
Abstract
In order to rescue neuronal function, neuroprotection should be required not only for the neuron soma but also the dendrites. Here, we propose the hypothesis that ephrin-B2-EphB2 signaling may be involved in dendritic degeneration after ischemic injury. A mouse model of focal cerebral ischemia with middle cerebral artery occlusion (MCAO) method was used for EphB2 signaling test in vivo. Primary cortical neuron culture and oxygen-glucose deprivation were used to assess EphB2 signaling in vitro. siRNA and soluble ephrin-B2 ectodomain were used to block ephrin-B2-Ephb2 signaling. In the mouse model of focal cerebral ischemia and in neurons subjected to oxygen-glucose deprivation, clustering of ephrin-B2 with its receptor EphB2 was detected. Phosphorylation of EphB2 suggested activation of this signaling pathway. RNA silencing of EphB2 prevented neuronal death and preserved dendritic length. To assess therapeutic potential, we compared the soluble EphB2 ectodomain with the NMDA antagonist MK801 in neurons after oxygen-glucose deprivation. Both agents equally reduced lactate dehydrogenase release as a general marker of neurotoxicity. However, only soluble EphB2 ectodomain protected the dendrites. These findings provide a proof of concept that ephrin-B2-EphB2 signaling may represent a novel therapeutic target to protect both the neuron soma as well as dendrites against ischemic injury.
Introduction
As part of neuronal cell injury, dendrite degeneration is an important mechanism of stroke pathogenesis. Ischemic stroke leads to destabilization of dendrite and spines on neurons, observed as reduced dendrite arbor size in the peri-infarct region in murine stroke models.1,2 Aberrant neuronal circuit function caused by dendrite degeneration in the penumbra significantly contributes to the neurological deficits after stroke. 3 Conversely, dendritic spine recovery is also associated with improved neurological outcome after stroke. 4 Neuronal function is ultimately dependent on the integrity of its vast network of dendritic connections.
Despite the importance of dendrite degeneration in stroke pathogenesis, very limited effort has been devoted to investigating dendrite protection in neuroprotection research. Technically, many of our commonly used screening methods such as lactate dehydrogenase (LDH) release or tetrazolium-based assays in primary neuron culture are not capable of assessing dendrite degeneration. Although neuron soma and dendrite function are closely linked, emerging evidence suggest that dendrite degeneration may be mediated in part through distinct pathways from those occurring in the soma. 5 For example, low dose hydrogen peroxide induces dendrite degeneration, but not soma loss.6–8 Moreover, flavopiridol and minocycline combination protects neuron soma but not dendrite following global ischemia. 9 A recent study showed that although the NMDA antagonist MK801 10 was able to improve neuronal viability as measured by a tetrazolium-based assay, it did not significantly protect dendrites. 11 Therefore, it would be essential to find ways to protect both neuronal soma and dendrites for neuroprotection.
Ephrins are membrane-bound molecules that mediate critical functions in the central nervous system (CNS) through binding their receptors, Eph.12,13 There are 3 members of ephrin-B subclass (B1-B3) that interact with their corresponding EphB receptors. Activated ephrin-B2-EphB2 has inhibitory effects on axon and dendrite growth during development, which is critical for specific dendrite arborization.14,15 Activated EphB2 suppresses Ras and PI3K signaling, leading to axon and dendrite retraction.15,16 Ephrin-B2-EphB2 signaling also modulates dendritic spine morphology and synaptic plasticity.17–19 Emerging evidence now implicate ephrin-B2-EphB2 signaling in neurological diseases. For example, in glaucoma, ephrin-B2 and EphB2 upregulation is associated with RGC axon loss. 20 A recent study showed that EphB2 signaling promotes neuronal excitotoxicity after ischemic stroke. 21 On the other hand, ephrin-B2 silencing ameliorates astroglial scar formation and improves axon growth after spinal cord injury. 22 In this study, we hypothesize that Ephrin-B2-EphB2 signaling is involved in dendrite degeneration in neurons after oxygen-glucose deprivation.
Methods and materials
Animals
All animal experiments were performed following protocols approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee and were conducted in compliance with the NIH Guide for the Care and Use of Laboratory Animals and the ARRIVE guidelines.
Ischemic focal stroke models
The standard intraluminal middle cerebral artery occlusion (MCAO) method was used in C57/Bl6 male mice aged 11–12 weeks (Charles River Laboratories, Wilmington, MA) to make a transient (45 min arterial occlusion) focal cerebral ischemia as we previously described.23–25 Briefly, anesthesia was induced with 1.5% isoflurane in a mixture of 70% nitrous oxide and 30% oxygen delivered by face mask. Focal cerebral ischemia was induced by introducing a silicone-coated 6–0 monofilament until it occluded the origin of the right MCA. The rectal temperature was maintained at 37 ± 0.5°C and the regional cerebral blood flow of the right front parietal cortex was continually monitored during the surgical procedure. The filament was withdrawn after 45 min to allow reperfusion of the ischemic hemisphere. The mice were given buprenorphine (0.05 mg/kg, subcutaneous injection) 30 min prior to the induction of ischemia. After surgery, the mice were placed in a heated recovery box for 2 h, and administered buprenorphine (0.05 mg/kg, subcutaneous injection) twice daily (every 12 h) for 3 days after stroke for pain relief. The animals were assessed with laser doppler flowmetry to confirm adequate induction of focal ischemia and successful reperfusion. The operator was blinded to the treatment status of the animals.
Primary neuron culture and OGD
Primary cortical neuron culture and oxygen-glucose deprivation (OGD) were performed following our published studies.11,26 Briefly, primary cortical neurons were isolated from 15-day embryonic cortex obtained from the pregnant female C57B/L mice. Neurons were isolated from each embryo cortex and seeded into multiple well plates with equal cell numbers (1.25 × 105 cells/well in 24-well plates, 5 × 105 cells/well in 6-well plate). The neurons were maintained in Neurobasal medium (NBM) with 3% B27 and 0.3 mM glutamine (Invitrogen). Medium was half changed every 3 days.
