determine if each experimental group’s imaging data distribution significantly diverged from the characteristics of a normal distribution. D’Agostino-Pearson and Jarque-Bera tests 69 were performed because as moment tests, they account for both sample skewness and kurtosis. D’Agostino-Pearon and Jarque-Bera have also been previously reported to have better power than Kolmogorov-Smirnov and chi-squared tests, including with data distributions that are long-tailed and approximately symmetric or moderately skewed, 70 such as those reported here. These tests confirmed that the data distributions did not reject the null hypothesis that the data can be considered to originate from a normal distribution (D’Agostino-Pearson SAH p = .0737 Sham p = .5559, SAH:PostC p = .3252, SAH:PostC + E527 p = .4359, SAH:Res p = .8569) (Jarque-Bera SAH p = .1139 Sham p = .6756, SAH:PostC p = .4456, SAH:PostC + E527 p = .5418, SAH:Res p = .7927). A 2-way repeated measures ANOVA (rm-ANOVA) containing one between-subjects variable (treatment) and one within-subjects variable (day) was used to evaluate differences in Neuroscore. To evaluate whether SAH altered connectivity, we performed a two-sample student’s t-test to compare mean bilateral FC scores at the 3-day timepoint between SAH and Sham animals. To assess whether the treatments altered the SAH FC, we compared the treatment groups’ mean FC scores values using the two-sample student’s t-test to compare SAH:PostC, SAH:PostC+E527, and RSV groups each to the SAH group. The same analysis was performed on pixelwise FC. A Bonferroni correction was performed on the global mean comparisons to account for the 3 separate comparisons between each of the 3 treatment groups and the SAH animals. A multiple comparison correction was also performed on the pixelwise bilateral FC maps based on the clustering of significant pixels, as previously described, 71 after the aforementioned Bonferroni correction that adjusted the significant p-value from 0.50 to 0.0167. Finally, a linear regression model was used to fit FC and Neuroscore under various conditions and a subsequent t-test was performed on the coefficient of determination.
Results
SAH causes FC deficit at 3 days post injury
To determine whether aneurysmal SAH induces FC deficits, wild-type mice underwent the cranial window placement procedure at a minimum of 3 days before baseline neurological testing and either sham (n = 19) or SAH (n = 29) surgery. Neuroscore behavioral testing was performed at 24 h increments until the day of imaging in order to confirm neurological deficits in SAH mice compared to sham (Figure 2(a), 2-way ANOVA for repeated measures (rm-ANOVA), F(1,46) = 68.715, p = 1e-10). Surgery was followed by fcOIS imaging at 3 days post-SAH, an experimentally reproducible time frame for the occurrence of DCI in mice across multiple animal models of SAH induction. 72 Bilateral homotopic connectivity maps displaying correlation between left and right pixels across whole-brain showed marked deficits in correlation patterns between Sham and SAH conditions conditions and a spatially extensive decrease in pixelwise bilateral connectivity across the cortex (Figure 2(b)). To further elucidate the differences between groups, histograms were used to compare the number of pixels displaying a given bilateral Pearson’s correlation coefficient, “r”, in the homotopic maps of the SAH and Sham conditions (Figure 2(c)). The histogram comparing the entire cerebral cortical surface (Figure 2(c), left) shows that sham mice have a globally stronger bilateral correlation than SAH mice, as seen by the right shift on the histogram. A region of interest (ROI) was manually traced along the area of greatest deficit in the SAH bilateral map (black, Figure 2(c), upper right corner). The histogram comparing this lower somatosensory and parietal cortical region (Figure 2(c), right) illustrates a network specific effect induced by SAH. To further illustrate this network specific effect, FC maps with seeded regions of interest were compared between SAH and sham (Supplemental Figure 1 A, top 2 rows). Seeds were specifically chosen within frontal (Fr), cingulate (Cing), motor (Mot), somatosensory (Ss), retrosplenial (Rs), visual (Vis), and audio (Aud) cortices in order to sample the major cortical networks within our field-of-view. These seeded maps displayed high similarity between SAH and sham groups for seeds placed in anterior brain regions (e.g. Fr, Cing, Mot). Loss of homotopic connections are visible in posterior seeds (e.g. Ss, Vis, Aud). Calculating the average bilateral Pearson correlation coefficient across the entire field-of-view resulted in significant differences between Sham and SAH global connectivity (Figure 2(d), two-sample t-test p = 5.052e-05). These large changes in global connectivity were correlated with behavioral deficits as quantified through Neuroscore (Figure 2(e)).
