
Abstract
Reductions of baseline cerebral blood flow (CBF) of ∼10–20% are a common symptom of Alzheimer’s disease (AD) that appear early in disease progression and correlate with the severity of cognitive impairment. These CBF deficits are replicated in mouse models of AD and recent work shows that increasing baseline CBF can rapidly improve the performance of AD mice on short term memory tasks. Despite the potential role these data suggest for CBF reductions in causing cognitive symptoms and contributing to brain pathology in AD, there remains a poor understanding of the molecular and cellular mechanisms causing them. This review compiles data on CBF reductions and on the correlation of AD-related CBF deficits with disease comorbidities (e.g. cardiovascular and genetic risk factors) and outcomes (e.g. cognitive performance and brain pathology) from studies in both patients and mouse models, and discusses several potential mechanisms proposed to contribute to CBF reductions, based primarily on work in AD mouse models. Future research aimed at improving our understanding of the importance of and interplay between different mechanisms for CBF reduction, as well as at determining the role these mechanisms play in AD patients could guide the development of future therapies that target CBF reductions in AD.
Keywords
Introduction
The metabolic demand of the brain is narrowly met by the cerebral blood flow (CBF) it receives, with very little local energy reserve, making brain function particularly sensitive to reductions in CBF. It is well acknowledged that disruption of normal blood supply, largely characterized by regional hypoperfusion, is an early and persistent symptom in the development of Alzheimer’s disease (AD) and other neurodegenerative diseases.1–4 In addition to such hypoperfusion, there are other dysfunctions of the cerebrovascular system that have also been linked to AD and other neurodegenerative diseases, such as disintegrated blood brain barrier (BBB) (reviewed in Sweeney et al. 5 ) and impaired autoregulation and neurovascular coupling (reviewed in Iadecola 6 ). These vascular contributions to cognitive impairment and dementia have recently received much attention. 7 While an increasing number of studies link vascular dysfunction, including baseline CBF reductions, to AD and cognitive decline,2,8 the mechanisms leading to these CBF reductions remain poorly understood. This review will focus on the causes and consequences of changes to baseline CBF in AD. Specifically, we review the alterations in CBF found in patients and mouse models of AD, the correlations of these CBF deficits with the progression or severity of the brain pathology and cognitive impacts, and the potential molecular and cellular mechanisms that may underlie the CBF reduction found in AD.
Cerebrovascular structure and physiology
Regulation and maintenance of CBF are crucial for proper brain function. The blood flow supply to the brain is fed by two internal carotid arteries that bifurcate from the common carotid arteries and the two vertebral arteries. These arteries feed the Circle of Willis, located at the base of the brain, which then gives rise to the anterior, middle, and posterior cerebral arteries that support a large proportion of the brain, including the cerebral cortex. We note, however, that there is significant variability between species and between individuals in the general pattern of large cerebral vessel arrangement.9,10 In the cortex, the three cerebral arteries give rise to a network of pial surface vessels that then branch into penetrating arterioles that plunge into the brain. These penetrating arterioles feed the capillary network, where most of the oxygen and nutrient exchange occurs. 11 The capillary beds have the smallest diameter vessels, such that blood cells (leucocytes and red blood cells (RBCs)) need to deform to flow through capillary segments, and they thus contribute the most to overall vascular resistance. 12 The capillaries converge on ascending venules that return blood to the cortical surface, where it is drained out of the brain by surface venules. 5 This review will mostly focus on the AD-related CBF disruptions in the cortex, where imaging tools in humans and animal models have enabled the most detailed studies.
Not only does the diameter and connectivity vary across different vessel classes, but also the cellular structure of the vessel wall and milieu of nearby associated cells, termed the neurovascular unit (NVU), 6 changes between arterioles, capillaries, and venules. 13 Endothelial cells line all blood vessels, are anchored by a continuous basal membrane, and are connected to each other by tight junction proteins, thereby forming the BBB. While small, lipid-soluble molecules (e.g. oxygen) can passively diffuse in and out of the brain through the BBB, the entry and exit of larger molecules is inhibited and transport proteins (e.g. insulin transporter) are required for BBB crossing. 5 The cells of the NVU found adjacent to the endothelium vary across vessel classes. Arterioles are surrounded by a tight layer of smooth muscle cells, astrocytic endfeet, and then the brain parenchyma (containing neurons, astrocytes, microglia, and other cells), whereas the capillaries are covered sparsely by pericytes, and then by astrocytic endfeet. 5 Capillaries are the blood vessels that are the closest, on average, to neurons and other brain cells. Venules are surrounded by a sparse layer of smooth muscle cells and astrocytic endfeet. Cerebral vessels are surrounded by a perivascular space, where extracellular fluid containing proteins and other solutes are transported. 14 In addition to different vessel classes having different cellular compositions, vascular cells show different transcriptional profiles in different vessel classes. This zonation of the cerebral vasculature was recently described in detail. 15
These differences in topology and cellular structure along the vasculature reflects varying functional roles of different vessel classes. The cerebral vascular system is very dynamic and actively regulates CBF. Several cell-types of the NVU, including neurons, glial cells (astrocytes and macrophages/microglia), mural cells (smooth muscle cells and pericytes), and endothelial cells, are implicated in brain blood flow regulation. 5 Changes in blood pressure are sensed in endothelial cells and drive vessel diameter changes that maintain constant blood flow – a process named autoregulation. 16 Endothelial cells can signal adjacent endothelial cells, as well as nearby smooth muscle cells, to coordinate such modulation of vascular tone through gap-junctions and nitric oxide signaling.17,18
CBF also increases locally in response to increases in neuronal activity – a process termed neurovascular coupling. 13 Smooth muscle cells surrounding arterioles and pericytes surrounding capillaries are the contractile cells around vessels that are critical for this flow regulation.19–22 Several studies have shown that neuronal activity causes Ca2+ release in astrocytic endfeet that drives vasodilation and/or vasoconstriction in arterioles, 23 while other studies suggest astrocytic Ca2+ dynamics can also influence capillary diameter through pericyte contractility.24–27 Thus, it is the interplay of sensing and signaling across multiple cell types in the NVU that coordinates the tight regulation of CBF in the brain. It is widely appreciated that these flow regulation mechanisms are disrupted in AD patients28,29 and in mouse models of AD. 6 In this review we will mostly focus on changes in baseline blood flow in AD and how this may impact disease progression.
