
Abstract
Only partial deficiency/inhibition of P-glycoprotein (P-gp, ABCB1) function at the blood-brain barrier (BBB) is likely to occur in pathophysiological situations or drug-drug interactions. This raises questions regarding the sensitivity of available PET imaging probes to detect moderate changes in P-gp function at the living BBB. In vitro, the half-maximum inhibitory concentration (IC50) of the potent P-gp inhibitor tariquidar in P-gp-overexpressing cells was significantly different using either [11C]verapamil (44 nM), [11C]N-desmethyl-loperamide (19 nM) or [11C]metoclopramide (4 nM) as substrate probes. In vivo PET imaging in rats showed that the half-maximum inhibition of P-gp-mediated efflux of [11C]metoclopramide, achieved using 1 mg/kg tariquidar (in vivo IC50 = 82 nM in plasma), increased brain exposure by 2.1-fold for [11C]metoclopramide (p < 0.05, n = 4) and 2.4-fold for [11C]verapamil (p < 0.05, n = 4), whereby cerebral uptake of the “avid” substrate [11C]N-desmethyl-loperamide was unaffected (p > 0.05, n = 4). This comparative study points to differences in the “vulnerability” to P-gp inhibition among radiolabeled substrates, which were apparently unrelated to their “avidity” (maximal response to P-gp inhibition). Herein, we advocate that partial inhibition of transporter function, in addition to complete inhibition, should be a primary criterion of evaluation regarding the sensitivity of radiolabeled substrates to detect moderate but physiologically-relevant changes in transporter function in vivo.
Keywords
Introduction
P-glycoprotein (P-gp, ABCB1) is an important efflux transporter of the ATP-binding cassette (ABC) transporter family, which is notably expressed at the vascular pole of endothelial cells forming the blood-brain barrier (BBB). P-gp selectively regulates the BBB transfer of many drugs, metabolites and endogenous compounds. 1 There is growing evidence that P-gp expression and function are impaired in various conditions, including normal ageing, Alzheimer’s disease or neuroinflammatory states. 2 Overexpression of P-gp has been reported in brains of rat epilepsy models 3 and in patients with epilepsy,4,5 suggesting a relationship with resistance to anti-seizure medication. Active P-gp-mediated efflux is therefore increasingly regarded as a dynamic biomarker of BBB function, which may be linked to the onset of neurological disorders. 2
Preclinical studies have shown that the brain distribution of many positron emission tomography (PET) imaging radioligands is restricted by P-gp function at the BBB.6,7 However, only few radiolabeled substrates have so far been used as dedicated probes to selectively assess P-gp function at the human BBB. These include [ 8 C]verapamil 9 (racemic or the (R)-enantiomer analogue of the calcium channel blocker verapamil), [11C]N-desmethyl-loperamide 10 (a radiolabeled metabolite of loperamide, an antidiarrheal opioid drug) and [11C]metoclopramide 11 (analogue of metoclopramide, a dopamine receptor antagonist used as an antiemetic drug). The particular characteristics of P-gp-mediated transport at the BBB level as compared to conventional neuroimaging targets (e.g. receptor proteins) have prompted the need for new criteria to describe and select effective PET probes for P-gp activity. 8 A first prerequisite is selectivity for P-gp over breast cancer resistance protein (BCRP, ABCG2), the other major ABC transporter at the BBB. Second, a limited contribution of radiometabolites to the brain PET signal is preferred for correct estimation of P-gp function at the BBB. Third, “avid” substrates were initially preferred as probes, with the aim to generate maximal contrast in the PET signal between the baseline condition, in which P-gp is fully functional, and a pharmacological inhibition challenge in which P-gp-mediated efflux is attenuated or abolished. Strains of P-gp deficient mice are useful tools to assess this last criterion in vivo.12,13 However, from a clinical perspective, complete deletion or genetic deficiency of P-gp expression is extremely rare in humans. 14 Indeed, only partial decreases in P-gp expression have been found in histology or proteomics studies of human specimens.2,15 This raises the important question of the sensitivity of available PET probes to detect the functional impact of moderate changes in P-gp expression at the BBB.
PET studies using either [11C]verapamil or [11C]N-desmethyl-loperamide have clearly shown the efficacy of pharmacological inhibitors to dose dependently decrease P-gp function at the human BBB.16,17 Tariquidar is a highly potent and specific P-gp inhibitor that can be safely used to achieve an almost maximal decrease in P-gp function at the human BBB.16–18 Many marketed drugs are described as P-gp inhibitors in vitro, although with a lower potency or efficacy than tariquidar.19,20 Clinical PET data using [11C]verapamil or [11C]N-desmethyl-loperamide as model P-gp substrates have been used as reference points to estimate the risk for P-gp-mediated drug-drug interactions at the human BBB. 21 These results led to the assumption that a high degree of P-gp inhibition by marketed drugs is very unlikely to occur, so that only partial inhibition can be achieved in clinical practice.21,22
[11C]Metoclopramide has been designed as an alternative P-gp probe for PET imaging, based on the need for a PET probe to detect induction of P-gp function at the BBB, starting from substantial baseline brain uptake, when P-gp is normally expressed.23,24 [11C]Metoclopramide corresponds to the definition of a “weak” P-gp substrate, thus mimicking the characteristics of many CNS active drugs including antiepileptic drugs, antidepressants, antipsychotics or opioids. 4 The overall increase in brain exposure to [11C]metoclopramide after a high degree or nearly complete P-gp inhibition was consequently lower than that observed with the “avid” substrates [11C]verapamil or [11C]N-desmethyl-loperamide.24–26 However, it may be hypothesized that [11C]metoclopramide with its lower transport rate and higher BBB permeability may show better sensitivity to detect partial P-gp inhibition compared with previous PET radioligands. 26 This would imply a higher “vulnerability” to clinically relevant drug-drug interactions with P-gp inhibitors, resulting in increased brain exposure.