For OGD experiment, at day 8–9 of primary neuron culture, NBM was replaced with deoxygenated, glucose-free extracellular solution–Locke's medium (154 mM NaCl, 5.6 mM KCl, 2.3 mM CaCl2, 1.0 mM MgCl2, 3.6 mM NaHCO3, 5 mM Hepes, pH 7.2), the cells were then put in a specialized, humidified chamber (Heidolph, incubator 1000, Brinkmann Instruments, Westbury, NY, USA) at 37°C, which contained an anaerobic gas mixture (90% N2, 5% H2, and 5% CO2). Neurons with medium change only were used as controls (replace culture medium with fresh NBM, incubate at normoxia for 4 hrs, then replace with previously saved old medium). The cells were treated with recombinant ephrin-B2 ectodomain during OGD. LDH release and MTT assay were used to test neurotoxicity and neuronal survival, respectively. Immunostaining for MAP2 was used to examine dendrite degeneration.26,27 The number of technical replicates in each experiment was 3.
Immunostaining
Coronal brain sections of mice (16 µm) or cultured neurons were fixed in 4% PFA and blocked with 3% bovine serum albumin (BSA) for 1 h and incubated at 4°C overnight with primary antibodies. The sections or cells were then washed and incubated for 1 h with fluorescence-conjugated secondary antibodies (Jackson ImmunoResearch). Vectashield mounting medium containing DAPI (Vector Laboratory, Burlingame, CA) was used to coverslip the slides or cells. Fluorescent signals were examined using Nikon Eclipse T300 fluorescence microscope.
Western blots
Western blot was performed as we previously described.23,28 Briefly, for mouse brain tissue, the mouse was cardio-perfused with saline at 3 days after stroke, and the brain was rapidly removed from the skull. The infarct was identified from the paleness of the cortex surface, and peri-infarct cortex was dissected as the approximately 1 mm surrounding tissue. The tissue was homogenized in lysis buffer (Cell Signaling, MA) in the presence of protease inhibitor cocktail (ThermoFisher). For primary cultured neurons, after treatment, neurons were washed twice with cold PBS, then homogenized using lysis buffer. The protein concentration was measured with Bradford reagent (Bio-Rad laboratories, Hercules, CA), and 30–50 µg protein from each sample was loaded onto an SDS gel for electrophoresis and transferred to nitrocellulose membranes. The blots were then reacted with primary antibodies, mouse anti-β-actin (1:5000, Millipore, Billerica, MA) was used as a control. After incubation at 4°C overnight, the membrane was incubated with the corresponding horseradish peroxidase-conjugated secondary antibody (1:2000) for 1 h at room temperature. An enhanced chemiluminescence system (Pierce; Thermo Fisher Scientific) was used for antibody detection.
EphB2 knockdown using siRNA
Minimum siRNA concentration for sufficient gene knockdown was first determined by titration. Briefly, serial concentrations 10, 20, 40, 80, 100 nM of siRNA (Santa Cruz Biotechnology, CA) were used to treat cells, protein levels were examined by Western blot to determine siRNA efficiency. The lowest working concentration was used for the following experiments.