During healthy aging, brain blood flow reaches its maximum value at the age of 4–6 years, and then decreases to about 60–70% of the maximum value by 50–60 years of age. 30 Some structural changes occur in cerebral blood vessels with aging, including increased collagen deposition and calcification in arterioles, leading to increased vessel wall thickness, decreased vessel elasticity, and overall increased resistance.31,32 These vascular changes are correlated with increased levels of reactive oxygen species (ROS) and vascular inflammation.33,34 There is also an age-dependent decrease in capillary density.35,36 These structural changes in vessels are found to be more severe in patients with neurodegenerative disease, including AD,6,37 and likely contribute to the increase in vascular resistance, hypoperfusion, and alterations in cerebrovascular regulation seen in AD 37 (Figure 1(a)).
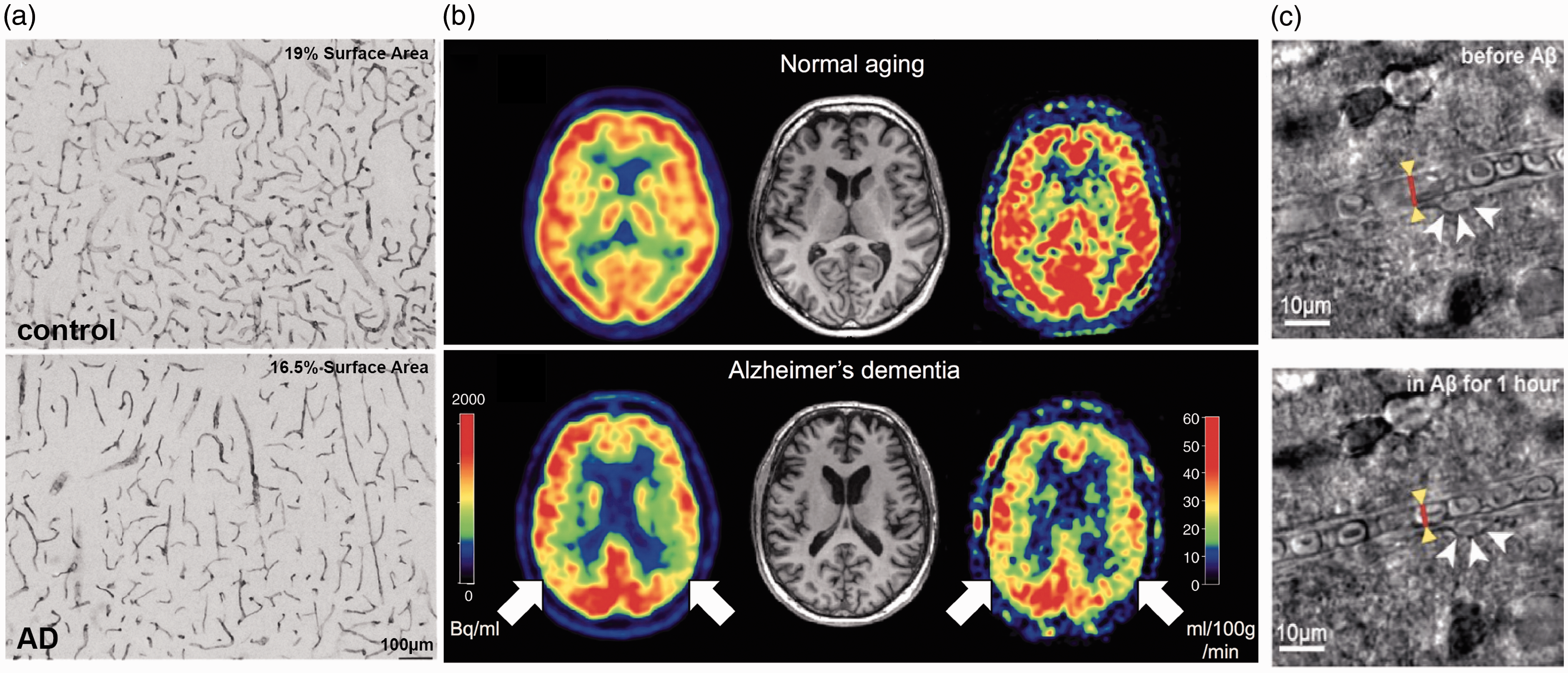
Vascular alterations and brain blood flow reductions in AD patients. (a) Heparan sulfate proteoglycan mmunohistochemistry in the cortex of a control subject (upper panel) and an AD patient (lower panel), showing reduced vascular density with AD. 36 (b) Images of brain perfusion using ASL-MRI from a normal aged subject (top images), and an AD patient (bottom images). Red colors represent higher perfusion, while blue colors represent lower perfusion. 167 (c) Bright field images of a capillary segment from a human brain slice before (left) and after (right) application of 72 nM Aβ1-42 onto the slice, which triggered activation of a pericyte (white arrows) and constriction of the capillary (red line and yellow arrowheads). 157
Cerebral blood flow reductions in patients with Alzheimer’s disease
Many studies have shown the correlation between reduced CBF, impaired cognitive function, and an increased probability of developing dementia, including AD, later in life. The blood flow reductions correlated with increased risk for dementia exceed the levels of flow reduction associated with normal, healthy aging. 38 Here, we focus on brain blood flow reductions in dementia, and in particular in AD, that are not associated with the incidence of a clinically-diagnosed stroke (which in AD patients is associated with more severe cognitive impairments, 39 and is reviewed in Pendlebury and Rothwell 40 ).
