
Abstract
White matter lesions (WMLs) caused by cerebral chronic hypoperfusion (CCH) may contribute to the pathophysiology of Alzheimer’s disease (AD). However, the underlying mechanisms and therapeutic approaches have yet to be totally identified. In the present study, we investigated a potential therapeutic effect of the free radical scavenger edaravone (EDA) on WMLs in our previously reported novel mouse model of AD (APP23) plus CCH with motor and cognitive deficits. Relative to AD with CCH mice at 12 months (M) of age, EDA strongly improved CCH-induced WMLs in the corpus callosum of APP23 mice at 12 M by improving the disruption of white matter integrity, enhancing the proliferation of oligodendrocyte progenitor cells, attenuating endothelium/astrocyte unit dysfunction, and reducing neuroinflammation and oxidative stress. The present study demonstrates that the long-term administration of EDA may provide a promising therapeutic approach for WMLs in AD plus CCH disease with cognitive deficits.
Introduction
White matter lesions (WMLs) have increasingly raised attention to the pathophysiological progress of ischemic stroke, 1 chronic cerebral hypoperfusion (CCH), 2 , 3 Alzheimer’s disease (AD), 4 and vascular dementia.5–7 This is partly due to damage of components of cerebral white matter (CWM) under pathophysiological conditions. 8 The majority of CWM is composed of oligodendrocytes (OLs), a lipid-rich myelin sheath, and neuronal axons which are not only susceptible to oxidative stress and inflammation, but are also an abundant source of reactive free radicals. 9 , 10 Our previous study reported that severe WMLs displayed a disruption of white matter (WM) integrity while inflammation occurred in the corpus callosum (CC) of a novel mouse model that combines AD with CCH. 11 , 12 However, specific molecular mechanisms associated with WMLs and effective therapeutic approaches to WMLs in the present AD plus CCH mouse model have not yet been fully identified.
The free radical scavenger, edaravone (EDA), was the first neuroprotective agent approved in Japan for clinical use in 2001. At present, EDA is widely used in Japan for acute ischemic stroke patients and amyotrophic lateral sclerosis (ALS) patients in clinics. 13 , 14 Besides the application of EDA in clinical ischemic stroke and ALS, experimental studies have demonstrated the neuroprotective effects of EDA in WMLs of CCH animal models via the improvement of disrupted WM integrity, inhibition of vascular endothelial injury, oxidative stress, and inflammation, as well as the promotion of proliferation of oligodendrocyte progenitor cells (OPCs) and oligodendrogenesis.15–18 Moreover, increasing evidence has demonstrated that EDA alleviated pathologies of AD and cognitive deficits in in vivo or in vitro studies by targeting multiple key pathways of AD such as amyloid beta, hyperphosphorylated tau, oxidative stress and inflammation in grey matter.19–21 Recently, our study reported the improvement of cognitive and motor deficits with pathological benefits in grey matter in the present AD plus CCH mouse model after the administration of EDA. 22 , 23 However, no information is available on the roles of EDA in WMLs of the present AD plus CCH mouse model.
Therefore, in the present study, we aimed to investigate the role of EDA in WMLs of the present AD plus CCH mouse model, focusing primarily on the mechanisms associated with WM integrity, oligodendrogenesis, endothelium/astrocyte units, inflammation, and oxidative stress.
Materials and methods
Experimental model and drug treatment
All animal experiments were performed in compliance with a protocol approved by the Animal Committee of the Graduate School of Medicine and Dentistry, Okayama University (OKU#2012325) and conducted in accordance with ARRIVE guidelines (https://www.nc3rs.org.uk) as well as the Okayama University guidelines on the Care and Use of the Laboratory Animals. The present study is a part of a larger project focusing on the effect of EDA in AD with CCH. Experiments were performed in male APP23 transgenic mice and its male littermate. Transgenic mouse APP23 overexpressing the human APP gene with the Swedish mutation driven by a Thy1 promotor was generated from B6, D2-TgN (Thy1-APPSwe), which is a proven valid animal model for AD. 24 , 25
We calculated the sample size based on a preliminary study and on the requirement of statistical power. 26 To detect a decrease in CBF between the vehicle group and the EDA group with a two-sided 5% level of significance and 80% power, a sample size of eight mice per group was necessary. Based on knowledge gained from our previous experiments (unpublished data), an approximate anticipated dropout rate of 30% in mice until they were 12 months (M) old was considered. Therefore, when 4 M old, 14 male mice were randomly assigned to each experimental group, giving a total of 56 male mice in this study, 16 of which were excluded based on the following exclusion criteria: mice died as a result of procedural problems during CCH surgery or died after surgery (n = 12), or mice failed to display a decrease of CBF after CCH treatment (n = 4). Finally, at 12 M, the number of sacrificed mice assigned to the four groups was: wild type (WT) group (WT + sham surgery, n = 10), APP23 group (APP23 + sham surgery, n = 12), CCH group (APP23 + CCH, n = 8), and EDA-treated group (APP23 + CCH + EDA, n = 10). The mice in the WT group were C57BL/6J mice.