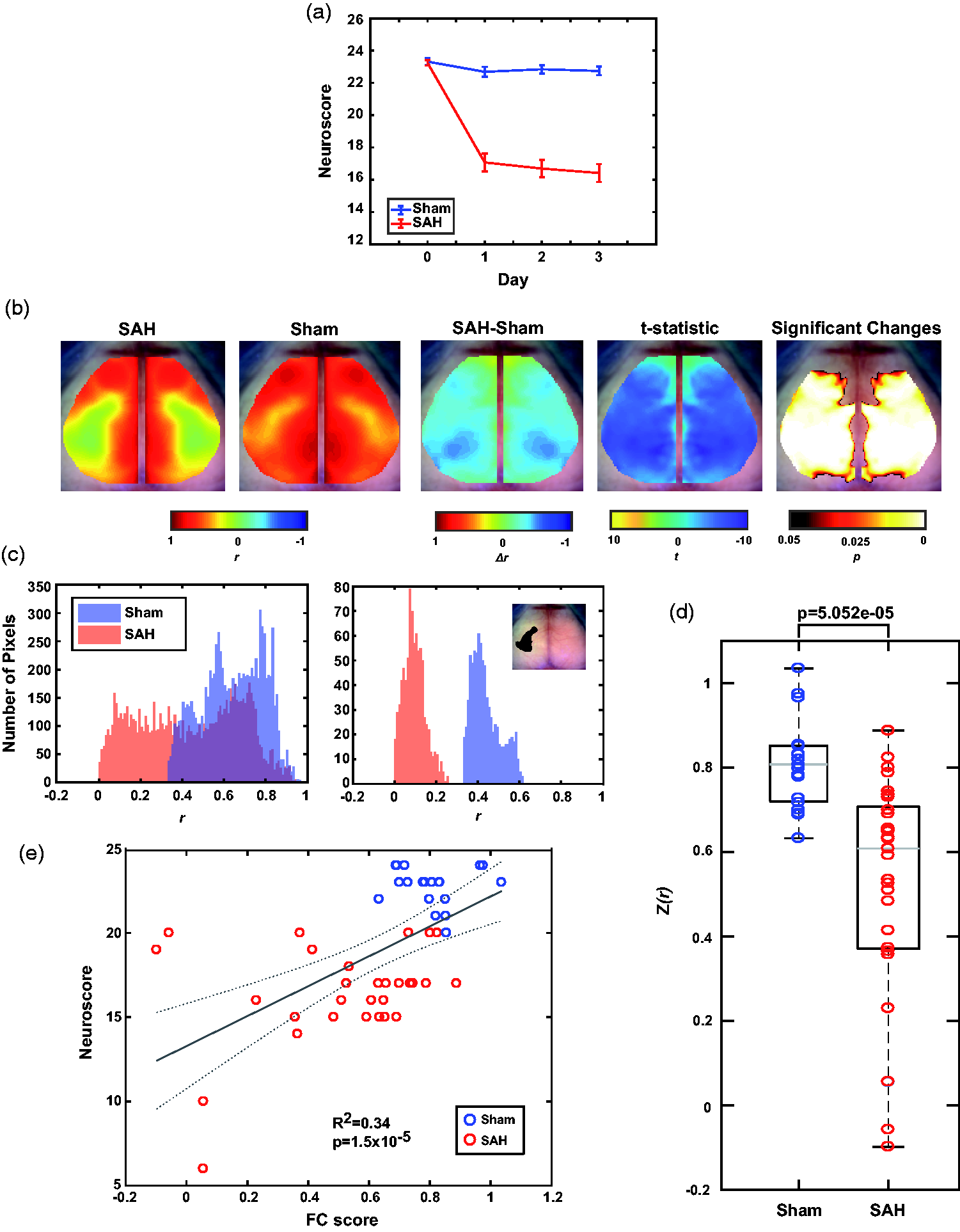
SAH results in decreased Neuroscore and FC score compared to Sham mice. (a) Average Neuroscore for Sham and SAH mice at baseline and Days 1–3. Error bars are SEM. (b) Group average bilateral homotopic connectivity maps for Sham and SAH, their difference when Sham is subtracted from SAH, and the t-statistic and significantly changed pixels after a cluster-based correction. (c) Left, every Pearson correlation coefficient value per pixel plotted between Sham and SAH groups. Right, only Pearson correlation coefficients within the black outline plotted between Sham and SAH groups. (d) Global average Pearson correlation coefficient from each mouse within each condition. Grey horizontal bar is the median, each box edge marks the 25th/75th percentile, whiskers extend to most extreme values not considered outliers (*p < 0.05, two sample student’s t-test). e) Linear fit of Neuroscore vs the global FC average plotted in d). Dotted grey line corresponds to confidence bounds (n = 19 Sham, 29 SAH).