The largest study of the correlation between CBF reductions and dementia comes from the ongoing Rotterdam Study. Data from 1,730 patients, aged 55 years or older, showed that higher baseline CBF, as measured by transcranial laser Doppler of the middle cerebral artery, was associated with lower risk of being diagnosed with dementia 6.5 years later in life, and also with slower cognitive decline over this time, as compared to patients with lower baseline CBF.41,42 Using single-photon emission computerized tomography to spatially resolve changes in CBF, lower flow within the medial parietal cortex was found to develop before other symptoms of AD, indicating that reduced CBF occurs at early stages of disease progression.43,44 In agreement with this, CBF reductions have been found to precede the onset of detectable memory deficits in patients at predementia stages who later developed AD.8,45,46 Arterial spin-labeled magnetic resonance imaging (ASL-MRI) has provided even greater spatial resolution for imaging CBF changes. With this approach, patients that had mild cognitive impairment (MCI) or AD showed progressively worse disease symptoms with more severe regional CBF reductions in the left hippocampus and right amygdala. 47 Interestingly, this study found variable impacts of MCI and AD on regional CBF in the inferior parietal, left lateral, left superior, and left orbitofrontal cortices, and also found an increase in perfusion within the anterior cingulate gyrus. Further studies have largely shown CBF reductions ranging from 2 to 32%, with regional variability, in patients with MCI and/or AD, as compared to healthy controls48–50 (Table 1 and Figure 1(b)). However, some studies report no changes in CBF or even hyperperfusion, for example in the hippocampus, putamen, caudate, lentiform, and thalamus, in patients with AD.51,52 These differing results might be due to subtleties of various CBF measurement techniques, the disease stage at which participants are evaluated, or the inclusion/exclusion criteria for selecting participants – it is known, for example, that cardiovascular risk factors can have complex effects on CBF.
CBF changes in patients with MCI and AD.
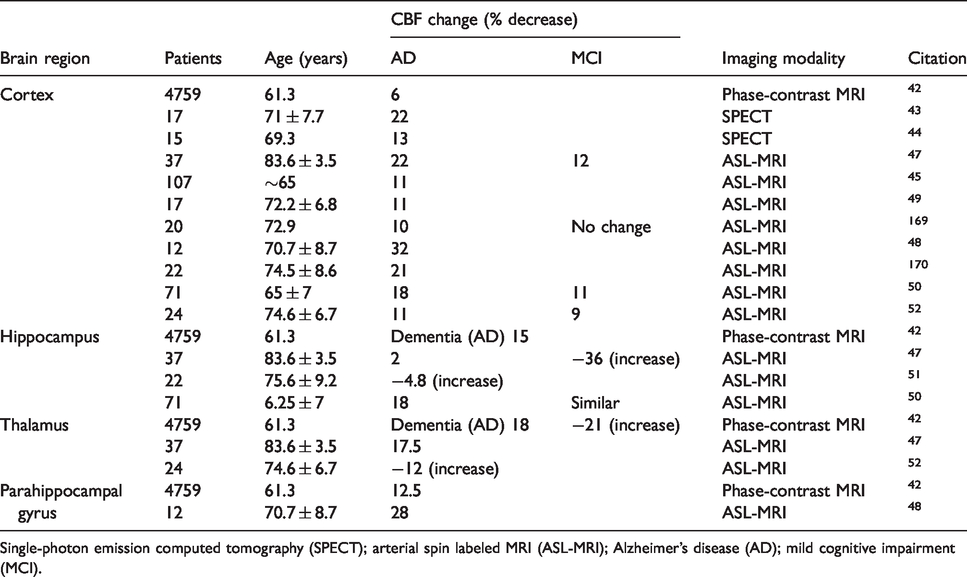
Single-photon emission computed tomography (SPECT); arterial spin labeled MRI (ASL-MRI); Alzheimer’s disease (AD); mild cognitive impairment (MCI).
The deposition of tau has also been found to influence CBF. A recent study including MCI and AD patients that analyzed the association of CBF and cognition with both tau and amyloid deposition showed that while increased tau deposition was linked to lower CBF and more severe cognitive impairment, this association was much stronger in patients with high amyloid burdens as compared to those with lower amounts of amyloid. 53
While characterization of structural changes in the vasculature associated with AD can be assessed using post-mortem tissue, there are few approaches that can be used to quantify hypoperfusion using such histological approaches. One approach is measuring the ratio of the concentration of myelin-associated glycoprotein to proteolipid protein 1, which has been found to decrease with chronic hypoperfusion. 54 This ratio was found to be reduced in the frontal cortex of tissue from AD patients, as compared to matched controls. 55 Scaling this approach could allow the use of large sample sizes from tissue banks to explore, for example, genetic, gender, age, or disease marker progression of regional brain hypoperfusion, as well as to correlate hypoperfusion with other pathological changes. Such data could complement the in vivo imaging data on CBF in AD patients that is available.
Reduced CBF has also been linked to increased levels of amyloid deposition and more severe brain pathology in AD patients. 56 Measuring brain amyloid levels using positron emission tomography with the amyloid-labeling Pittsburgh Compound B (PiB) revealed that higher levels of Aβ deposition was correlated to global CBF reductions, 57 and that even regional CBF deficits were linked to regional increases in Aβ deposition. 58 In addition, a study by Huang et al. has shown a correlation between brain regions with CBF deficits and brain atrophy in AD patients. 59 The severity of white matter hyperintensities, areas of white matter damage linked to hypoperfusion that are identified through MRI, is exacerbated in AD patients. 60 These studies show that there are correlations between reduced regional CBF and AD pathology.