Ameroid constrictors (0.75 mm internal diameter; Research Instruments NW, Lebanon, OR, USA,) were applied to induce a CCH in AD mice. Our previous studies reported that cerebral blood flow (CBF) gradually and progressively decreased, and CCH was successfully established by ameroid constrictors in APP23 mice. 11 , 27
In order to conduct the surgery of CCH, at least one week before surgery, mice were kept under constant light, humidity, and temperature conditions to acclimatize them to the experimental room, housed at one mouse per cage. Mice were fasted for less than 6 h prior to surgery, but were allowed free access to water. All animals were randomly assigned to the experimental groups before surgery.
During surgery, briefly, experimental mice at 4 M of age were anesthetized with a mixture of nitrous oxide/oxygen/isoflurane (69%:30%:1.5%, respectively) using an inhalation box. Once anesthetized, which was defined by the lack of a response to a toe pinch, experimental mice were subjected to cervical incision, and ameroid constrictors were implanted in bilateral common carotid arteries (BCCAs).
After surgery, mice were placed in a 37 °C chamber to allow them to recover from the surgery, then placed in standard mice cages with free access to food and water under standard rearing conditions. In addition, buprenorphine (0.05 mg/kg, 0.015 mg/ml) was administered intramuscularly to relieve pain.
As for the EDA treatment, mice in the APP23 + CCH + EDA group were treated with an intraperitoneal (i.p.) injection of EDA (3 mg/ml; Mitsubishi Tanabe Pharmaceutical Co. Ltd., Osaka, Japan) every other day at 50 mg/kg body weight. The administration of EDA was initiated at 4 M and ended before sacrifice at 12 M, over a period of 8 months.
CBF was measured with a laser-Doppler flowmeter (FLO-C1, Omegawave, Tokyo, Japan) in mice at 12 M before sacrifice. A laser doppler flowmetry probe was fixed perpendicular to the skull 1 mm posterior and 2.5 mm lateral to the bregma where CBF values were measured five times. The mean CBF value was recorded.
Tissue preparation and immunohistochemistry
At 12 M, mice in the four groups were deeply anesthetized by i.p. injection of pentobarbital (40 mg/kg), and transcardially perfused with 20 ml of ice-cold phosphate-buffered saline (PBS) and then 20 ml of ice-cold 4% paraformaldehyde (PFA) in 0.1 mol/L phosphate buffer. The brains were removed and post-fixed in 4% PFA overnight. Floating coronal sections were sliced to a thickness of 50 µm with a vibrating blade microtome (LEICA VT1000S; Leica, Nussloch, Germany). The morphological and pathological changes in WM were detected in the CC in the present study. To evaluate WMLs, mice brain sections were stained with luxol fast blue (LFB). The LFB Stain Kit was purchased from ScyTek Laboratories, Inc. (LBC-1; Logan, Utah, U.S.A.). The LFB staining method was performed according to our previous report. 12
For immunohistochemical staining by the diaminobenzidine (DAB) reaction, brain sections were immersed in 0.6% periodic acid to block intrinsic peroxidase and treated with 5% bovine serum in 50 mM PBS (pH 7.4) containing 0.1% Triton X-100 to block any non-specific antibody responses then were incubated with primary antibodies. The amino acid sites were probed with the following antibodies: rabbit anti-myelin basic protein (MBP) antibody (1:500, ab40390; Abcam, Cambridge, U.K.), mouse anti-myelin-associated glycoprotein (MAG) antibody (1:200, sc-166849; Santa Cruz Biotechnology, San Jose, CA, U.S.A.), mouse anti-SMI312 antibody (1:200, 837904; Biolegend, San Diego, CA, U.S.A.), rabbit anti-GST-π antibody (1:500, ADI-MSA-102-E; Enzo Life Sciences, Nassau, NY, U.S.A.), rabbit anti-Iba-1 antibody (1:500, NCNP24; Wako, Osaka, Japan), mouse anti-4-HNE antibody (1:50, MHN-020P; JaICA, Shizuoka, Japan), and 8-OHdG (1:50, MOG-020P; JaICA, Shizuoka, Japan). The negative control contained no primary antibody. Immunoreactions were visualized using horseradish peroxidase-conjugated secondary antibodies and then developed in horseradish 1 peroxidase streptavidin-biotin complex solution (PK-6104; Vectastain ABC kit; 1 Vector Laboratories, Burlingame, CA, U.S.A.) with the DAB reaction.