Hypoxic post-conditioning affords SIRT1-mediated protection against SAH-induced FC deficits
In addition to establishing a model for SAH injury and visualizing both global and regional-specific deficits in FC, the effects of hypoxic postconditioning on FC were examined in wild-type mice with (n = 12) and without (n = 14) the SIRT1 inhibitor EX527. Cranial window and SAH surgery followed the same timeline as above (Figure 1(a)). Neuroscore was performed at 24 h increments until the day of imaging in order to confirm that hypoxic postconditioning effectively protected against SAH-induced neurological deficits, and that EX527 removed this protection (Figure 3(a), 2-way rm-ANOVA, Sham vs Hypoxia, F(1,31) = 10.893, p = 0.002, Sham vs Hypoxia + EX527 F(1,29) = 102.1, p = 5e-11, Hypoxia vs Hypoxia + EX527, F(1,24) = 49.589, p = 2e-7, SAH vs Hypoxia, F(1,41) = 30.125, p = 2e-6, SAH vs Hypoxia + EX527, F(1,39) = 0.029, p = 0.86). Bilateral homotopic connectivity maps show an improvement in global FC in the hypoxic postconditioning group, providing evidence that hypoxic postconditioning is protective against FC deficits in SAH mice (Figure 3(b) vs Figure 2(b)). A pixel-wise evaluation of the SAH and Hypoxia groups did find significant increases in bilateral connectivity in retrosplenial, somatosensory and motor cortices (Figure 3(b)). Bilateral FC analysis also shows that EX527 removes the protective effect provided by hypoxic postconditioning, suggesting that the protection is SIRT1-mediated (Figure 3(b) vs Figure 2(b)). Visualized using the same histogram ROI-based analysis as described above, we can see that there is a specific protective effect in the highly effected somatosensory/parietal regions provided by hypoxic postconditioning, as exhibited by the right shifted mean correlation coefficient that is similar to the sham group (Figure 3(c)). The group that underwent hypoxic postconditioning with administration of EX527 displayed similar FC characteristics to the SAH group, with strong deficits in somatosensory/parietal specific regions (Figure 3(c)). FC maps with the same six seed regions (detailed above) exhibit clear differences between SAH mice treated with hypoxic postconditioning, and SAH mice treated with hypoxic postconditioning and co-treated with EX527, across the posterior seed regions (e.g. Ss, Vis, Aud). Seed maps also display similarities between SAH and Hypoxia + EX527 groups, as well as between Sham and Hypoxia groups, across all regions (Supplemental Figure 1 A, rows 1–4). Comparing the global correlation coefficients, hypoxia treatment produced a mean increase of FC score (Figure 3(e), two-sample t-test p = .0430) and a significant increase in pixelwise connectivity as previously mentioned, but EX527 treatment did not significantly alter either global or pixelwise SAH outcomes. Adding the EX527 co-treatment, global FC was not significantly different from the SAH group in global FC metrics (two-sample t = test p = .6775), and pixelwise bilateral FC comparisons did not display any significant differences between SAH and EX527 groups (Figure 3(c)). These changes in global connectivity were correlated with behavioral deficits as quantified through Neuroscore (Figure 3(e)). This data strongly indicates that hypoxic postconditioning is protective against FC deficits induced by aneurysmal SAH injury, and that this protection is primarily mediated via SIRT1.