These reductions in CBF in AD patients are frequently correlated with the development of vascular pathology. Cerebral amyloid angiopathy (CAA) is defined as Aβ deposition around the vessel walls of cortical arteries, arterioles, capillaries, and, rarely, veins.61,62 CAA is present in more than 80% of patients diagnosed with AD, 63 and is linked to pathological changes in the vasculature and increased incidence of microhemorrhages and microinfarcts. 64 It is thought that CAA contributes to impaired neurovascular regulation and perivascular flow by reducing the elasticity of vessels, 6 and by disrupting smooth muscle cell function.65,66 Only a few studies have correlated CBF with CAA burden in AD patients, and they found that the presence of CAA was associated with more severe CBF reductions.67,68 Microvascular pathologies, including pericyte loss, BBB breakdown, and reduced vascular density (Figure 1(a)), have also been found at higher incidence in AD patients, as compared to controls.69,70 The brain microinfarcts and microhemorrhages that are consequences of microvascular occlusions and ruptures are also found in increased number in AD patients as compared to controls. 71
The apolipoprotein E (APOE) ε4 allele increases the risk of AD by 3–8-fold and lowers the onset age of AD symptoms by 7–15 years.72,73 The ε4 allele of APOE is also linked to cerebral hypoperfusion. A study by Thambisetty M, et al., showed that APOE ε4 carries have 2–6% reduction in CBF across several brain regions, as compared to non-carries, as well as impaired memory function. 73 Similar results were observed in other studies of APOE ε4 carries,57,74 although newer findings have suggested APOE ε4 carries have regional brain hyperperfusion in mid-life. 75
Some of the cerebrovascular pathology seen in AD may be exacerbated by the presence of cardiovascular risk factors. Indeed, most cardiovascular risk factors, including hypertension, type 2 diabetes, hypercholesterolemia, as well as metabolic syndrome and obesity (especially when present in mid-life) have been shown to increase the risk for developing AD and severity of symptoms in AD patients.76,77 Cardiovascular risk scores taken during mid and late age in patients can predict cognitive decline later in life.42,55 In a prospective study, the Framingham cardiovascular risk score was predictive of the degree of cognitive decline over a year in a cohort of 254 patients with AD, 78 while other studies have correlated the Framingham score with cognitive decline in the middle-age general population. 79 Patients with hypertension frequently also have decreased brain perfusion and an increased probability of developing AD later in life. 80 A recent meta-analysis studied this correlation in more depth and showed that, in particular, stage one (BP > 140 mm Hg/90 mm Hg (systolic/diastolic)) or two (BP > 160 mm Hg/95 mm Hg) systolic hypertension, but not diastolic hypertension (> 90 mm Hg distolic), was associated with more severe AD. 81 These cardiovascular risk factors are all associated with brain blood flow reductions and increased microemboli, 82 and it may be, in part, through these flow reductions that these cardiovascular risk factors influence AD progression. In addition, cardiovascular disease, such as coronary heart disease or small vessel disease, is associated with brain hypoperfusion (largest in watershed regions, including the basal ganglia, white matter, and hippocampus83–85) and has been shown to increase the risk of developing MCI and dementia. 86 CBF increase, however, is not always associated with improved cognitive function. For example, while moderate exercise is consistently associated with improved cognitive performance, this is not always accompanied by an increase in CBF.87–89 While these studies were not conducted in AD patients, they suggest a complex interplay between CBF and cognitive performance.
In addition to alterations in baseline CBF, the dynamic regional and global regulation of brain blood flow may be impaired in AD patients. While there have been many reports of regional and global differences in blood oxygen level dependent (BOLD) functional MRI measurements in AD patients as compared to controls,90,91 only a few studies have linked performance on a memory-encoding task during functional MRI imaging sessions to the degree of functional hyperemia in the brain regions functionally linked to the task. These studies showed muted CBF increases during task performance in AD patients, as compared to healthy individuals, indicating a deficit in CBF regulation.92–94 More studies will be needed on task- and context-dependent CBF changes between healthy controls and AD patients to shed light on the mechanisms causing these impairments in neurovascular regulation.
In summary, MCI and AD patients tend to:
have reduced cerebral blood flow across a variety of brain regions show correlation between the severity of hypoperfusion and cognitive impairment have microvascular pathology and brain microinfarcts exhibit defects in CBF regulation as well as baseline flow
The presence of cardiovascular risk factors, some years earlier, has been shown to increase the risk and severity of AD and this exacerbation of AD may be related, in part, to the CBF deficits often associated with these risk factors. Broadly speaking, the mechanisms underlying the reductions in CBF seen in AD patients remain poorly understood and remain difficult to study since the origin is likely multifactorial.
Cerebral blood flow reductions in mouse models of AD
Mouse models of AD offer the opportunity to elucidate the mechanistic links between CBF reductions and AD pathology. Hypoperfusion has been repeatedly demonstrated in multiple different mouse models of amyloid precursor protein (APP) overexpression, including PS2APP, TgCRND8, APP/PS1dE9, 5xFAD, Tg2576, and J20 mice (Table 2).95–99 In addition, CBF deficits were found in knock-in mouse models that drive mutant human APP under the endogenous mouse APP promoter. 100 Flow deficits in AD mice relative to wild-type animals varied from 13% to 55% across these studies. High field ASL-MRI (7–11.4 T) allows regional CBF to be assessed in mice, and several brain regions in AD mouse models have been shown to exhibit reduced brain blood flow using this approach, including the occipital cortex,101,102 cerebral cortex, hippocampus, and thalamus.97,103,104 However, one study showed increased CBF in the frontoparietal cortex and thalamus, 105 while another found no deficits in CBF at eight months of age in APP/PS1 mice, 106 but a reduction at 15 months of age compared to age-matched wild-type mice.106,107 The APOE4 allele is the largest risk factor for AD, and targeted replacement knock-in mouse models carrying the human APOE allele are associated with CBF reductions in the cortex, hippocampus, thalamus, and the white matter.108–110 The CBF reductions associated with APOE manipulation were found to correlate with pericyte loss, 111 decreased capillary density, 112 and BBB breakdown via the CypA-MMP9 pathway,109,113 suggesting the possibility of mechanistic ties between different microvascular dysfunctions. In patients who carry the APOE4 allele, increased pericyte loss and BBB breakdown was also observed, as well as more severe cognitive decline.114,115
CBF changes in AD mouse models.