The present study aimed at comparing the “vulnerability” of [11C]verapamil, [11C]N-desmethyl-loperamide and [11C]metoclopramide to partial P-gp inhibition. To this end, standardized in vitro uptake assays using cells expressing human P-gp and in vivo PET experiments in rats were performed with these 3 substrates, in the absence or presence of increasing doses of tariquidar. Based on these data, we propose new and important criteria to describe and compare the sensitivity of radiolabeled substrate probes to detect moderate changes in P-gp function at the BBB.
Materials and methods
Chemicals and radiotracers
Tariquidar used for P-gp inhibition was purchased from Eras Labo (France). Tariquidar solutions for intravenous injection were freshly prepared at the selected concentration on the day of the experiment by dissolving tariquidar dimesylate in dextrose solution (5%, w/v), followed by dilution with sterile water.
Three radiolabeled P-gp probes suitable for human use were compared. Ready-to-inject [11C]metoclopramide, racemic [11C]verapamil and [11C]N-desmethyl-loperamide were synthetized as previously described27–29 starting from cyclotron-produced [11C]carbon dioxide (Cyclone-18/9 cyclotron; IBA, Belgium) by [11C]methylation of nor-metoclopramide (Toronto Chemicals, Canada), racemic nor-verapamil (ABX advanced biomedical compounds, Germany) or in-house synthesized di-desmethyl-loperamide 28 using a TRACERLab FX CPro synthesizer (GE Healthcare, France). Quality control was performed by radio-high-performance liquid chromatography (HPLC) to assess the identity, radiochemical and chemical purities of the radiotracers and their molar activities.
Cells
Culture media and buffers were obtained from Fischer Scientific, France. Stably transfected MDCKII-MDR1 cells were obtained from Dr. Alfred Schinkel (National Cancer Institute, The Netherlands) and were grown under a controlled atmosphere at 37 °C, 5% CO2. The culture medium was composed of DMEM Glutamax (Dulbecco’s Modified Eagle Medium, 4.5 g/L dextrose, 1 mM pyruvate) supplemented with 10% fetal bovine serum and 1% antibiotics (penicillin and streptomycin 5000 U/mL).
Uptake assay in P-gp overexpressing cells
P-gp-mediated transport of [11C]verapamil and [11C]N-desmethyl-loperamide and [11C]metoclopramide was compared in cells expressing human P-gp. Cells were seeded in 24-well plates (30,000 cells per well) in 500 μL culture medium. Cells were grown to confluence (∼2 days). On the day of the experiment, culture medium was removed and replaced by 200 μL of incubation buffer (10% HBSS (Hanks’ Balanced Salt Solution) + 1.26 mM CaCl2 + 0.49 mM MgCl2) containing 1 mM pyruvate and 10 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 37 °C). The incubation buffer contained the tested radiolabeled P-gp substrate (∼37 MBq/40 mL corresponding to <1 µg/40 mL of the unlabeled compound) and tariquidar at the selected concentration. Tariquidar was dissolved in DMSO and tariquidar concentrations ranged from 0 to 200 nM (1% v/v final DMSO concentration). After 30 minutes of incubation at 37 °C, buffer was removed and cell monolayers were rapidly washed with 300 μL of ice-cold buffer Dulbecco’s phosphate buffer. Cells were then lysed with 500 μL of NaOH (10 mM, 10 min). Then, 400 μL of cell lysate was collected from each well (n = 4 wells per condition) and gamma-counted using a Cobra Quantum (Perkin-Elmer, France).
Animals
Thirty-six male Sprague Dawley rats (Janvier, France) were used for the study (mean weight: 354 ± 76 g). Animals were housed and acclimatized for at least 3 days before the experiments. Rats had free access to chow and water. All animal experiments were in accordance with the recommendations of the European Community (2010/63/UE) and the French National Committees (law 2013-118) for the care and use of laboratory animals. The experimental protocol was approved by a local ethics committee for animal use (CETEA) and by the French ministry of agriculture (APAFIS#7466-20 1611 04 1 7049220 v2). Samples size for each group was based on previous studies.24,30 Reporting of animal data is in compliance with the ARRIVE (Animal Research: Reporting in Vivo Experiments) guidelines.
PET experiments
PET protocol
PET acquisitions were performed using an Inveon microPET scanner (Siemens, Knoxville, TN, US). Anesthesia was induced and then maintained using 3.5% followed by 1.5-2.5% isoflurane in pure oxygen. Thirty-minute dynamic scans were acquired, starting with intravenous bolus injection of [11C]verapamil (40 ± 8 MBq; 2.0 ± 1.5 µg), [11C]N-desmethyl-loperamide (35 ± 5 MBq, 1.7 ± 1.3 µg) or [11C]metoclopramide (35 ± 5 MBq, 3.4 ± 1.3 µg), via a catheter inserted in the caudal lateral vein.
P-gp inhibition protocol
Tariquidar was used to modulate P-gp function in rats. Tariquidar doses up to 8 mg/kg were shown to be well tolerated with a negligible impact of tariquidar on the arterial input function and peripheral metabolism of [11C]verapamil, 31 [11C]N-desmethyl-loperamide 30 or [11C]metoclopramide. 24 Tariquidar was injected in a volume of 200–300 µL into the caudal vein at 15 minutes before radiotracer injection. For [11C]metoclopramide, a range of different tariquidar doses was tested (vehicle-0, 1, 2, 3, 4 or 8 mg/kg). PET acquisitions using [11C]verapamil or [11C]N-desmethyl-loperamide were performed after injection of vehicle and 1 or 8 mg/kg of tariquidar (n = 4 per condition).