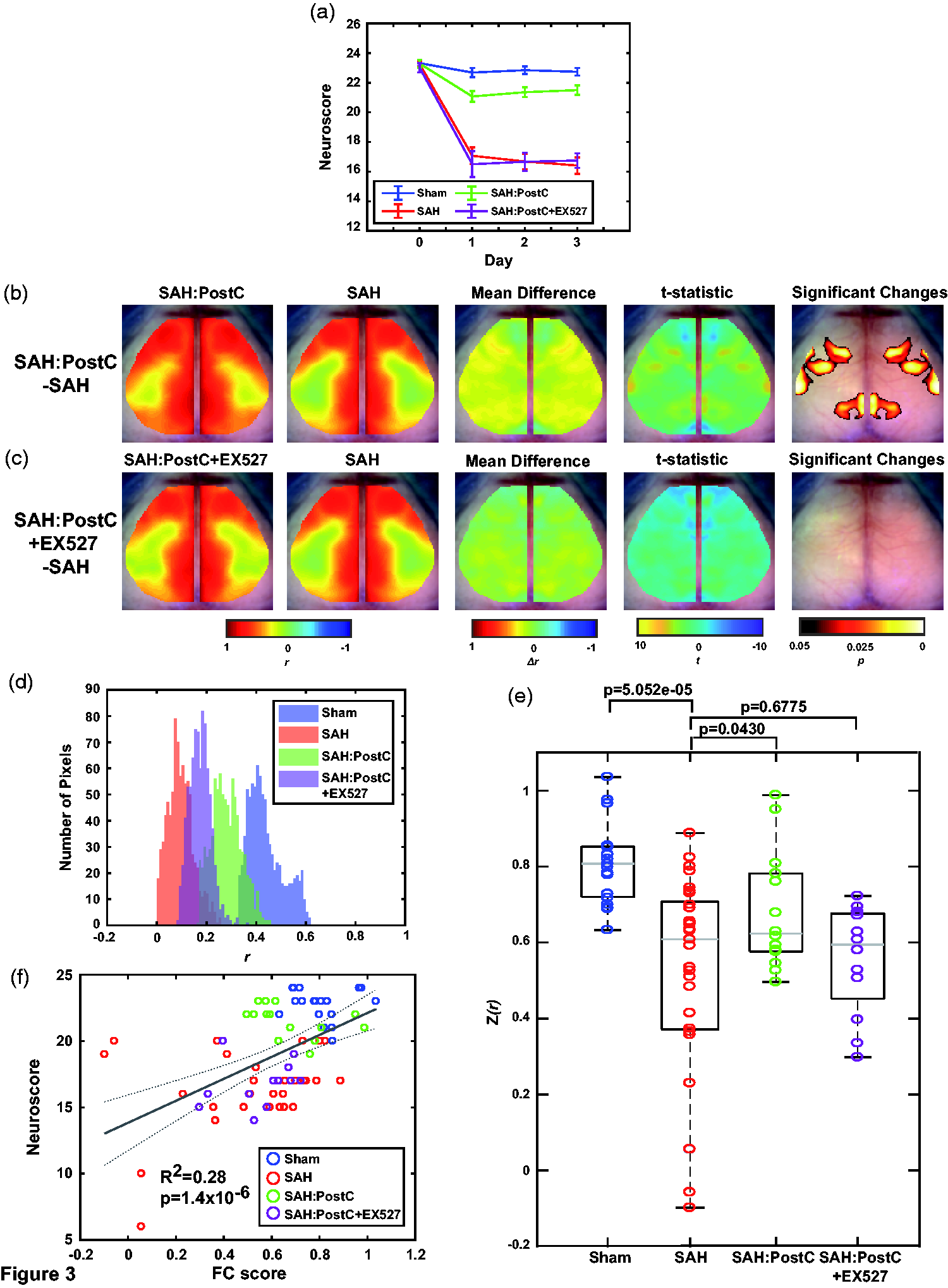
Hypoxic conditioning protects against SAH through a SIRT1 mediated mechanism. (a) Average Neuroscore for Sham and SAH mice compared to SAH mice that have been treated with Hypoxic conditioning with or without the SIRT1 inhibitor EX527 at baseline and Days 1–3. Error bars are SEM. (b) Group average bilateral homotopic connectivity maps for Hypoxia and SAH, their difference when SAH is subtracted from Hypoxia, and the t-statistic and significantly changed pixels after a cluster-based correction. (c) Group average bilateral homotopic connectivity maps for Hypoxia + EX527 and SAH, their difference when SAH is subtracted from Hypoxia + EX527, and the t-statistic and significantly changed pixels after a cluster-based correction. (d) Pearson correlation coefficient values per pixel plotted between Sham, SAH, Hypoxia, and Hypoxia + EX527 groups using the same ROI delineated in Figure 2(c). (e) Global average Pearson correlation coefficient from each mouse within each condition. Grey horizontal bar is the median, each box edge marks the 25th/75th percentile, whiskers extend to most extreme values not considered outliers. (f) Linear fit of Neuroscore vs the global FC average plotted in e). Dotted grey line corresponds to confidence bounds (n = 19 Sham, 29 SAH, 14 SAH:PostC, 12 SAH:PostC + EX527).