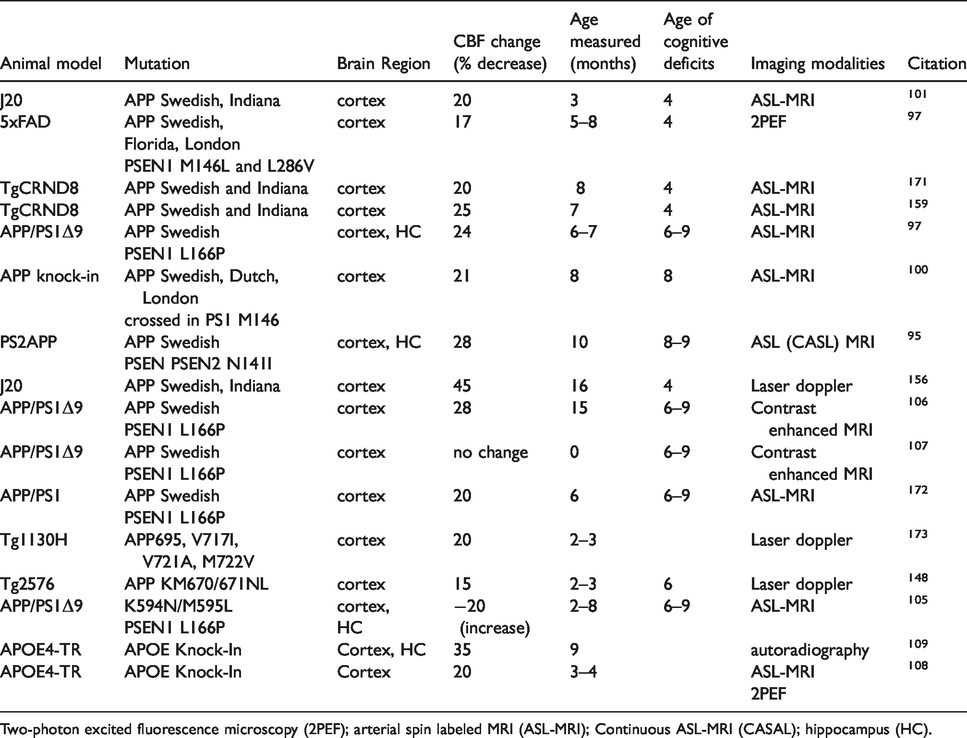
Two-photon excited fluorescence microscopy (2PEF); arterial spin labeled MRI (ASL-MRI); Continuous ASL-MRI (CASAL); hippocampus (HC).
Overall, these data suggest a broad consensus that hypoperfusion occurs in mouse models of AD, but the degree of hypoperfusion and the brain regions most affected are very heterogeneous across the published literature. This variability, including findings of normoperfusion or hyperperfusion, could be a result of differences in age, sex, genetic background, transgenic mouse strain, controls, and imaging modality, much of which is underreported in the existing literature.
Impact of chronic hypoperfusion on AD-like pathology and behavior in mice
Chronic hypoperfusion has been shown, on its own, to cause cognitive deficits in mice, as well as to recreate some of the brain inflammation associated with AD, although the hypoperfusion sufficient to cause these effects is more severe than that found in AD patients or mouse models. We briefly review these findings because they suggest how CBF deficits may exacerbate AD-related pathology. To induce chronic hypoperfusion in mice, common methods include the unilateral common carotid artery occlusion (UCCAO) and the bilateral carotid artery stenosis (BCAS) model, which lead to steady-state reductions of between ∼30% and ∼70%, depending on coil diameter, although often with more severe hypoperfusion just after coil implantation.18,116,117 After 1–4 months, these CBF decreases lead to deficits in short-term memory.116–118 Such chronic cerebral hypoperfusion also induced hyperphosphorylation of the mouse tau protein after 2.5 months. 117 When hypoperfusion is induced in mouse models of AD, the pathological phenotypes associated with the AD-related pathology are accelerated and the cognitive impact is worsened.119,120 For example, in 9-month old J20 mice, one month of chronic hypoperfusion led to more severe deficits in spatial short-term memory measured using a Barnes maze, as compared to sham surgery controls. 121 Interestingly, this study showed a decrease in the density of diffuse amyloid plaques and a decrease in the concentration of Aß(1-42) in the hypoperfused mice. 121 In APP23 mice, hypoperfusion was also found to cause more severe cognitive deficits, but the hypoperfusion was associated with an increase in the density of amyloid plaques. 122 In 10–11 months old female Tg2576 AD mice 8 weeks of chronic hypoperfusion led to impaired learning in the Morris water maze. 123 In the PS1V97L mouse model of AD chronic hypoperfusion led to increased BBB permeability, caused by a reduction in tight junction proteins, which was attributed to enhanced oxidative stress. 124 Similarly, an increase in the concentration of amyloid species was found in APP/PS1 mice with induced hypoxia, 125 although the impact of microvascular obstructions on the aggregation state and density of Aß can be complex. 126 Several studies have also suggested that cerebral hypoperfusion exacerbates CAA in the Tg-SwDI mouse model.127,128 However, there are differences in the reported impact of hypoperfusion across AD mouse models that might be explained by the underlying disease-driving mutations, promoters driving these transgenes, or the strategies that were used to reduce brain blood flow. In addition, the magnitude of CBF reduction caused by these chronic induced hypoperfusion models is larger than that typically seen in AD patients, 129 and these more severe CBF reductions could activate pathogenetic mechanisms that are not involved with the more modest CBF reductions found in AD. Recognizing this, new models that use either larger diameter coils or alternative surgical approaches (e.g. asymmetric common carotid artery surgery) have been developed. 129 These models caused CBF reductions of about 30% and this CBF deficit was still associated with memory deficits, as measured using a Y-maze, after 28 days. 130
Taken together, these findings indicate that chronic reductions in brain blood flow in wild-type mice cause AD-like memory deficits and brain pathologies, although this has generally required more severe CBF deficits than seen in AD patients. In addition, inducing hypoperfusion in mouse models of AD leads to an acceleration of disease progression, as characterized by amyloid burden, brain pathology, and cognitive deficits.