Determination of tariquidar in plasma
Blood samples (∼2 mL) were collected under anesthesia at the end of the PET acquisition (i.e. at 65 ± 2 min after tariquidar injection) by intracardiac puncture. Afterwards animals were killed using 180 mg/kg i.v. pentobarbital (Dolethal®, France). Samples were centrifuged and plasma was collected and stored at −80°C until analysis. Determination of tariquidar concentration in plasma samples was performed using a validated HPLC method using elacridar as an internal standard. 32 Calibration curves for the analysis of plasma samples were generated by analyzing different dilutions of a tariquidar solution in drug-free human plasma (obtained from Etablissement Français du Sang, France).
Data analysis and statistics
In vitro data
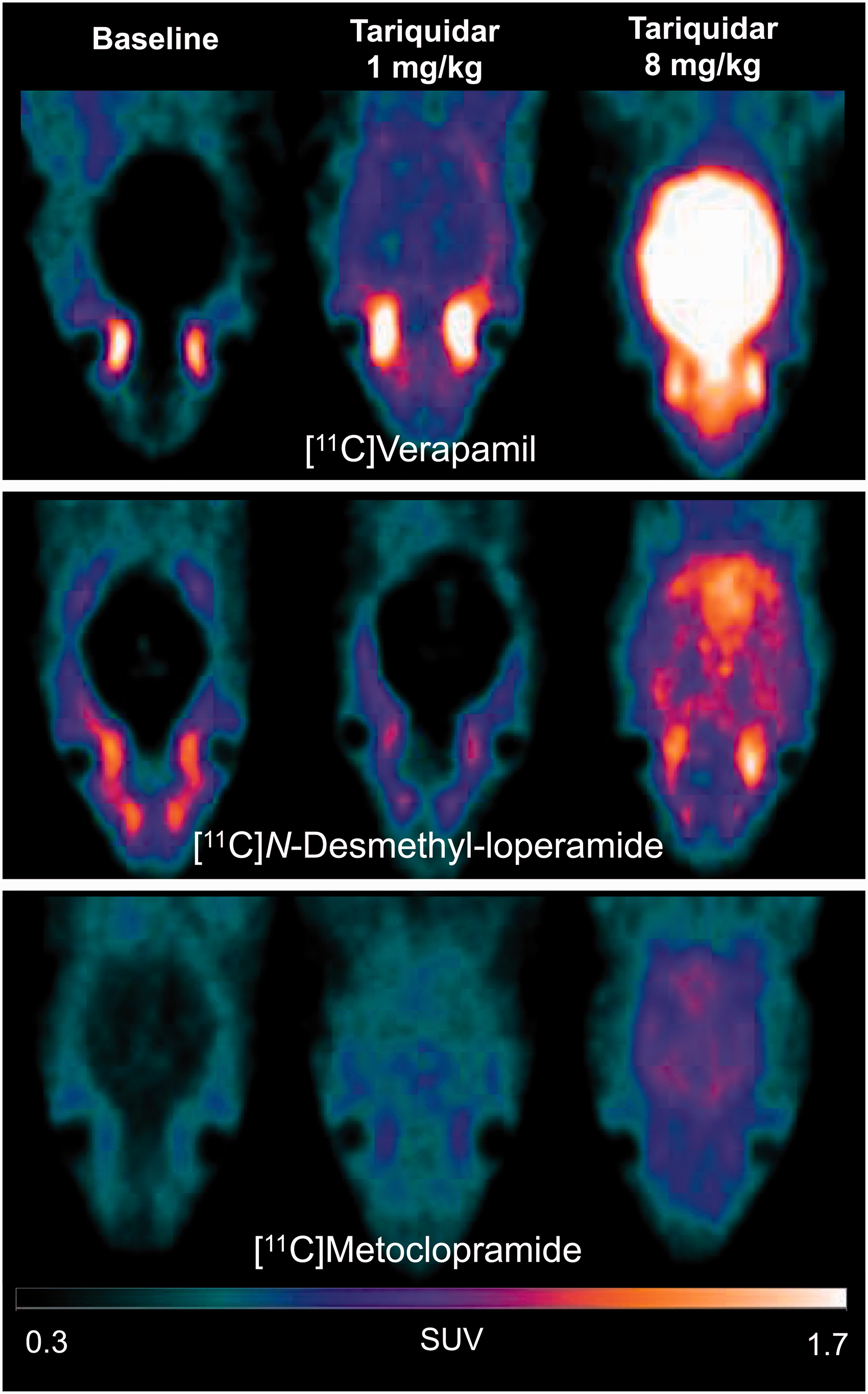
Counting values obtained in each well (R) were normalized using either the concentration of radioactivity in the incubation buffer (%C0 = 100 × R/C0) or using the following equation:
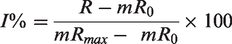
The in vitro half-maximum inhibitory concentration (IC50) was estimated by non-linear regression using the “One-site binding Hill equation” function in Graphpad Prism software (V8.0, San Diego, CA, USA) with maximal uptake constrained to 100%. IC50 values obtained for each radiotracer were considered different when their 95% confidence interval [CI95%] did not overlap. The uptake ratio was calculated as Rmax/mR0 (mean ± S.D) for each P-gp substrate. Normality of the data was checked using the Shapiro-Wilk’s test and homoscedasticity using the Brown-Forsythe’s test. Uptake ratios were then compared using one-way ANOVA and Tukey’s post hoc test.
PET data analysis
Images were reconstructed with the Fourier rebinning algorithm and the 3-dimensional ordered-subset expectation maximization algorithm including normalization, attenuation, scatter, and random corrections. Image analysis and quantification of radioactivity uptake were performed using PMOD software (version 3.9; PMOD Technologies, Zürich, Switzerland). A region of interest was drawn over the whole brain to generate time–activity curves (TACs) with time frame durations of 0.25 min, 2 × 0.5 min, 0.75 min, 4 × 1 min, 1.5 min, 4 × 2 min, 3 × 2.5 min, 3 × 3 min and 3.5 min. Brain radioactivity was corrected for 11C decay, injected dose and animal weight, and expressed as the standardized uptake value (SUV).