SIRT1 agonist resveratrol provides protection against SAH-induced FC deficits
After providing strong evidence that SIRT1 provides a protective effect against FC deficits following SAH, the next step was to examine whether this protection could be mediated pharmacologically with the SIRT1 activator Resveratrol (RSV). Mice underwent cranial windowing, SAH surgery, and RSV treatment following the experimental timeline (Figure 1(a) and (b), n = 16). Neuroscore assessment was performed at 24 h increments until the day of imaging to confirm the protective effect of RSV against SAH (Figure 4(a), 2-way rm-ANOVA, Sham vs RSV, F(1,33) = 14.833, p = 5e-4, SAH vs RSV, F(1,43) = 12.098, p = 0.0011). Bilateral homotopic connectivity maps show a mean increase in global FC in the RSV group when compared to the SAH group (Figure 4(b) vs Figure 2(b)). Pixelwise bilateral comparisons display significant increases in retrosplenial FC (Figure 4(b)). Somatosensory/parietal site-specific histogram comparisons display that RSV has a protective effect when compared to the SAH group in this highly effected area, as evident by the right shift (Figure 4(c)). While this effect is not as strong as hypoxic postconditioning, it is still indicative of the protection afforded by SIRT1 against FC deficits post-SAH. This protective phenomenon can be observed in the posterior seed maps comparing SAH, Sham, and RSV, as there are strong differences between sham and SAH groups, while the RSV group improves in comparison to SAH, but is still different when compared to sham (by comparison, the seed maps for hypoxic postconditioning showed fewer differences from sham, Supplemental Figure 1 A). Also noted is the trending increase in global connectivity in RSV compared to SAH (Figure 4(d)); the global FC comparisons display a mean increase in RSV compared to SAH (two-sample t-test p = .0777), that corresponds to the significant pixel-wise bilateral increases observed in Figure 4(b). These changes in connectivity were correlated with behavioral deficits as quantified through Neuroscore (Figure 4(e)). This data is evidence that RSV exhibits a protective effect against FC deficits after SAH. These results also suggest that SIRT1, while heavily involved, may not be the only mediator of the protection afforded by hypoxic postconditioning.
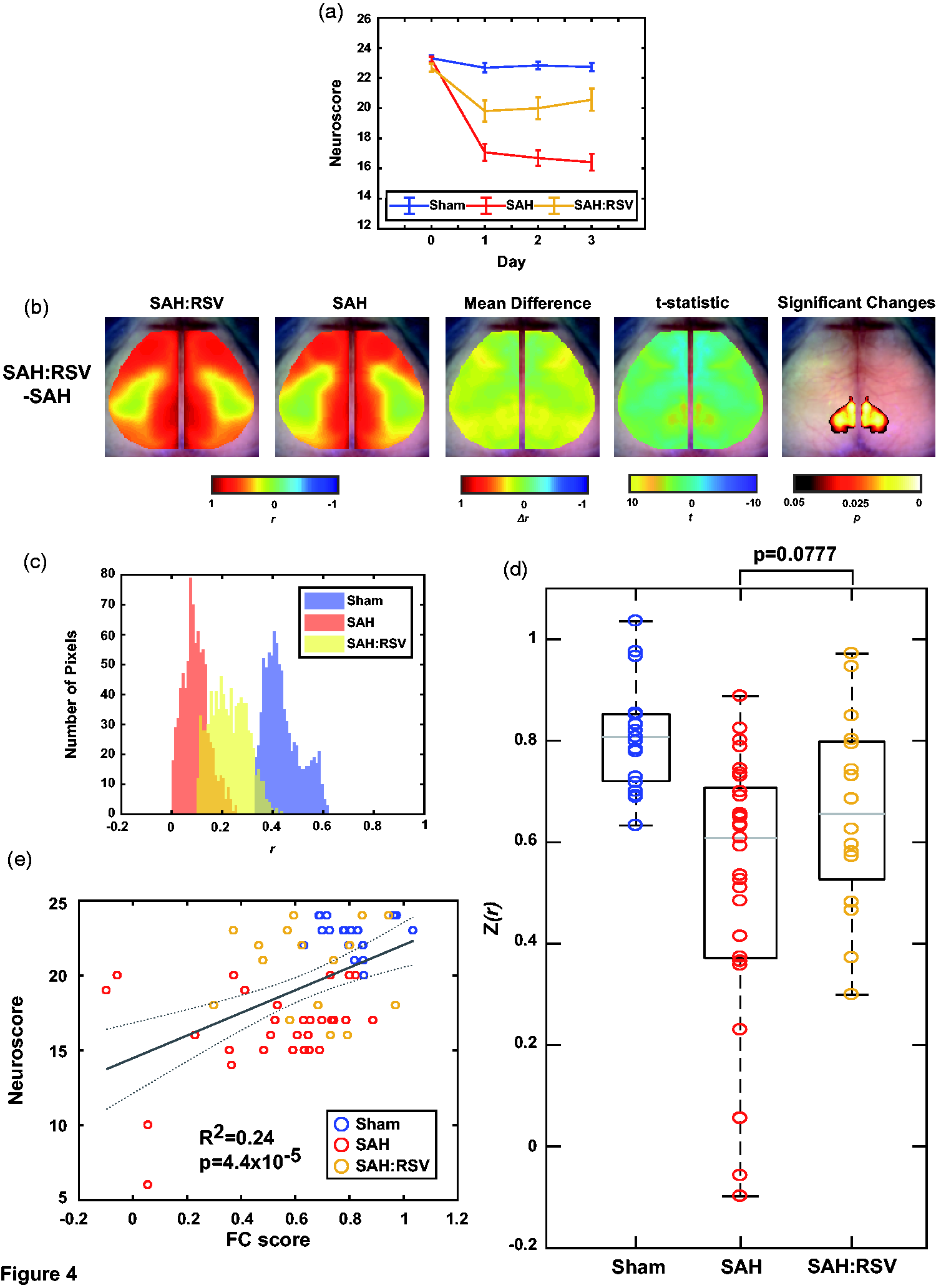
SIRT1 agonist Resveratrol protects against FC deficits in SAH. (a) Average Neuroscore for Sham and SAH mice compared to SAH mice that have been treated with RSV at baseline and Days 1–3. Error bars are SEM. (b) Group average bilateral homotopic connectivity map for RSV and SAH, their difference when RSV is subtracted from SAH, and the t-statistic and significantly changed pixels after a cluster-based correction. (c) Pearson correlation coefficient values per pixel plotted between Sham, SAH, and RSV groups using the same ROI delineated in Figure 2(c). (d) Global average Pearson correlation coefficient from each mouse within each condition. Grey horizontal bar is the median, each box edge marks the 25th/75th percentile, whiskers extend to most extreme values not considered outliers (*p < 0.05, two sample student’s t-test). (e) Linear fit of Neuroscore vs the global FC average plotted in d). Dotted grey line corresponds to confidence bounds (n = 19 Sham, 29 SAH, 16 SAH:RSV).
Discussion