Potential mechanisms contributing to reduced CBF in AD that have emerged from mouse model studies
While several mechanisms have been proposed as contributing to brain blood flow reductions in AD, the physiological cause remains poorly understood. Disease-related structural changes in vessels as well as variance in the anatomy of large vessels can be important factors in CBF changes linked to neurodegeneration. For example, the hippocampal blood supply is provided by the collateral branches of the posterior cerebral artery and the anterior choroidal artery. Recent studies have shown that variations in the anatomy of the anterior choroidal artery in individuals led to impaired vascular reserve that was associated with increased risk for neurodegeneration and more precipitous cognitive decline with age.131,132 In addition, several studies from human AD patients have shown reduced vascular density and structural vascular alterations that could increase flow resistance, such as increased tortuosity, 133 vessel wall thinning, 134 string vessel formation. 135 In mouse models, the data on structural changes in cerebral vessels is less homogenous. Several studies found decreased density of capillaries in the APP23 transgenic AD model 136 and in the hippocampus of 12–15 month old APP/PS1 mice. 137 Another study has also shown that capillary density decreases in close proximity to amyloid plaques in the Tg2576 model, suggesting direct Aß-mediated damage to the vasculature. 138 Mouse models of APP overexpression also exhibit increased tortuosity in cortical ascending venules, increasing vascular resistance. 139 In contrast, mice that overexpress a human mutant tau protein linked to frontotemporal dementia and which leads to the formation of neurofibrillary tangles in neurons were found to have increased capillary length, diameter, and density, as compared to controls. 140 Overexpression of non-mutant tau, which does not aggregate into neurofibrillary tangles, did not cause such vascular changes. In this same study, APP overexpression models showed no difference in these measures of vascular structure as compared to controls, indicating different impacts of Aß and tau overexpression on vascular structure (Figure 2(a)). 140
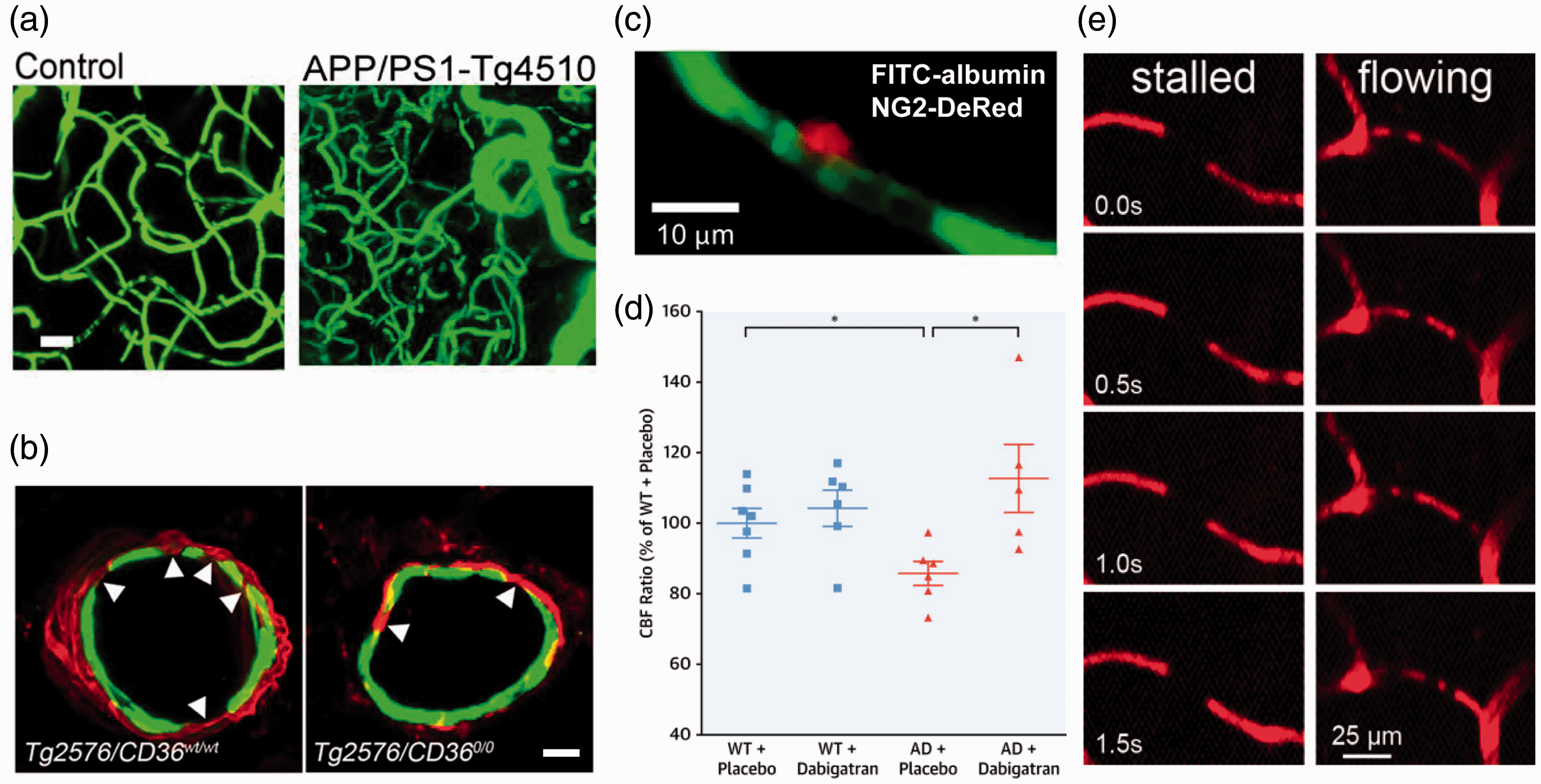
Mechanisms contributing to CBF reductions in mouse models of Alzheimer’s disease. (a) Images from 15 month old WT (left) and APP/PS1-Tg4510 mice (right; overexpresses mutant APP and mutant tau), showing clearly increased vascular volume in this AD mouse model containing both APP and tau-related mutations (Scale bar, 20 μm). 140 (b) Fragmentation of smooth muscle cells (arrows) along a cortical arteriole in Tg2576 mice (left image), which is attenuated by CD36 deletion (right image) (Scale bar, 10 μm; anti-α-actin: green, anti-Aβ: red). 168 (c) Example image of a pericyte (red) constricting around a cortical capillary (blood plasma shown in green), with blood cell flow blocked, from an AD mouse. 157 (d) Cerebral perfusion was maintained in TgCRND8 mice treated with dabigatran. 159 (e) Image sequences showing stalled (left panels) and flowing (right panels) capillaries, where the darker spots in vessels are due to unlabeled blood cells within the fluorescently-labeled blood plasma. Stalled capillaries, where blood cells do not move frame-to-frame, occurred at higher incidence in APP/PS1 mice, as compared to controls. 97