In rats, the determination of an arterial input function during the PET scan is very challenging. The impact of P-gp inhibition was therefore estimated from the area under the brain TAC, which was determined from 10 to 30 minutes after radiotracer injection (AUC10-30 min) to limit the pharmacokinetic variability associated with the bolus injection procedure. First, the doses that inhibited 50% (ID50%/Meto) and 100% (ID100%/Meto) of the P-gp-mediated transport of [11C]metoclopramide were estimated by fitting plots of brain AUC10-30 min versus the injected dose of tariquidar (n = 1–4 animals per dose) with the non-linear one-site binding Hill equation. Brain AUC10-30 min was also plotted versus the plasma concentration of tariquidar to estimate IC50%/Meto, corresponding to the plasma concentration of tariquidar that inhibited 50% of the P-gp-mediated efflux of [11C]metoclopramide at the BBB. Then, PET experiments were performed using [11C]verapamil and [11C]N-desmethyl-loperamide at baseline and after administration of tariquidar at ID50%/Meto or ID100%/Meto (n = 4 per condition).
Graphical and statistical analysis were performed using Graphpad® prism software. Normality of the data was checked using the Shapiro-Wilk’s test and homoscedasticity using the Spearman’s test. Brain AUC10-30 min values obtained under all tested conditions were compared using 2-way ANOVA analysis and compared with the baseline condition using the Dunett’s post-hoc test.
Results
In vitro experiments
The in vitro P-gp inhibitory potency of tariquidar was tested in P-gp overexpressing cells using either [11C]verapamil, [11C]N-desmethyl-loperamide or [11C]metoclopramide as substrate probes. The fitted concentration-inhibition curves for each radiotracer are shown in Figure 1. Cellular uptake of the tested substrates was not significantly higher in the presence of 200 nM vs 100 nM tariquidar suggesting that maximal inhibition was achieved using 200 nM tariquidar. The estimated IC50 of tariquidar was 44 nM [33–53 nM] for [11C]verapamil, 19 nM [13-25 nM] for [11C]N-desmethyl-loperamide and 4 nM [2-8 nM] for [11C]metoclopramide. The CI95% of the IC50 values of the different tested substrates did not overlap, suggesting that each substrate’s “vulnerability” to partial P-gp inhibition was significantly different (Figure 1). Uptake ratios were 1.4 ± 0.09 for [11C]verapamil, 3.9 ± 0.64 for [11C]N-desmethyl-loperamide and 1.4 ± 0.07 for [11C]metoclopramide. For [11C]metoclopramide, maximal inhibition was achieved with ∼50 nM of tariquidar. At this concentration, only 58% of the P-gp-mediated transport of [11C]verapamil and 70% of the transport [11C]N-desmethyl-loperamide was inhibited (Figure 1).