The key novel findings from the present study are as follows: 1) The endovascular perforation model of SAH induction causes profound global deficits in FC; 2) Hypoxic conditioning initiated at a clinically relevant time point after SAH induction provides robust protection against neurological deficits and FC deficits; 3) Hypoxic postconditioning-induced protection against FC deficits is critically dependent on SIRT1; 4) A therapeutic strategy designed to activate SIRT1 mimics the protective effects of hypoxic postconditioning against FC deficits. These data strongly suggest that conditioning, or molecular therapies based on conditioning, carry great promise as a novel strategy against SAH-induced brain injury and neurological deficits. They also suggest SAH-induced deficits in FC may represent a measurable imaging biomarker that could be leveraged when assessing efficacy of new therapeutics in SAH including conditioning-based agents.
DCI is the most common treatable cause of secondary neurovascular injury following aneurysmal SAH. 73 Our first objective was to determine whether or not experimental SAH causes deficits in functional connectivity (FC) that correspond to the timeline of DCI onset. Mice underwent SAH or sham surgery and were imaged 3 days later using fcOIS, corresponding to an experimentally reproducible timeline for the occurrence of DCI in multiple mouse models. 72 Significant neurological deficits in mice that underwent SAH induction were noted. Functional neuroimaging displayed deficits that mirrored this behavioral readout, providing strong evidence that experimental SAH causes both global and region-specific deficits in mouse FC. Using a translational therapeutic approach, we then sought to use this FC readout to monitor whether hypoxic postconditioning would provide functional neuroprotection following experimental SAH. Hypoxic postconditioning has been proven to mitigate major deleterious effects of DCI and protect against both large artery vasospasm and microvascular thrombosis in a SIRT1-mediated fashion. 31 Here, we demonstrated that moderate repetitive hypoxia, when initiated at the clinically relevant time point of 3 hours post-SAH, provided robust protection against the FC deficits caused by SAH while also improving the neurological outcome. We also showed that co-treatment with the SIRT1 inhibitor, EX527, blocked the protective effect on both FC and neurological deficits, validating the hypothesis that the protection afforded by hypoxic postconditioning is mediated primarily by SIRT1.
Next, we sought to determine whether the SIRT1 activator, resveratrol, mimics the protection afforded by hypoxic postconditioning against FC deficits. Following the same experimental timeline, but replacing hypoxic postconditioning with resveratrol administration, we discovered that resveratrol also provided significant protection against both global and regional-specific FC deficits caused by SAH, but that the magnitude of this protective effect was less than that seen with hypoxic postconditioning. The reason for this differential level of protection is likely due to the fact that the protective effects of hypoxic postconditioning on SAH-induced FC deficits may be multifaceted, with SIRT1 serving as a key inducer, but other molecules also playing important secondary roles in the overall protective effect.