Quantification of dendrite length after immunostaining
The length of dendrites in cultured neurons was measured by immunostaining of MAP2. The dendrites were traced and the length quantified using ImageJ-Neurite Tracer in a blinded manner.
Recombinant protein
Recombinant ephrin-B2-ectodomain (Eb2-ECD) was obtained from Thermo Fisher Scientific.
Statistical analysis
The power calculation was performed using information collected from our preliminary studies that was conducted under the same conditions. The data were normally distributed (normality tested using Shapiro-Wilk test in Prism). Non-parametric Kruskal-Wallis followed by Mann-Whitney tests were performed to compare protein levels, cell death and dendrite length between groups. Data were expressed as mean ± SD, p < 0.05 is considered statistically significant. All studies were randomized, and all procedures and analyses were fully blinded.
Results
Ephrin-B2-EphB2 signaling is activated after cerebral ischemia and neuronal oxygen-glucose deprivation
Our overall hypothesis states that ephrin-B2-EphB2 signaling is involved in dendrite degeneration after ischemia. So, we sought evidence for this potential phenomenon in vivo and in vitro. Mice were subjected to 45 min transient focal ischemia and brains were removed for analysis at 3 days. In peri-infarct cortex, western blots showed that phospho-EphB2 levels were significantly increased, while total EphB2 levels were not obviously changed (
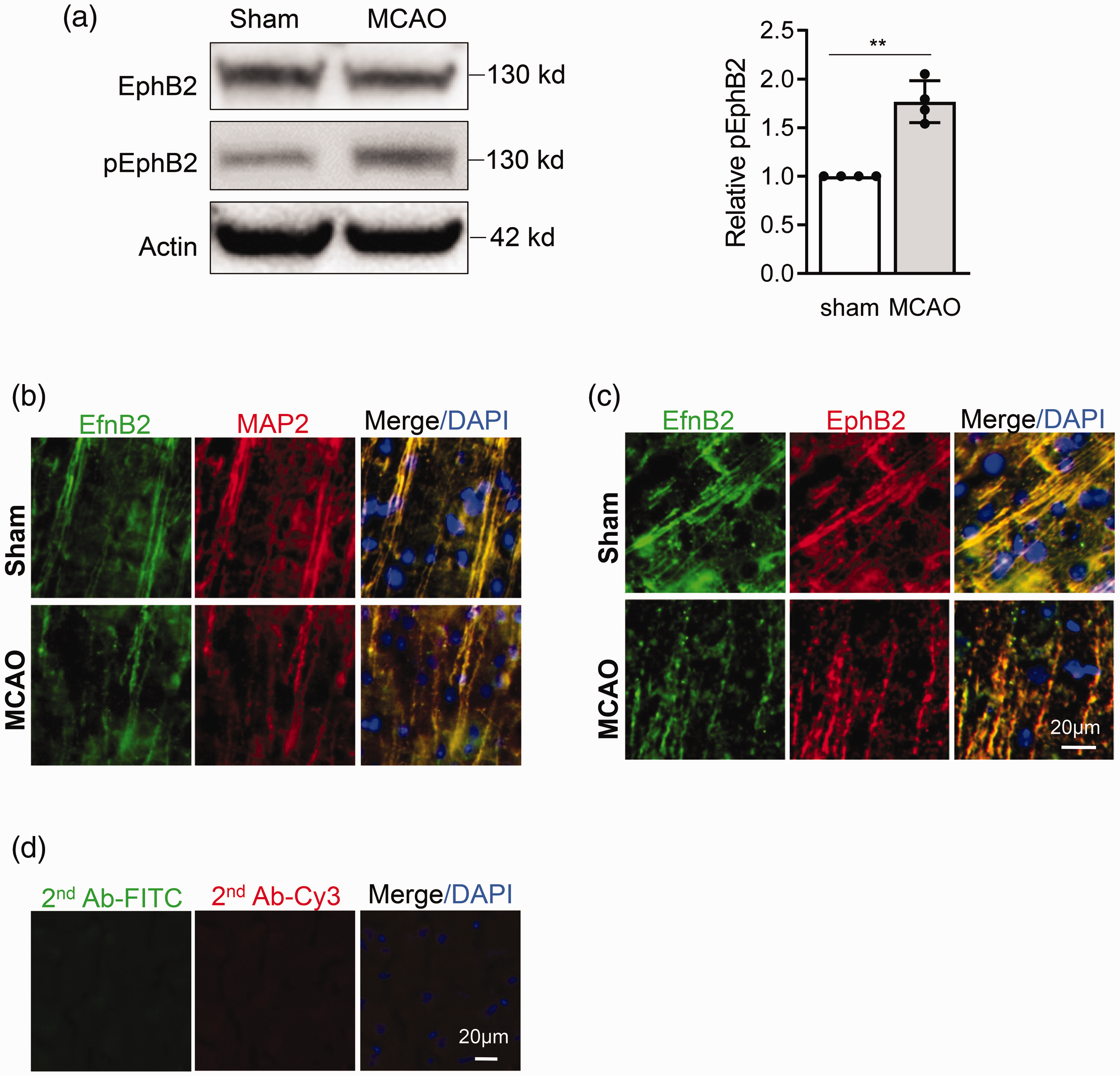
Ephrin-B2-EphB2 activation in mice brain after stroke. Mice were subjected to MCAO, 3 days later, eprhin-B2 and EphB2 activation in peri-infarct cortex were examined by Western blot and immunostaining. (a) Representative Western blot image and quantification for pEphB2 in peri-infarct cortex. Data are expressed as mean ± SD, **p < 0.01 (n = 4); (b and c) Immunostaining shows that EfnB2 co-localizes with dendrite (MAP2) (b) and EphB2 (c), and become more clustered after stroke in mouse brain. (d) Control immunostaining in the absence of primary antibodies.
Next, we asked whether we could also observe this pathway in neuron cultures after oxygen-glucose deprivation (OGD). Immunostaining showed that ephrin-B2 and EphB2 were co-localized along dendrites, and both were more clustered after 6 hr OGD (
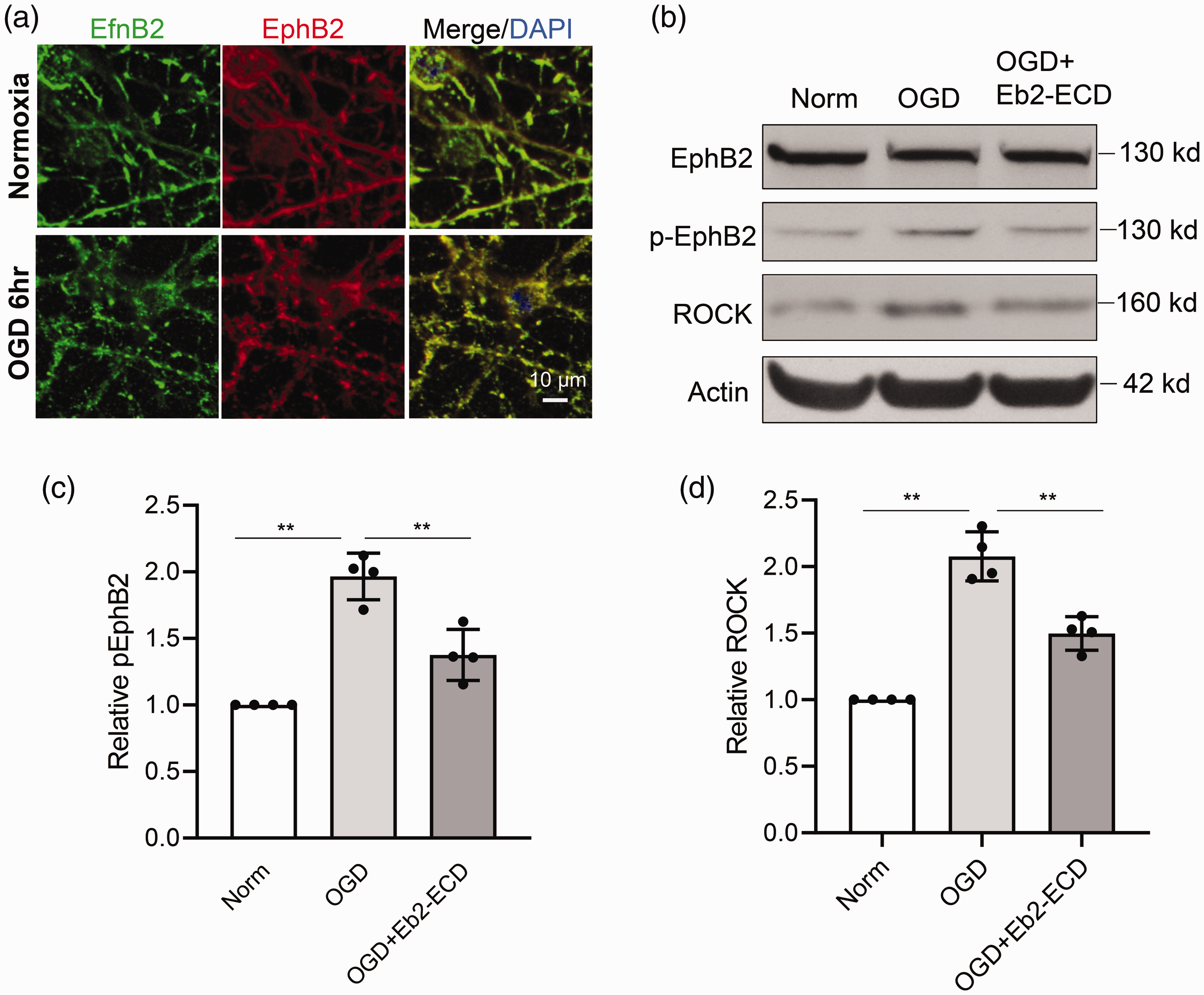
Ephrin-B2-EphB2 activation in primary neurons after OGD. Primary neurons were subjected to OGD for 6 hrs, eprhin-B2 and EphB2 activation were examined by immunostaining and Western blot. (a) Immunostaining show the clustering of ephrin-B2 and EphB2 after OGD. (b–d) Representative Western blot image and quantification for pEphB2 (c) and ROCK (d) in primary neurons after OGD. Data are expressed as mean±SD, **p<0.01 (n=4).