Uptake of [11C]verapamil, [11C]N-desmethyl-loperamide and [11C]metoclopramide in MDCKII-MDR1 cells in presence of increasing doses of tariquidar (TQD). Intracellular uptake is expressed as the percentage of radioactivity in the cell lysate relative to the incubation buffer (%C0, in A) and as the percentage of maximal inhibition (I%, assuming maximal inhibition was achieved using tariquidar 200 nM, in B). Lines in B represent fits of Hill model employed to estimate the in vitro half-maximum inhibitory concentration (IC50) with corresponding 95% confidence interval (CI95%), n = 4 per condition, reported in C. In D, uptake ratios are reported as mean ± S.D (n = 4 per condition). ****p < 0.0001, ns = not significant (one-way ANOVA with Tukey’s post-hoc test for multiple comparisons).
In vivo experiments
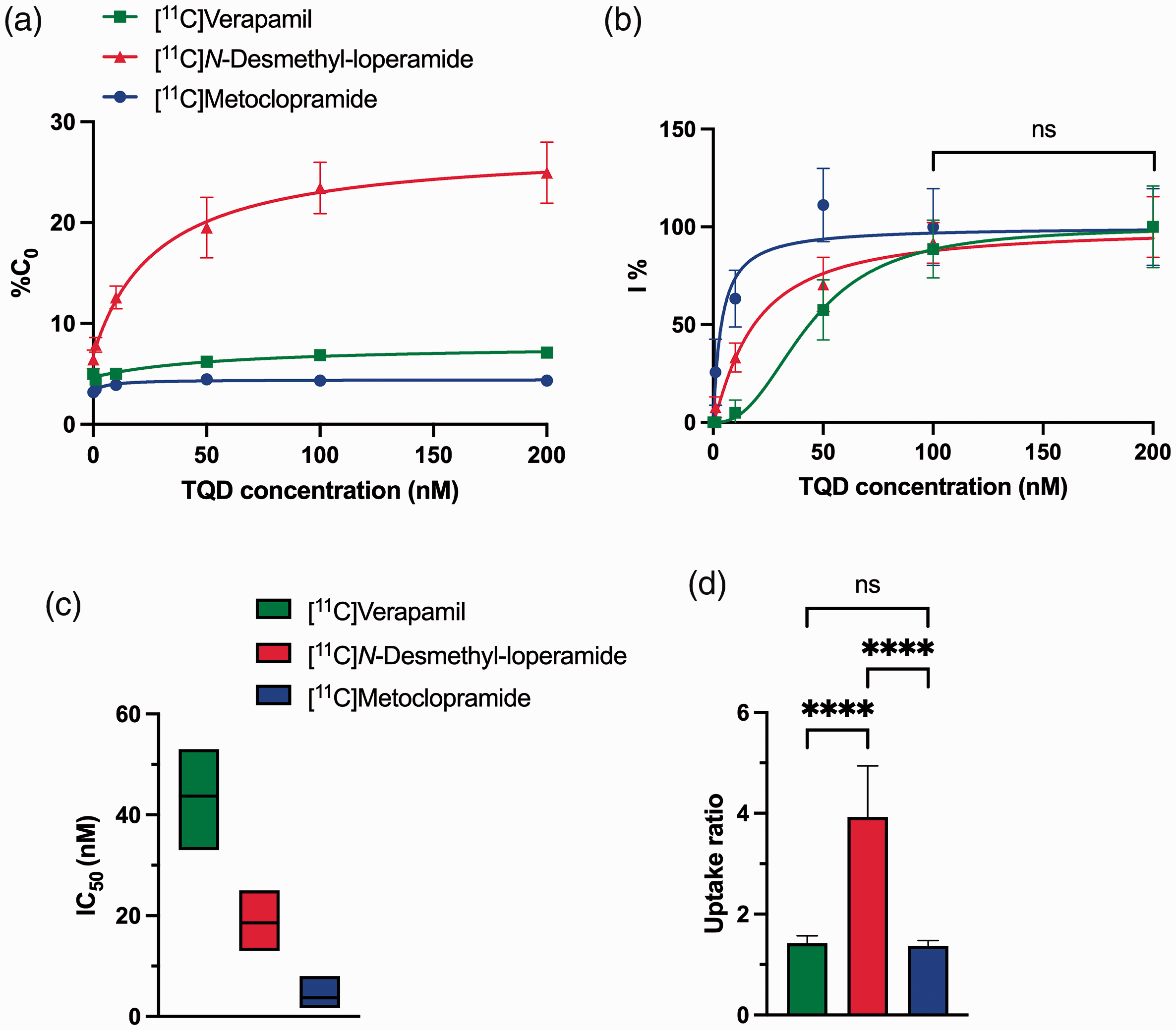
Under the baseline condition, [11C]metoclopramide PET signal in the rat brain was lower than that in tissues surrounding the skull (Figure 2). The PET signal reached similar levels as peripheral tissues in rats injected with 1 mg/kg of tariquidar. Tariquidar doses >1 mg/kg dose dependently increased the brain PET signal with a limited impact on radioactivity in peripheral tissues (Figure 2). This is consistent with a localization of P-gp at the BBB and a negligible impact of tariquidar on the peripheral pharmacokinetics of [11C]metoclopramide. 24
Dose- and concentration-response of tariquidar (TQD) for inhibition of P-gp-mediated transport of [11C]metoclopramide at the rat BBB. In A, representative summed brain PET images are shown for each condition. Brain exposure (AUC10-30 min) of [11C]metoclopramide as a function of administered tariquidar dose (B) or tariquidar plasma concentration measured at the end of the PET scan (C). Dotted lines in B and C represent fits of the employed Hill model.