For immunofluorescent staining, brain sections were treated with 5% bovine serum in 50 mM PBS (pH 7.4) containing 0.1% Triton X-100 to block any non-specific antibody responses, then were incubated with primary antibodies. The following primary antibodies were used: mouse anti-Caspr antibody, clone K65/35 (1:100, MABN9; Merck Millipore, Burlington, MA, U.S.A.), rabbit anti-Nfasc186 antibody (NF186, 1:100, ab31719; Abcam, Cambridge, U.K.), goat anti-PDGFRα antibody (1:100, AF1062; R&D Systems, Minneapolis, MN, U.S.A.), rabbit anti-Ki67 antibody (1:500, ab15580; Abcam), rabbit anti-GFAP antibody (1:500, Z0334; Dako, Santa Clara, CA, U.S.A.), rat anti-CD31 antibody (1:100, 553371; BD Pharmingen, Bergen, NJ, U.S.A.), and negative control was obtained without primary antibody. Each primary antibody was detected by appropriate secondary antibodies conjugated with Alexa Fluor 488 or 555™ (Molecular Probes, Waltham, MA, USA). The sections were mounted with 4’,6-diami dino-2-phenylindole (DAPI) medium (H-1200, Vector Laboratories) or antifade mounting medium (H-1000, Vector Laboratories).
Quantitative analysis
Individual immunohistochemical sections were digitized with a digital microscope camera (Olympus BX-51; Olympus Optical Co., Tokyo, Japan). The immunofluorescent sections were digitized with a confocal laser microscope (LSM780; Zeiss., Oberkochen, Germany). Three levels of sections per brain were taken into consideration from the caudate putamen (1.0, 0.5, and 0 mm rostal to the bregma) and 3–6 randomly regions per section were selected to take photos for analysis (i.e., n = 9–18 measurements per mouse). For the semiquantitative evaluation of MBP, MAG, LFB, SMI 312, CD31, Iba-1, 4-HNE, and 8-OHdG staining, the average pixel intensity of signals in the CC were measured. For the semiquantitative analyses of GST-π, PDGFRα, Ki67, and GFAP staining, the number of positive cells in the CC were counted. The length of NF186 and the number of nodes were measured or counted in the CC by double immunolabeling (NF186/Caspr). The length of the gap between Caspr signals was measured by contrasting with the nodal signal (NF186). To analyze endothelium/astrocyte remodeling in the neurovascular unit (NVU) in the CC, the inner diameter of CD31/GFAP double positive vessels were evaluated in the CC. Additionally, the thickness and area of the CC were also measured to better quantitatively assess WM atrophy or loss by analyzing MAG staining. All immunostaining data were analyzed by image processing software (Image J; National Institutes of Health, Bethesda, MD, U.S.A.).
Statistical analysis
During data collection and analysis, the investigators were blinded to the experimental groups. All results were presented as mean ± SD. Statistical comparisons were tested using one way ANOVA based on a Tukey-Kramer post comparison. p < 0.05 was considered statistically significant.