Cardiovascular risk factors have also been shown to accelerate disease progression in mouse models of AD, including by exacerbating cerebrovascular dysfunction.141,142 A reduction in vascular density was found in APP/PS1 mice with hypertension induced by angiotensin II infusion, as compare to normotensive APP/PS1 mice. 143 Interestingly this study did not show a difference in vascular density at baseline between wild-type and APP/PS1 mice. 143 The links between AD and cardiovascular risk factors may be bidirectional; a one-month infusion of Aβ1-40 was found to induce hypertension in Wistar rats and lead to a reduction in carotid artery blood flow. 144
Loss of appropriate regulation of vascular diameter, either in the resting state or in response to vasoactive mediators, such as hypercapnia, blood pressure change, or increased neural activity, can also lead to CBF deficits. Superfusion of monomeric species of Aβ1-40 onto the somatosensory cortex of wild-type mice was found to induce vasoconstriction.145,146 In the Tg2576 mouse model of AD, autoregulation and neurovascular coupling were impaired early in disease progression, before the appearance of amyloid-beta deposits.99,147,148 However, other studies have found no differences in neurovascular regulation between Tg2576 and age-matched wild-type mice when the mice were young, with the AD mice showing impairment only later in disease progression when amyloid deposition along vessels was noted. 149 In vitro experiments show that Aβ application to endothelial cells causes an increase in reactive oxygen species (ROS), 150 and subsequent work has implicated ROS in the impairment of neurovascular regulation. In particular, the scavenger receptor CD36 has been found to bind Aβ and mediate activation of NOX2-containing NAPDH oxidase leading to the production of ROS, which not only causes endothelial dysfunction and impairs neurovascular regulation, but also drives smooth muscle cell degeneration in cortical arterioles later in disease progression (Figure 2(b)).151,152 Interestingly, another NOX protein, NOX1, has been implicated as a dominant source of ROS within the hippocampus in a mouse model of vascular dementia, raising the possibility that different brain regions and/or different disease states involve distinct, but related, molecular players that cause ROS-mediated vascular dysfunction.153,154 In the J20 mouse model of AD, treatment with simvastatin restored neurovascular coupling and this was associated with improvements in spatial memory, as measured by the Morris water maze. 155 In stark contrast, Nicolakakis N et al. used the peroxisome proliferator-activated receptor-gamma agonist pioglitazone in J20 mice, which similarly restored neurovascular coupling, but led to no improvements in spatial memory. 156 These findings suggest that rescuing neurovascular coupling may not be sufficient to improve cognition in this mouse model. While the magnitude of the change in CBF due to neural activation was restored with these two drugs in J20 mice, the impact on baseline CBF was not assessed.