Dose-response and concentration-response curves of tariquidar for inhibition of P-gp-mediated transport of [11C]metoclopramide at the rat BBB are shown in Figure 2. The estimated ID50%/Meto of tariquidar was 1 mg/kg [0.78–1.5 mg/kg] (IC50%/Meto ∼82 nM). Maximal P-gp inhibition was observed for tariquidar doses ≥ 2 mg/kg (IC100%/Meto ∼300 nM) using [11C]metoclopramide as a substrate probe.
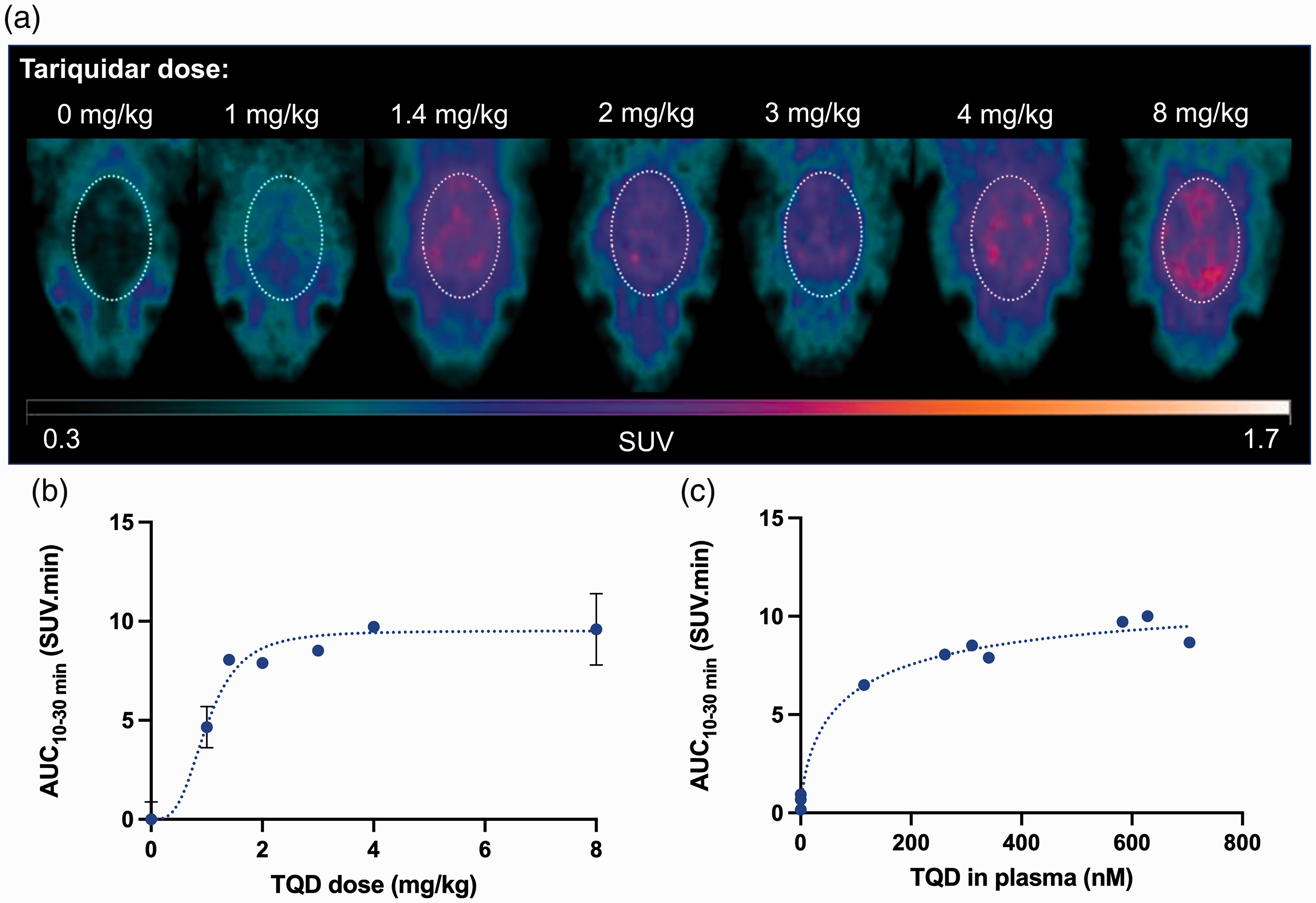
The 1 mg/kg dose (ID50%/Meto) and the 8 mg/kg dose (ID100%/Meto) of tariquidar were then selected to test the impact of partial and maximal P-gp inhibition using [11C]verapamil and [11C]N-desmethyl-loperamide as probes. Figure 3 shows representative [11C]verapamil, [11C]N-desmethyl-loperamide and [11C]metoclopramide PET images obtained at baseline and after injection of either 1 or 8 mg/kg of tariquidar. Maximal P-gp inhibition using 8 mg/kg tariquidar visually enhanced the brain PET signal (Figure 3) and significantly increased AUC10-30 min compared to baseline (p < 0.0001) for all three radiotracers (Figure 4(d)). The rank order of increases was [11C]verapamil (7.4-fold) > [11C]N-desmethyl-loperamide (6.8-fold) > [11C]metoclopramide (3.3-fold). The intermediate dose of tariquidar (1 mg/kg) visually increased the brain PET signal for [11C]verapamil and [11C]metoclopramide with a limited impact on brain uptake of [11C]N-desmethyl-loperamide (Figure 3). The corresponding increase in AUC10-30 min relative to baseline was significant for [11C]verapamil (2.4-fold, p < 0.05) and [11C]metoclopramide (2.1-fold p < 0.05), but not for [11C]N-desmethyl-loperamide (1.8-fold, p > 0.05, Figure 4(d)).
PET images obtained with [11C]verapamil, [11C]N-desmethyl-loperamide and [11C]metoclopramide after selected doses of the P-gp inhibitor tariquidar. Representative PET images are summed from 0 to 30 min and were obtained at baseline, after partial P-gp inhibition (1 mg/kg tariquidar) and complete P-gp inhibition (8 mg/kg tariquidar).
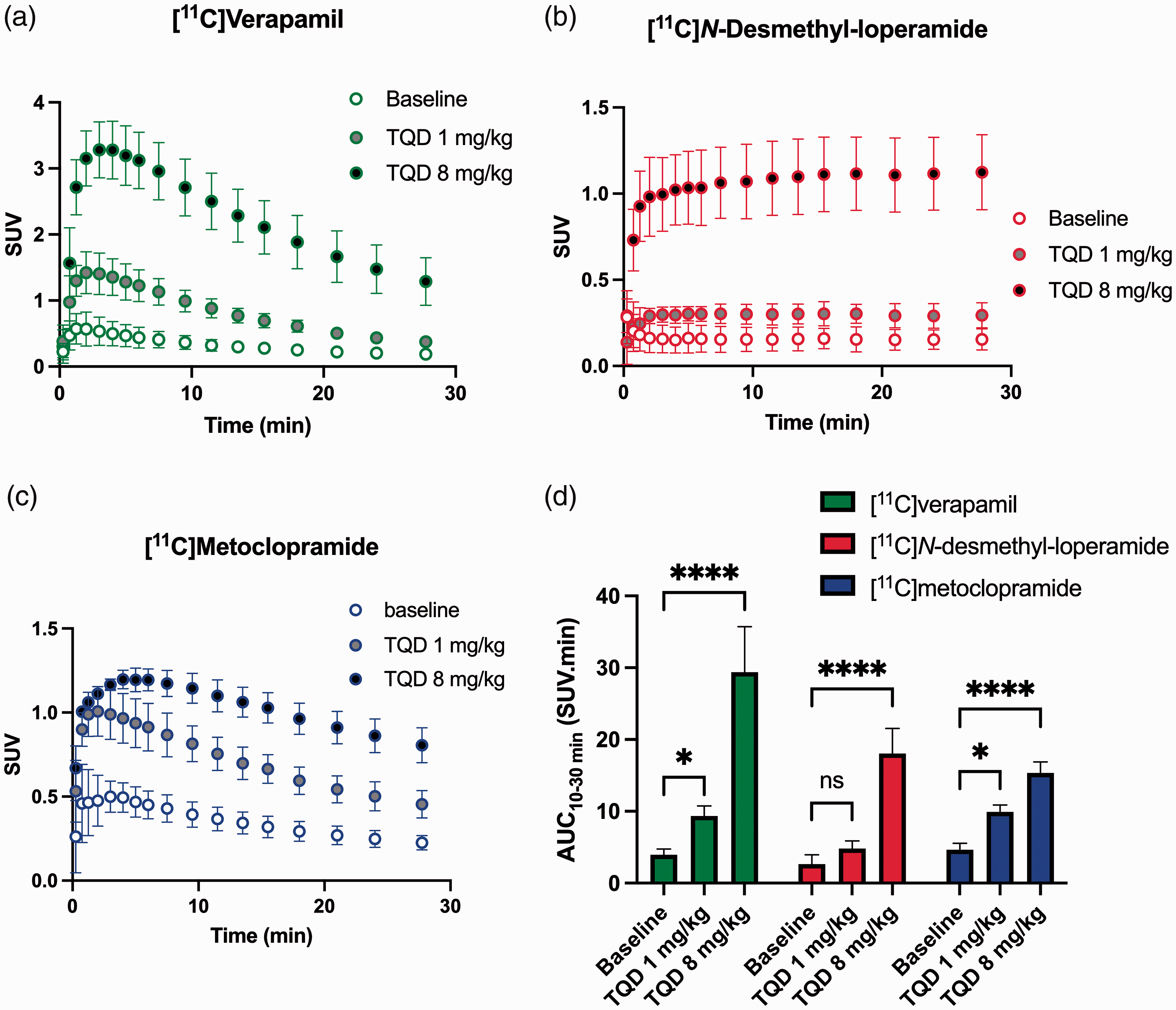
Impact of P-gp inhibition of the brain kinetics of radiolabeled substrates. Time-activity curves obtained with [11C]verapamil (A), [11C]N-desmethyl-loperamide (B) and [11C]metoclopramide (C) in presence of selected states of P-gp inhibition. In D, the corresponding brain exposures (AUC10-30 min) are shown. Data are reported as mean ± S.D with n = 4 per condition. *p < 0.05, ****p < 0.0001, ns = not significant (2-way ANOVA with Dunett’s post-hoc test for comparison with baseline).