Blockade of ephrin-B2-EphB2 signaling protects dendrites after neuronal oxygen-glucose deprivation
Since ephrin-B2-EphB2 signaling appears to be activated in dendrites after ischemia and oxygen-glucose deprivation, we asked whether blockade of this pathway can protect dendrites. RNA silencing against EphB2 (siEphB2) effectively reduced EphB2 protein levels in neurons without affecting cell viability (
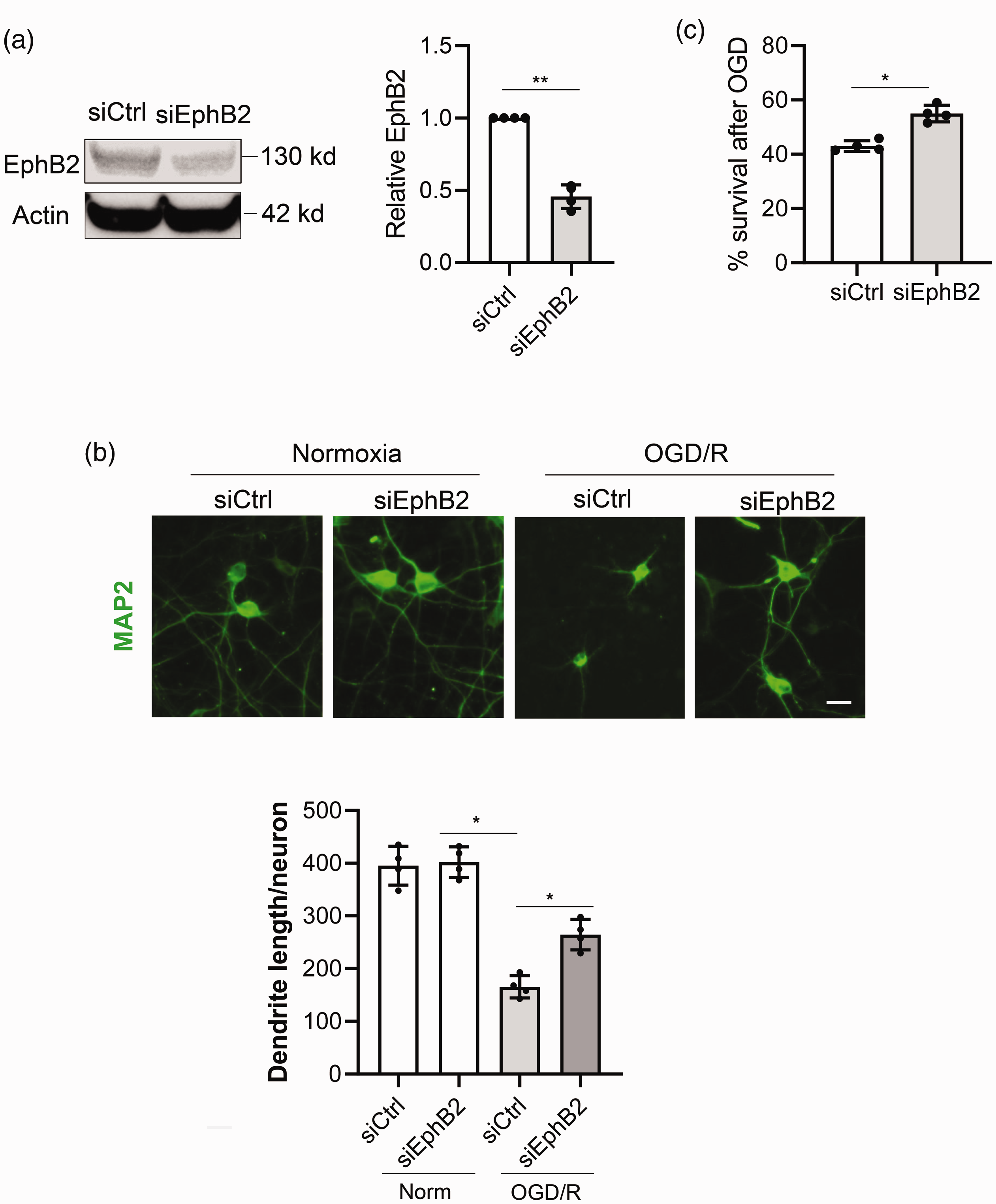
Blockade of Ephrin-B2-EphB2 signaling protects dendrite after neuronal oxygen-glucose deprivation. EphB2 was knocked down using siRNA before being subjected to 3 h OGD and 21 h reoxygenation. (a) EphB2 protein level after siRNA treatment measured by Western blot. (b) Dendrite examined by immunostaining for MAP2 and quantification of dendrite length using ImageJ-Neurite Tracer. (c) Neuron survival measured by LDH release assay. *p < 0.05, **p < 0.01 (n = 4). Scale bar = 20 µm.