The use of in vivo microscopy tools has enabled the microvascular contributions to CBF reductions in AD to be explored. Recently, three different cellular mechanisms acting at the microvascular level have been proposed to contribute to baseline CBF reductions in AD. First, a recent study showed, using human brain tissue taken from neuro-oncological biopsies, that Aβ1-42 oligomers induced ROS production that led to release of endothelin-1 and constriction of capillaries via pericyte contraction, likely contributing to increased vascular resistance and thus reduced CBF (Figure 1(c) and 2(c)). 157 The authors also analyzed capillary diameters near pericyte locations in APPNL-G-F mice, 157 a knock-in mouse model that expresses mutant human APP under the endogenous mouse APP promoter, and found that diameters were smaller in APPNL-G-F mice, as compared to wild-type controls, further implicating Aβ-induced capillary constriction as a potential mechanism of CBF reduction of AD (Figure 2(c)). 157 Interestingly a recent study has shown that, in the capillary bed of the retina and cortex, pericytes communicate via tunneling nanotubes. These tunnels were implicated in coordinated pericyte constrictions in response to neural activation, likely contributing to neurovascular coupling. 158 These nanotubes could contribute to the orchestration of pericytes mediated capillary constrictions seen in the neuro-oncological biopsies of AD patients and in APPNL-G-F mice. 157
Second, AD mouse models show hypercoagulability and increased stability of fibrin clots that could cause vascular obstructions that reduce CBF.159,160 For example, inducing fibrin clot formation in TgCRND8 mouse models of AD by injections of FeCl3 led to earlier and more occlusions at lower doses, as compared to wild-type mice. Another study by the same group demonstrated, in female TgCRND8 mice, that treatment for one year with dabigatran, a reversible thrombin inhibitor, lead to improvements in cognition and reductions in inflammation and amyloid load that were correlated with increases in CBF (Figure 2(d)). 159
Third, we have recently found a small but significantly increased number of capillaries with transiently stalled blood flow in the cortex of APP/PS1 mice as compared to wild-type controls. 97 A similar increase in stalled capillaries was also recently described in a mouse model of vascular dementia. 161 In the APP/PS1 mice, the stalled capillaries were found to be caused by neutrophils that became stuck and plugged flow in individual capillary segments. Administration of antibodies that unstuck the neutrophils lead to a ∼20–30% increase in CBF within minutes. Strikingly, this CBF increase was accompanied by a rapid restoration of short-term memory within hours (Figure 2(e)). 162 There is currently no data establishing whether or not such capillary stalls occur in human patients with AD or whether they share the same cellular mechanism. Interestingly, in patients with diabetic retinopathy leucocyte adhesion has been shown to occlude retina capillaries, with low-level vascular inflammation implicated as the cause. 163 Finding leukocyte-mediated obstruction of capillaries in human retina lends credence to the possibility that similar capillary obstructions could contribute to the CBF reduction in AD patients, as we have shown in AD mouse models. Because the retina is, in some sense, a “window into the brain,” looking for capillary obstructions in the retina of AD patients may be the first path to establishing whether they occur in AD patients. In fact, the Alzheimer’s Association has launched an initiative to further explore the possibility of following progression of AD pathology, including blood flow deficits, in the eye. 164 It is likely that all three of these mechanisms contribute to CBF reductions in AD, and that these mechanisms reinforce each other.
These mouse models do not, of course, capture the full spectrum of pathogenic factors that contribute to AD. For example, these studies rely on mouse models that overexpress the mutant form of APP or tau, and results may be challenging to transfer in the clinic. The mice also do not have contributions from cardiovascular risk factors, although these have been layered on to AD mice in some studies. 165 New, second generation mouse models of AD have also been generated to improve the quality of disease modeling by, for example, knocking in human mutant APP at the mouse APP locus to provide more normal APP expression and regulation than in overexpression models. 166
Conclusion
CBF deficits are frequently found in AD patients, with some inconsistency across studies (Table 1). Generally, larger CBF deficits appear to be correlated with poorer cognitive performance in AD patients. CBF also decreases in healthy aging, although not as much as in AD patients, and these flow decreases do not seem sufficient to cause cognitive impairment. This suggests that CBF deficits are not the sole driver of cognitive impairment and, more likely, multiple pathogenic factors contribute to the cognitive decline seen in AD. This does not imply, however, that treating CBF deficits in AD patients would not lead to cognitive improvement. More longitudinal data are clearly needed to further investigate CBF changes over disease progression and to determine the correlation with cognitive deficits. The regional differences in blood flow in the brain of AD patients, as well as how CBF deficits differ in AD patients according to sex, race, ethnicity, cardiovascular risk factors, and other relevant factors needs to be further explored. Increased transparency in reporting details of subjects and methods would likely help to resolve remaining inconsistencies.
Mouse models of APP overexpression replicate the CBF deficits found in AD patients, including CBF decreases early in disease progression and ∼20% CBF reductions, on average (Table 2). Recent studies link these CBF deficits to a few potential cellular mechanisms, including tonic constriction of capillaries by activated pericytes, hypercoagulability that leads to vascular obstructions, and increased stalling of capillary segments by circulating white blood cells. Substantial evidence points toward some of these cellular mechanisms being a result of ROS-mediated dysfunction in microvascular cells. More research is needed to determine the role of, and interactions between, different cell types within the NVU that may drive these cellular mechanisms that contribute to CBF deficits in AD, and may cause other microvascular dysfunction, such as BBB failure and neurovascular dysregulation. In AD mouse models, there remains a lack of data on CBF deficits from brain regions other than the cortex, as well as on differences in CBF deficits with sex and across different mouse models of AD. In addition, the contribution of aggregated tau to brain blood flow reductions in AD is severely understudied. Still, given the dramatic structural impacts on the brain vasculature caused by tau aggregation, it is critical to understand its impacts on CBF, as well as the interplay with mutant APP expression.
The correlation of CBF deficits with more severe cognitive outcomes in AD patients as well as the improvement in memory performance in AD mouse models with rescued blood flow both suggest that improving CBF could be a potential symptomatic treatment for AD, especially in early phases of disease progression. Elucidation of the detailed molecular and cellular mechanisms that underlie the CBF deficits in AD could suggest novel therapeutic targets. However, there remains only sparse data analyzing the mechanisms of CBF reduction in AD, and much more research is warranted to fully understand these mechanisms and their links to other aspects of cerebrovascular dysfunction in AD. Recent advances in optical imaging modalities and genetically-targeted molecular labeling and manipulation tools make some of these critical studies a possibility.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health grants AG049952, NS108472 (CBS), and NS097805 (LP), the BrightFocus Foundation (CBS), and the DFG German Research Foundation (OB). We thank Costantino Iadecola, Lianne Trigiani, and Nancy E. Ruiz-Uribe for critical reading of this manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.