Discussion
PET imaging using radiolabeled P-gp substrates has convincingly demonstrated the role of this efflux transporter in limiting the brain distribution of its substrates across the BBB in humans.11,16,18,33,34 Efforts have been made to develop optimized PET probes to quantitatively address the importance of P-gp function at the BBB in neurological disorders with good sensitivity.8,35,36 Previous studies have preferentially classified radiolabeled probes as “avid” or “weak” P-gp substrates which refers to their maximal capacity of transport. However, a direct in vitro and in vivo comparison of all three PET probes currently available for human use, performed in parallel conditions, has not yet been performed. Moreover, the present work focused on the impact of partial rather than complete P-gp inhibition at the BBB on the brain distribution of radiolabeled P-gp substrates with the aim to reflect clinically relevant situations of a moderate decrease in P-gp function. Strikingly, our results point to significant differences in the “vulnerability” of radiolabeled substrates to P-gp inhibition at the BBB, which contributes to their “sensitivity” to detect changes in P-gp function using PET imaging.
Differences in the substrate properties of [11C]verapamil, [11C]N-desmethyl-loperamide and [11C]metoclopramide were first compared in vitro, using a standardized uptake assay in cells overexpressing human P-gp. Maximal P-gp inhibition, using 200 nM tariquidar, allowed determining the uptake ratios of the tested substrates. The results confirmed that [11C]metoclopramide and [11C]verapamil are “weaker” P-gp substrates than [11C]N-desmethyl-loperamide (Figure 1). The magnitude of the uptake ratios was consistent with the transport ratios of these compounds measured using conventional bidirectional transport assays, which were 2.1 for verapamil, 3.8 for loperamide and 1.4 for metoclopramide. 37 Of note, the uptake assay employed in our work is more suitable to test compounds radiolabeled with short-lived isotopes such as carbon-11 than the conventional bidirectional transport assay. In vitro, [11C]metoclopramide was identified as one of the “weakest” P-gp substrates (lowest uptake ratio, similar to that of [11C]verapamil) but showed the highest “vulnerability” to P-gp inhibition (lowest IC50 of tariquidar). Interestingly, [11C]verapamil appeared to be a “weaker” P-gp substrate than [11C]N-desmethyl-loperamide (lower uptake ratio) but showed a lower “vulnerability” to P-gp inhibition (higher IC50). Importantly, these comparative in vitro data suggest that the “avidity” and the “vulnerability” of P-gp substrates are independent factors that are not directly or inversely correlated.
PET experiments were needed to compare the “vulnerability” of the three probes to P-gp inhibition in the living brain. Several factors other than P-gp function may additionally influence the PET signal obtained with the investigated radiotracers in the living brain. For instance, lysosomal trapping was shown to account for the irreversible uptake of [11C]N-desmethyl-loperamide in the brain. 38 The [11C]verapamil brain PET signal comprises a considerable proportion of radiometabolites. 39 [11C]Metoclopramide shows a significant brain-to-blood efflux component that contributes to limit brain exposure. 24 These particular kinetic properties may also contribute to the in vivo sensitivity of radiolabeled P-gp substrates to detect moderate changes in P-gp function using PET. 39 Dedicated kinetic models have been developed to estimate P-gp function, taking the individual properties of each radioligand into account.24,30,40–42 However, these models do not apply to all 3 radioligands. Tariquidar was selected as an inhibitor because doses up to 8 mg/kg were shown to not significantly impact the peripheral pharmacokinetics and metabolism of the tested radioligands in rats.24,30,31 Using kinetic modeling and an arterial input function, brain AUC has been previously validated as a simplified parameter to describe the response to tariquidar for [11C]metoclopramide 32 and [11C]N-desmethyl-loperamide 16 and (R)-[11C]verapamil (personal communication, Dr. Oliver Langer). We therefore used the brain AUC as a model-independent parameter to describe the increase in the brain exposure obtained after different degrees of P-gp inhibition, assuming negligible changes in the plasma kinetics of the investigated radioligands in the presence of increasing doses of tariquidar up to 8 mg/kg.