The Eph receptor is activated by clustered ephrin, and inhibited by soluble ephrin.29,32 In line with this, recombinant soluble ephrin-B2 ectodomain (Eb2-ECD) has been widely used in experimental studies as a tool to inhibit ephrin-B2-EphB signaling.33,34 Therefore, we tested the effects of soluble Eb2-ECD in primary neurons after OGD. Neurons were subjected to 14 hrs OGD, and different doses of soluble Eb2-ECD were administered during OGD. Soluble Eb2-ECD at 50 nM significantly improved neuronal survival compared to controls (
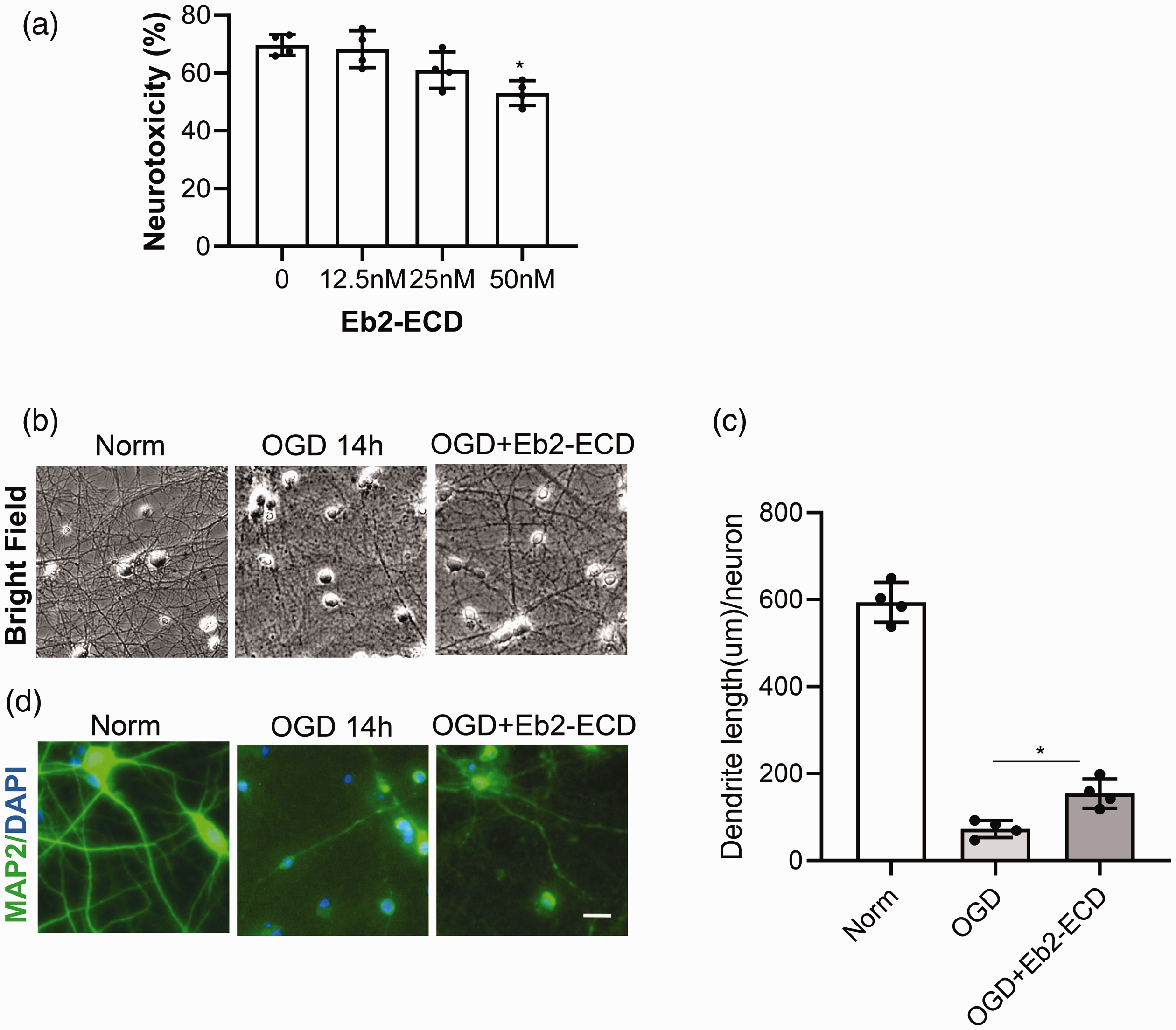
Soluble ephrin-B2-ectodomain (Eb2-ECD) protects against OGD-induced dendrite degeneration. Neurons were subjected to 14 hrs OGD and treated with soluble Eb2-ECD during OGD. (a) Different doses of Eb2-ECD were administered and neurotoxicity was measured by LDH release assay after OGD; (b and c) Neurons were treated with optimized dose (50nM) of Eb2-ECD, dendrite morphology was examined by bright field imaging (b) and by MAP2 immunostaining (c); (d) Relative dendrite length quantified by ImageJ-Neurite Tracer. Data expressed as mean±SD. *p < 0.05 (n = 4). Scale bar = 20 µm.