Results
Edaravone improved CBF in 12-month-old APP23 mice with CCH
There were no significant differences in CBF examined by LDF between the WT and APP23 groups at 12 M (Supplemental Figure 1). However, the value of CBF in the APP23 + CCH group was significantly lower than that in the APP23 group at 12 M (Supplemental Figure 1, ##p < 0.01 vs APP23). Comparatively, EDA significantly decreased CBF in mice in the APP23 + CCH group (Supplemental Figure 1, &p < 0.01 vs APP23).
Edaravone improved the disruption of white matter integrity in the damaged corpus callosum of APP23 mice with CCH
Both MBP and MAG labeling were observed in the CC myelin of all groups (Figure 1(a)). MAG staining showed that, compared with the APP23 group, the thickness and area of the CC significantly decreased in the APP23 + CCH group. And EDA treatment significantly improved the thickness of the CC relative to the APP23 + CCH group, indicating the overall impact of EDA therapy on WM loss (Supplemental Figure 2, #p < 0.05 vs APP23; &p < 0.05 vs APP23 + CCH). LFB staining positively marked the CC myelin in all groups (Figure 1(a)). Vacuoles and the disappearance of myelinated fibers were obviously observed in the CC of the APP23 + CCH group, which was significantly improved by EDA therapy (Figure 1(a)). SMI312 labeling was mainly observed in neurofilaments of the CC neuronal axon in all groups (Figure 1(a)). Some axon debris was observed as a bulk pattern in the APP23 + CCH group in the CC, which was improved to some degree following the administration of EDA (Figure 1(a)). NF186 was localized in the node of Ranvier, which was flanked by paranodel protein Caspr in the CC of all groups (Figure 1(a)).
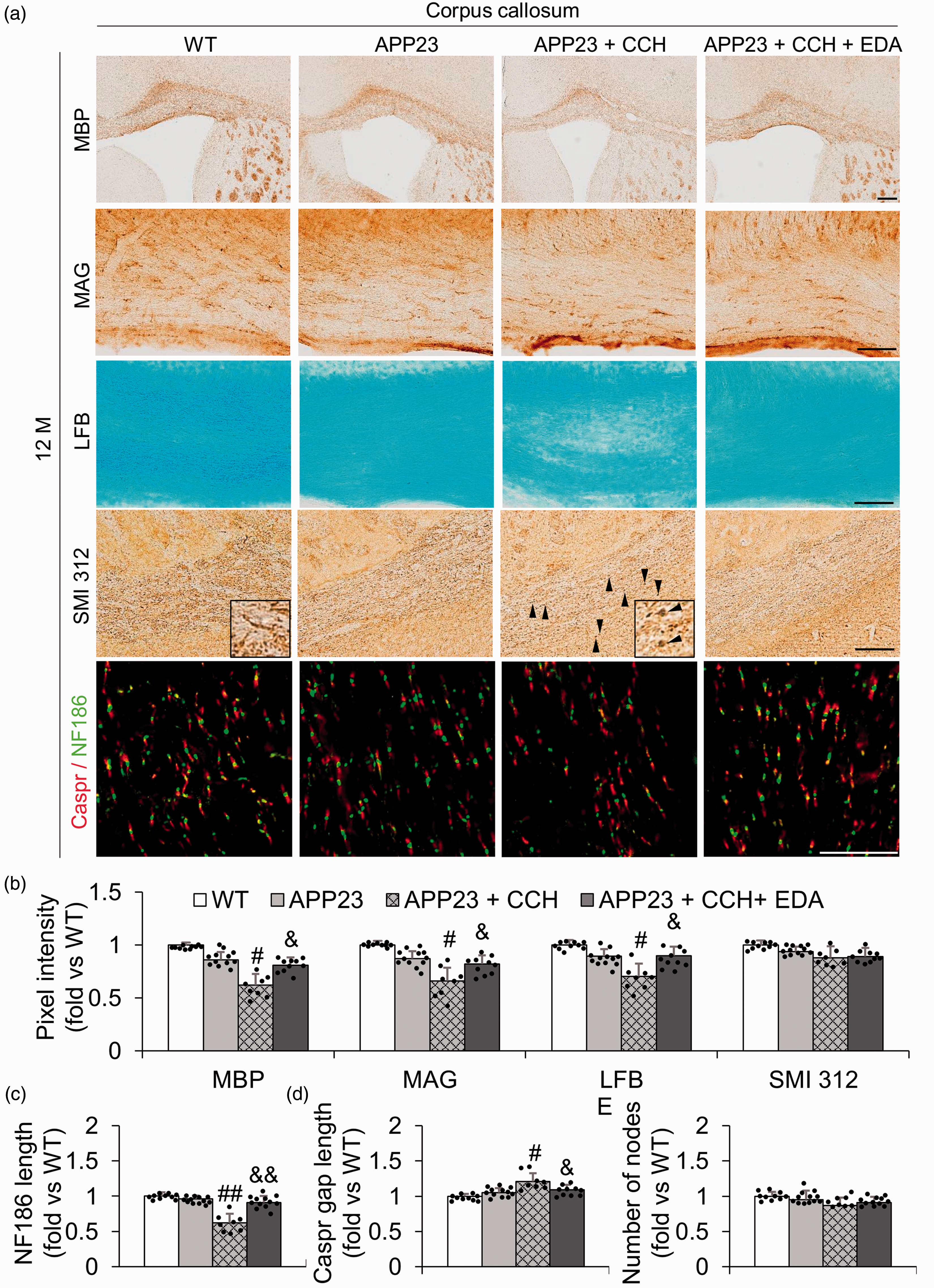
Representative immunohistochemical photomicrographs of myelin basic protein (MBP), myelin-associated glycoprotein (MAG), LFB, SMI 312, Caspr/NF186 in the corpus callosum (CC) at 12 M (a), and associated quantitative histograms (b–e). Note the significant relative recovery of pixel intensities of MBP, MAG, and LFB with EDA compared with the APP23 + CCH group at 12 M, except for SMI 312 (b). Note the significant relative recovery of NF186 