Recently, Chung et al. used the prechiasmatic injection model of SAH induction to explore FC deficits after experimental SAH injury. 60 This is a novel study that provides evidence of both early (4 days) and persistent (3 months) deficits in FC after SAH when compared to sham animals. This study also describes a significant decrease in global functional connectivity that corresponds behavioral deficits in mice in both Morris water maze and Y-maze testing. 60 Our study builds on these findings. We provide evidence of significant global FC deficits in the endovascular perforation model of SAH induction and show a correlation between neurological deficits and FC deficits. We also present a novel, molecular-targeted therapeutic strategy for prevention against these deficits in a mouse model of experimental SAH.
While the Chung et al. study was the first to examine FC in the context of experimental SAH, other studies have examined FC deficits in ischemic stroke, a cerebrovascular injury that is one of the leading causes of death and disability in adults. 74 Bauer et al. first examined ischemic stroke using fcOIS in 2014, imaging before and 72 h after transient middle cerebral artery occlusion (tMCAO). 58 This study demonstrated that FC patterns within the regions supplied by the middle cerebral artery (MCA) were most negatively affected by ischemic stroke, and that tMCAO causes homotopic FC metrics to incrementally decline towards zero with increasing infarct severity. 58 The largest infarcts resulted in no significant homotopic correlations over the entire field-of-view, suggesting that temporal synchrony between homotopic brain regions is globally affected by ischemia; similar to the severe decline in homotopic FC we present 72 h post-SAH. Mice that experienced ischemic stroke with moderate and large infarctions also seemed to have more motor and visual cortex deficits 72 h post-injury, while SAH mice seem to have more severe somatosensory deficits. Other studies examining stroke using fcOIS looked at prothrombotic (PT) stroke 14 days post-injury; while 14 days is not as relevant in DCI, which has a more acute time course, brain hemispheres 14 days post-PT still showed a strong decrease in homotopic FC in two separate studies.75,76 Additionally, one study by Hakon et al. displayed that mice given access to an enriched environment that provided multisensory stimulation (through cages equipped with toys, tubes, chains, ladders, and platforms) after ischemic stroke had improved FC in the motor cortex after 14 days, but not in the somatosensory cortex. 75 We have shown that SIRT1-mediated therapies can improve FC in the somatosensory cortex post-SAH in 3 days, and it may be worth exploring this same SIRT1 therapy method in the context of ischemic stroke. The similarities in the anatomical effects of SAH and ischemic stroke (i.e. cell death) also gives potential insights into the underlying mechanistic causes for the bilateral FC decrease observed in this study’s SAH mice. The death of cortical tissue results in no typical hemodynamic-related synchronized signal that can be correlated with the affected region’s homotopic cortex in the opposite hemisphere. Therefore, the logical results of cell death or severe structural damage in on region but not its homotopic counterpart would be a loss of firing synchrony that manifests as a decrease in bilateral FC.
Functional connectivity is just one of the many endpoints that have been explored in the context of hypoxic conditioning and SAH, along with vasospasm and microvascular thrombosis30,31 One thing that separates FC from other endpoints is that it is a viable option to examine the effects of SIRT1-mediated therapies in humans through fMRI. It has already been demonstrated using fMRI that cognitive impairments due to aneurysmal SAH can be characterized by alterations in FC, with seed-based FC maps showing significant differences in impaired SAH patients in the frontoparietal network, left thalamus, left parahippocampal gyrus, and left inferior temporal gyrus.77,78 Memory impairment after aneurysmal SAH has also been examined using fMRI, with one study suggesting that SAH-induced memory deficits may be related to disruption of critical functional connections involving the medial temporal lobe.78,79 Recently, it was also demonstrated that FC examined using magnetoencephalography (MEG) also displays differences in FC patterns between controls and aneurysmal SAH patients. 80 We therefore propose that the FC analysis presented here builds upon previously explored experimental endpoints of DCI, such as large artery vasospasm 81 and microvascular thrombosis, 68 and provides a promising future for the utilization of novel therapies, such as SIRT1 activation, for SAH in human populations.