Comparison of ephrin-B2-EphB2 blockade with the NMDA antagonist MK801
Although our primary hypothesis was that blockade of ephrin-B2-EphB2 signaling would protect dendrites, our results so far suggest that overall improvement of neuronal cell survival may also occur. Therefore, we systematically compared the total dendrite and neuroprotective efficacy of the soluble ectodomain Eb2-ECD against the NMDA antagonist MK801 as a standard neuroprotectant. An LDH release assay showed accumulating neuronal death after 6 to 14 hrs of OGD. Both MK801 and Eb2-ECD significantly ameliorated the progression of neurotoxicity (
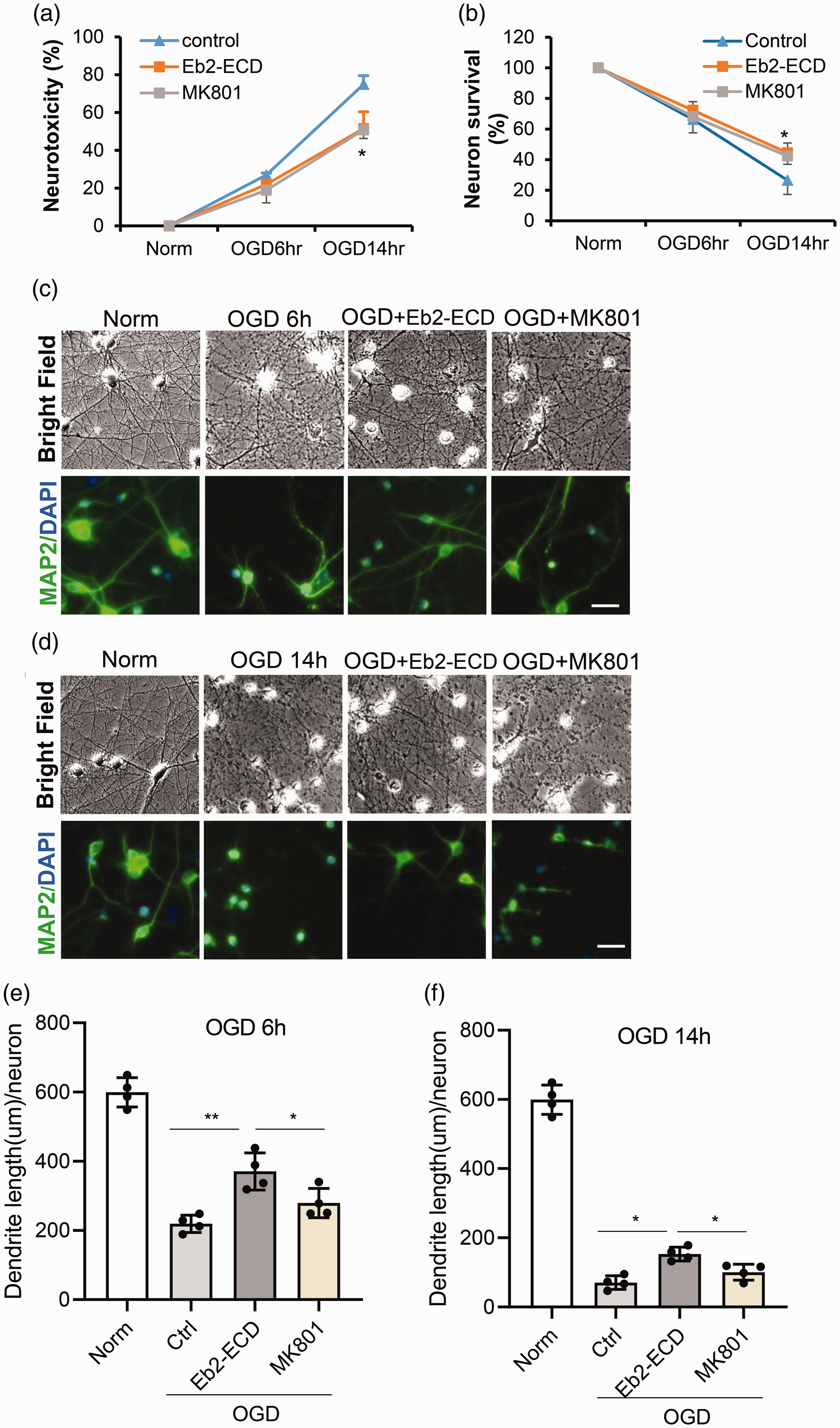
Comparison of the dendrite protective effect by ephrin-B2-EphB2 blockade and MK801 after OGD. Neurons were subjected to 6 hrs or 14 hrs OGD, and treated with soluble Eb2-ECD or MK801 during OGD. (a) Neurotoxicity was assessed by LDH release assay; (b) Neuron survival assessed by MTT assay. *p<0.05 between “MK801” vs “control” or “Eb2-ECD” vs “control” (n=4); (c) Dendrite morphology after 6 hrs OGD was examined by bright field imaging and MAP2 immunostaining; (d) Dendrite morphology after 14 hrs OGD was examined by bright field imaging and MAP2 immunostaining. (e and f) Quantification of dendrite length after 6 hrs (e) and 14 hrs (f) OGD. Data expressed as mean±SD. *p < 0.05, **p < 0.01 (n = 4). Scale bar = 20 µm.