Mice that are heterozygous for the genes encoding P-gp (Abcb1a/1b(+/−)) were shown to have approximately 50% lower P-gp expression levels at the BBB than wild-type mice. 43 Interestingly, no significant increases in the brain uptake of (R)-[11C]verapamil and [11C]N-desmethyl-loperamide were observed in Abcb1a/1b(+/-) mice as compared with wild-type mice. 44 This suggested that (R)-[11C]verapamil and [11C]N-desmethyl-loperamide lack the sensitivity to detect the consequences of a moderate decline (<50%) in P-gp function at the BBB in vivo. In our study, the dose of tariquidar that inhibited 50% of the P-gp-mediated transport of [11C]metoclopramide at the rat BBB increased the brain uptake of [11C]verapamil, but not that of [11C]N-desmethyl-loperamide. From a quantitative PET imaging perspective, the in vivo response to partial inhibition was similar for both [11C]metoclopramide and [11C]verapamil, although the latter is a much more “avid” substrate of P-gp at the rat BBB (Figure 4). A direct comparison remains however difficult because radiometabolites may contribute to the brain PET signal for [11C]verapamil but not for [11C]metoclopramide.24,39 Nevertheless, this suggests that the “weak” P-gp substrate [11C]metoclopramide offers an adequate sensitivity to detect moderate decreases in P-gp function, which is probably linked to its higher “vulnerability” to P-gp inhibition. 24
In rats, [11C]N-desmethyl-loperamide behaved as an “avid” and “refractory” substrate, with a limited response to partial P-gp inhibition. Given its excellent metabolic stability in humans and its favorable neuropharmacokinetic properties, [11C]N-desmethyl-loperamide should be preferably employed to explore situations in which a great degree of inhibition/repression of P-gp function is expected, with an expected large increase in the PET signal.16,30,45,46 Altogether, our results suggest that imaging “sensitivity” of PET probes to detect partial inhibition of P-gp at the BBB in vivo does not only depend on the “avidity” (i.e. the ability to generate contrast between situations of full and abolished P-gp function, related to the uptake ratio), as it also depends on the “vulnerability” of the substrate (i.e. the ability to reach nearly maximal inhibition, related to the IC50).
[11C]Metoclopramide and [11C]verapamil PET imaging in rats showed similar sensitivity (i.e. enhancement of the brain PET signal) to partial P-gp inhibition, although [11C]metoclopramide is a “weaker” substrate than [11C]verapamil in vivo, as revealed by the lower response to maximal P-gp inhibition (Figure 4). The in vivo IC50 of tariquidar using [11C]verapamil as probe has not been determined in this work to enable a direct comparison with the IC50 obtained with [11C]metoclopramide. It may nonetheless be hypothesized that the higher in vivo “vulnerability” of [11C]metoclopramide may compensate its lower “avidity” compared with [11C]verapamil to offer similar sensitivity to partial inhibition. Our preclinical data should be interpreted with caution with respect to their clinical translatability. Differences in P-gp expression at the BBB have been reported between humans and rodents.47,48 Moreover, many compounds show disparate P-gp-mediated transport between human and rodent P-gp in vitro.37,49,50 However, our comparative in vitro experiments, performed using cells overexpressing human P-gp, pointed to a higher “vulnerability” of [11C]metoclopramide to P-gp inhibition compared with [11C]verapamil and [11C]N-desmethyl-loperamide.
The neuropharmacokinetic consequences of partial P-gp inhibition on the brain exposure to “weak” but “vulnerable” P-gp substrates in humans remain to be further explored. Using [11C]metoclopramide PET imaging in rats, we show here that partial P-gp inhibition may have a relatively higher impact for brain exposure compared with more “avid” but “inhibition-refractory” substrates like [11C]N-desmethyl-loperamide. At the molecular level, the factors that account for the “avidity” and “vulnerability” of P-gp substrates remain poorly understood. This may imply the complex molecular interaction of substrates and inhibitors at multiple binding domains of P-gp. 51 From a PET imaging perspective, this suggests a higher sensitivity of [11C]metoclopramide to explore situations of moderate changes in P-gp function at the BBB. From a neuropharmacological perspective, our results suggest that partial inhibition/deficit of P-gp at the BBB may contribute to clinical variability in efficacy of CNS-targeted “vulnerable” P-gp substrates, which may deserve further attention.4,26
Bauer et al. have recently shown a significant decline in P-gp function at the BBB of elderly subjects using [11C]metoclopramide PET imaging. 52 Interestingly, differences in brain distribution between elderly and young subjects were greater for [11C]metoclopramide than for (R)-[11C]verapamil. 53 This is consistent with a higher “vulnerability” of [11C]metoclopramide to moderate and physiological changes in P-gp function at the human BBB. 26 Interestingly, differences between young and elderly subjects could be observed using (R)-[11C]verapamil in the presence of partial P-gp inhibition (3 mg/kg tariquidar). 54 This suggested that partial P-gp inhibition enhanced the “vulnerability” of (R)-[11C]verapamil, thus improving the sensitivity of the technique. Such a partial inhibition protocol is difficult to implement because tariquidar is no longer available for human use and may cause drug-drug interactions with concomitant treatments in patients. This supports the use of [11C]metoclopramide as a “change-sensitive” PET probe to safely explore P-gp function at the human BBB.
Conclusion
The sensitivity of PET probes to detect moderate changes in P-gp function at the BBB appeared different among clinically validated radiolabeled substrates. Our results suggest that the “weak” but “vulnerable” P-gp substrate [11C]metoclopramide offers good sensitivity as a PET probe to detect moderate changes in P-gp function in vivo. We advocate that the future development of radiolabeled substrates for imaging the function of P-gp or other ABC efflux transporters at the BBB should also include an evaluation under conditions of partial transporter inhibition. This may improve our understanding of the impact of clinically relevant situations of a moderate decline in transporter function at the BBB.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Louise Breuil received funding from the joined AP-HP/CEA grant. This work was performed on a platform partially funded by the France Life Imaging network (grant ANR-11-INBS-0006).
Acknowledgements
We thank Maud Goislard, Thierry Lekieffre, Christine Coulon and Kevin Pansavath for helpful technical assistance.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
Nicolas Tournier, Louise Breuil and Oliver Langer designed the research; Solène Marie, Sébastien Goutal, Charles Truillet, Wadad Saba and Fabien Caillé performed the research; Louise Breuil, Sylvain Auvity and Sébastien Goutal analyzed the data; Louise Breuil and Nicolas Tournier drafted the paper. All authors revised the manuscript critically for important intellectual content and approved the final version.