Future experimental studies
This study has established that aneurysmal SAH causes FC deficits in wild-type C57/B6 mice, and that hypoxic postconditioning and resveratrol effectively protect against these deficits in a SIRT1-mediated fashion. As increasing evidence suggests that protection through SIRT1 is effective in SAH, an alternative method of confirming this protection would be through experiments using genetically modified mice. Tamoxifen-inducible whole-body Sirt1-null mice (Cre-ert2+/−; Sirt1Δex4/Δex 4 ) could be used to further examine the SIRT1-mediated protective effects of hypoxic postconditioning against FC deficits after SAH. Constitutive whole-body Sirt1-overexpressing transgenic mice (Sirt1-Tg) on a pure C57BL/6J background could also be used to examine the effect of increased SIRT1 expression on FC deficits after SAH. Further, crossing these models with a mouse expressing a genetically encoded calcium indicator (GECI) driven by a Thy1 promotor (GCaMP6, JAX #024276) would allow for mesoscopic, neural specific imaging across the same field-of-view as presented here.82,83 Using concurrent fluorescent (provided by GCaMP6) and OIS imaging, we could obtain a direct neural calcium-based and hemoglobin-based readout. This approach would allow for further applications such as examining neurovascular coupling or determining if functional deficits are vascular or neural based, throughout SAH. Using forepaw stimulation-induced vessel dilation measurements by laser Doppler flowmetry, Balbi and colleagues recently demonstrated a difference in neurovascular coupling between sham and SAH mice one month after experimental SAH. 84 Utilizing concurrent fluorescent imaging with OIS in awake mice with forepaw stimulation would build upon this study by permitting serial temporal assessment of neurovascular coupling.
Limitations
Our study is not without limitations. 1) We measured FC under anesthesia, rather than in awake mice, as the feasibility of optical measured of FC in awake animals is still under investigation. 85 Future studies with assessment of FC in awake animals would strengthen our findings. 2) We only used male mice in our study. 3) As stated in the introduction, the optical approach is limited to imaging the surface of the mouse cerebral cortex and is therefore not as complete as fMRI when it comes to assessing the connectivity to deeper structures. This is important, as subarachnoid hemorrhage through the endovascular perforation model happens at the subcortical level. 3) We only used male mice in our study. 4) We only used one animal model of SAH induction. There are now studies with two separate animal models that thoroughly investigate the effects of SAH on FC in a mouse model. As there is no perfect animal model of SAH induction, further studies using a variety of SAH induction techniques in mice and other animals will no doubt improve our understanding of this devastating disease process. 5) We used resveratrol as a SIRT1 activator. While this is well-documented activator of SIRT1, it, like many pharmacologic agents, also has off-target effects. Future studies utilizing genetic means for augmenting SIRT1 expression will be needed to cross-validate our findings with resveratrol.
Conclusion
This study demonstrates that experimental SAH causes significant deficits in functional connectivity, and that hypoxic postconditioning, as well as the pharmacological approach resveratrol, exhibits a strong protective effect against these deficits along with robust improvement in neurological outcome after SAH. These protective effects are critically dependent on SIRT1. These results raise the possibility that SIRT1-directed therapies may provide protection against functional connectivity deficits in other cerebrovascular conditions, such as ischemic stroke. They also provide further evidence that the delay between SAH and DCI is a window of opportunity for a SIRT1-based postconditioning strategy. Future studies should not only continue to examine the effect that conditioning-based therapies have on FC in rodents, but also explore the possibility of testing these therapies in humans using FC.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X221079902 - Supplemental material for SIRT1 mediates hypoxic postconditioning- and resveratrol-induced protection against functional connectivity deficits after subarachnoid hemorrhage
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X221079902 for SIRT1 mediates hypoxic postconditioning- and resveratrol-induced protection against functional connectivity deficits after subarachnoid hemorrhage by Julian V Clarke, Lindsey M Brier, Rachel M Rahn, Deepti Diwan, Jane Y Yuan, Annie R Bice, Shin-ichiro Imai, Ananth K Vellimana, Joseph P Culver and Gregory J Zipfel in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institute of Neurological Disorders and Stroke (grant numbers R01NS091603 [to G.J.Z.], R01NS099429 [to J.P.C.], P01NS080675 [to J.P.C.]), the National Institute on Aging (grant number F30AG061932 [to L.M.B.]) and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (grant number U54HD087011 [to J.P.C.])).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
Clarke, JV and Brier, LM contributed to experimental and research design, performed experiments, contributed analytic tools, analyzed data, and wrote the initial manuscript. Rahn, RM contributed analytic tools, analyzed data, and contributed to experimental research and design. Diwan, D contributed to experimental design, data analysis, and paper writing. Yuan, JY and Bice, AR contributed to performing experiments and data analysis. Imai, S contributed significantly to experimental and research design. Clarke, JV, Imai, S, Vellimana, AK, and Zipfel, GJ conceived of the presented idea. Culver, JP and Zipfel, GZ contributed to experimental design and asked critical questions to guide the research process from inception to completion. Vellimana, AK and Culver, JP contributed significantly to critical assessment and editing of the initial manuscript. Zipfel, GJ also contributed to writing and editing the initial manuscript. All authors lent their critical thinking and assessment to the final manuscript.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.