Finally, we measured Bcl-2 and Nmnat2 as two representative molecular mediators for neuron soma and dendrite protection, respectively. The anti-apoptotic Bcl-2 has been reported to protect against neuron soma loss,36,37 whereas Nmnat2 exerts protective effects in dendrites and axon degeneration.38,39 After OGD, levels of both mediators were decreased. Consistent with the overall improvement in neuronal survival, both MK801 and Eb2-ECD restored levels of Bcl-2 (
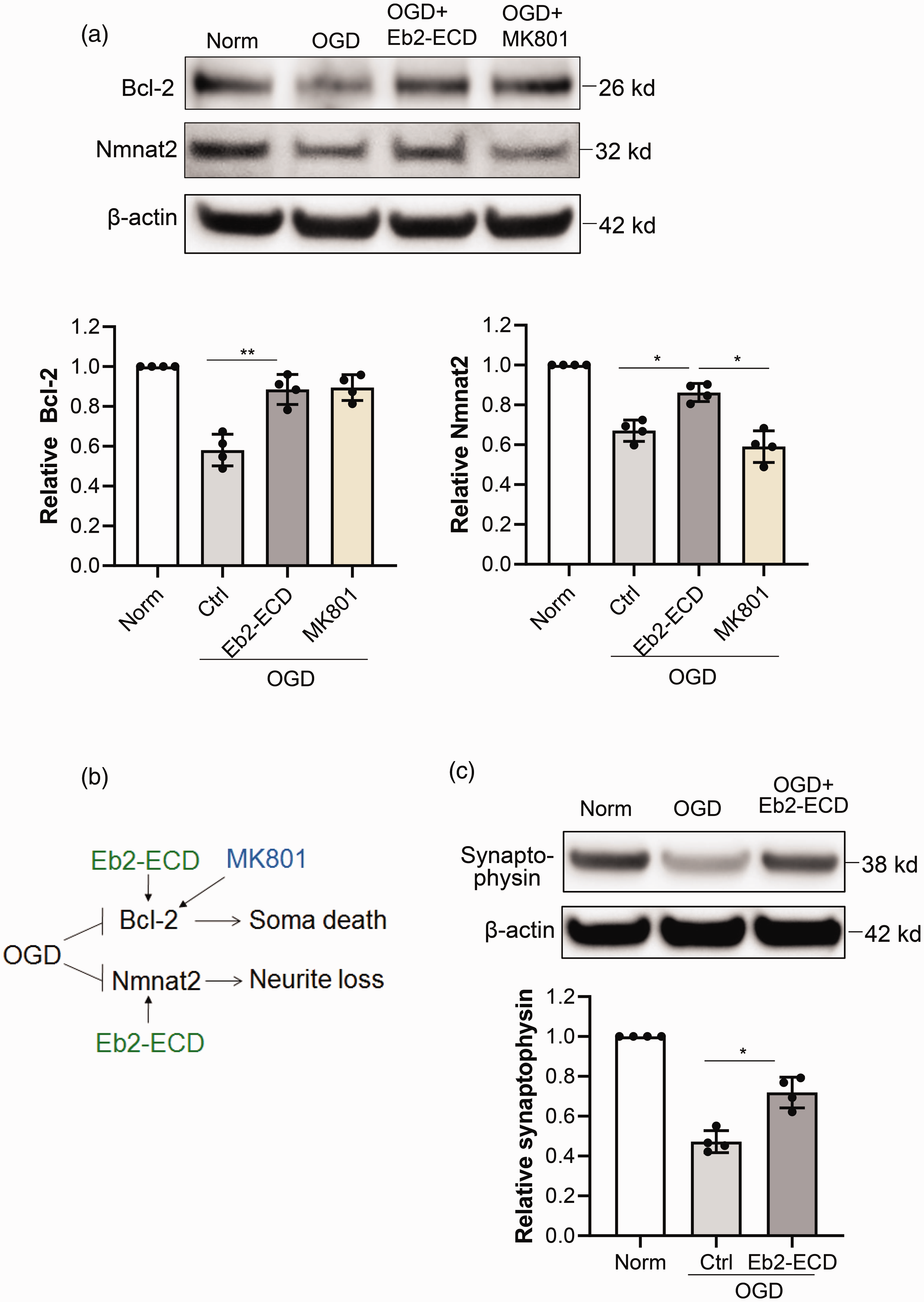
Comparison of neuroprotective mechanisms of ephrin-B2-EphB2 blockade and MK801. Primary neurons were subjected to 6 hrs OGD and treated with soluble Eb2-ECD or MK801. Western blot was performed to test the proteins involved in apoptosis and dendrite plasticity. (a) Representative Western blot images and quantification of Bcl-2 and Nmnat2 in primary neurons after OGD. (b) Diagram showing the differences of Eb2-ECD and MK801 in regulating soma and dendrite survival pathways. (c) Western blot and quantification of Synaptophysin protein level after OGD and Eb2-ECD treatment. Data expressed as mean±SD. *p < 0.05, **p < 0.01 (n = 4).
Discussion
Dendrite degeneration significantly contributes to stroke pathogenesis. However, very limited effort has been devoted to investigating dendrite protection in neuroprotection research. Technically, many of the popular methodologies in experimental stroke research, such as LDH release or tetrazolium-based assays to evaluate neurotoxicity in cultured neurons or animal stroke models, are not capable of assessing dendrite degeneration. Many studies indeed show that infarct volume does not always correlate with neurological outcomes of stroke; some of these discrepancies may be partially attributed to different levels of dendrite degeneration and protection.40–42 It is therefore critical to not only assess infarct volume or neuron soma death in neuroprotection research, but also examine dendrite integrity. In order to develop more effective stroke treatments it may be essential to protect both neuronal soma as well as dendrites.
In the present study, we aimed to investigate the roles of ephrin-B2-EphB2 signaling in dendrite protection after stroke. Our results suggest that: (1) ephrin-B2-EphB2 signaling is activated after stroke in mice or after OGD in primary cultured neurons; (2) EphB2 blockade, either by siRNA or by soluble ephrin-B2 ectodomain, may protect against both neuron soma and dendrite degeneration after oxygen-glucose deprivation in primary cultured neurons. This strategy is superior to MK801 as the latter has less dendrite protection effect. These findings may improve our understanding of the mechanisms of dendrite degeneration after stroke.
As membrane-anchored ligands, ephrins play significant roles in a wide range of developmental and pathological processes in central nervous system through coupling with their receptors, Eph. 12 Ephrins can be divided into Ephrin-A’s (5 members) and Ephrin-B’s (3 members) subclasses, and Eph receptors divided into EphA (9 members) and EphB (5 members) subclasses. Previous studies of ephrin-B2-EphB2 mostly focus on neural development, demonstrating that ephrin-B2-EphB2 activation is able to inhibit axon and dendrite growth.14,15 Recent studies further suggest important implications of ephrin-B2 signaling in the pathogenesis of neurological diseases. For example, upregulation of ephrin-B2 and EphB2 is associated with RGC axon loss in glaucoma, suggesting they might be involved in glaucoma progression. 20 Moreover, EphB2 signaling has been reported to promote neuronal excitotoxicity after ischemic stroke. 21 In this study we found ephrin-B2-EphB2 signaling can be activated in neurons after oxygen-glucose deprivation, while inhibition of ephrin-B2-EphB2 signaling can protect against both soma death and dendrite degeneration. These findings may broaden our understanding of the implications of ephrin-B2-EphB2 signaling in neurodegenerative diseases, and suggest this might be a novel target for the development of stroke treatment.
It should be noted that each ephrin ligand can bind multiple Eph receptors. For ephrin-B2, in addition to EphB2, it can also bind to EphB1, EphB4 and EphA4 and induce downstream signaling.12,43 These receptors may also engage important functions in the pathogenesis of stroke. For example, EphB1 can restrict axon outgrowth by inhibiting growth cone formation during neural development. 44 EphA4 knockdown in transgenic mice enhanced motor recovery after stroke, 45 and also improved the survival of primary cultured neurons after OGD. 46 On the other hand, each Eph receptor also has multiple binding ligands. For EphB2, both ephrin-B2 and ephrin-B1 are binding ligands that can activate EphB2 signaling. Ephrin-B2 and ephrin-B1 share some common functions in EphB2 signaling, e.g, both are required for EphB2 dependent presynaptic development. 47 Ephrin-B1 and EphB2 signaling is also implicated in neurite outgrowth. 48 There is a high probability that ephrin-B1-EphB2 signaling, or ephrin-B2 signaling through EphB1 and EphA4, may also regulate the dendrite degeneration after stroke, which warrants further investigation in the future.
In this study we used soluble Eb2-ECD to inhibit ephrin-B2 signaling, because it is well accepted that Eph receptor can be inhibited by soluble ephrin ligands,29,32 and recombinant soluble Eb2-ECD is widely used in experimental studies as a tool to inhibit ephrin-B2-Eph signaling.33,34 Recent studies revealed that Eb2-ECD can be cleaved by ADAM10 in vascular systems including endothelial cells, mesenchymal cells and pericytes, which may play critical roles in embryonic development and adult fibrotic diseases in mice.49,50 How endogenous Eb2-ECD in the brain may contribute to dendrite protection will be an important subject for future study.
In addition to prevention of dendrite degeneration in this study, another important strategy for neuron recovery is the potential dendrite regeneration after stroke. There have been a number of reports using invertebrate peripheral neurons to study endogenous or treatment-induced dendritic regrowth after injury,51,52 whereas studies using mammalian CNS neurons are rare. 53 The possibility of regenerating dendrites and axons after stroke awaits comprehensive investigation, which may significantly contribute to stroke recovery. As eprhin-B2 signaling regulates neurite growth during development,14,32 manipulating ephrin-B2 signaling may also impact neurite regrowth after stroke. Our pilot in vitro study using primary cultured neurons showed that dendrites can regrow after OGD, while treatment with Eb2-ECD improved dendrite regrowth (unpublished data). Whether ephrin-Eph signaling may influence dendritic regrowth in vivo need to be further investigated.
Although we focused on neuron and dendrite protection in this study, it is highly likely that ephrin-B2-EphB2 signaling may influence neurovascular and blood-brain barrier responses as well. Other studies documented that ephrin-B2 can be expressed in endothelial cells and this pathway plays an essential role in vascular function and angiogenesis during development.54–56 Indeed, we have some pilot data showing that EfnB2-ECD can ameliorate BBB leakage in in vitro study testing the permeability of cultured endothelial monolayer (unpublished data). Moreover, a recent study indicated that Ephrin-B2 signaling may enhance angiogenesis after stroke in hypertensive rats. 57 The roles of Ephrin-B2 signaling in vascular dysfunction and angiogenesis after stroke await further study in the future.
Our study implies that ephrin-EphB2 signaling might be a potential target in therapeutic development for stroke, however, cautions must be taken because ephrin-Eph system has a wide range of roles in CNS function including emotion. For example, EphB2 signaling in amygdala neurons plays important roles in fear response in neonatal brain. 58 EphB2 cleavage by neuropsin in amygdala neurons is also required for stress-induced anxiety response. 59 Thus to gain a comprehensive understanding of EphB2 functions in the brain, future studies should also assess psychiatric responses after stroke, and should also focus on specific subpopulations of neurons.
Given the successful development of thrombectomy devices for ischemic stroke patients in the past few years,60,61 there is now an opportunity to pursue combination therapies in the new era of reperfusion. 62 In light of the 30-year failure of neuroprotective strategies in clinical trials, the stroke community have developed consensus recommendations that protection for all cell types in the neurovascular unit should be required after stroke, i.e. brain cytoprotection instead of only neuroprotection. 63 Our study here may provide an added aspect to be considered. Saving only the neuronal soma may not be enough. Rescuing dendrites is also necessary for stroke treatment.
In summary, we found that ephrin-B2-EphB2 signaling is activated after ischemia and oxygen-glucose deprivation in neurons, and blockade of ephrin-B2-EphB2 signaling may protect both neuron soma and dendrites. These findings provide a proof of concept that ephrin-B2-EphB2 signaling may be a novel therapeutic target for protecting the entire neuronal network against ischemic injury.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X20973119 - Supplemental material for EphrinB2-EphB2 signaling for dendrite protection after neuronal ischemia in vivo and oxygen-glucose deprivation in vitro
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X20973119 for EphrinB2-EphB2 signaling for dendrite protection after neuronal ischemia in vivo and oxygen-glucose deprivation in vitro by Zhanyang Yu, Wenlu Li, Jing Lan, Kazuhide Hayakawa, Xunming Ji, Eng H Lo and Xiaoying Wang in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This work was in part supported by American Heart Association grant 15SDG25550035 (to ZY).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
ZY, XW and EHL designed the study; ZY, WL and JL performed the experiments, collected data and analyzed the data; KH, XJ, XW and EHL participated in data analysis and interpretation. ZY and EHL drafted the manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.