length with EDA, Caspr gap length with EDA, and no significant difference in the number of nodes between APP23 + CCH and APP23 + CCH + EDA groups at 12 M. Arrowheads represent axon debris in the APP23 + CCH group (a). Values are means ± S.D. Scale bar = 100 um. (#p < 0.05 vs APP23, ##p < 0.01 vs APP23; &p < 0.05 vs APP23 + CCH, &&p < 0.01 vs APP23 + CCH).
Although the pixel intensities of MBP, MAG, LFB, and SMI312 were not different between WT and APP23 groups at 12 M in the CC (Figure 1(b)), pixel intensity of MBP, MAG, LFB was significantly reduced in the APP23 + CCH group compared with the APP23 group at 12 M in the CC (Figure 1(b), #p < 0.05 vs APP23), showing a significant recovery following the administration of EDA (Figure 1(b), &p < 0.05 vs APP23 + CCH). However, no significant change was observed in pixel intensity of SMI312 between the APP23 and APP23 + CCH groups (Figure 1(b)). On the other hand, CCH significantly decreased the length of NF186 and Caspr gap length in the CC at 12 M with a remarkable recovery after the administration of EDA (Figure 1(c) and (d), #p < 0.05 vs APP23, ##p < 0.01 vs APP23; &p < 0.05 vs APP23 + CCH, &&p < 0.01 vs APP23 + CCH). However, the number of nodes in the CC was not significantly different between any two groups at 12 M (Figure 1(e)).
Edaravone enhanced the number of oligodendrocytes and the proliferation of oligodendrocyte progenitor cells
Compared with the APP23 group, CCH greatly decreased the number of GST-π-positive OLs in the CC at 12 M (Figure 2(a) and (c), #p < 0.05 vs APP23). However, a significant recovery was observed in the APP23 + CCH + EDA group relative to the APP23 + CCH group (Figure 2(a) and (c), &p < 0.05 vs APP23 + CCH). The double immunofluorescence data of the OPC marker (PDGFRα) and cell proliferation marker (Ki67) confirmed that the administration of EDA dramatically increased the number of proliferating cells, especially enhancing the proliferation of OPCs (Figure 2(b), (d), and (e), &&p < 0.01 vs APP23 + CCH). No large differences in the number of Ki67-positive cells and Ki67/PDGFRα double-positive cells were observed between the WT and APP23 groups (Figure 2(d) and (e)).
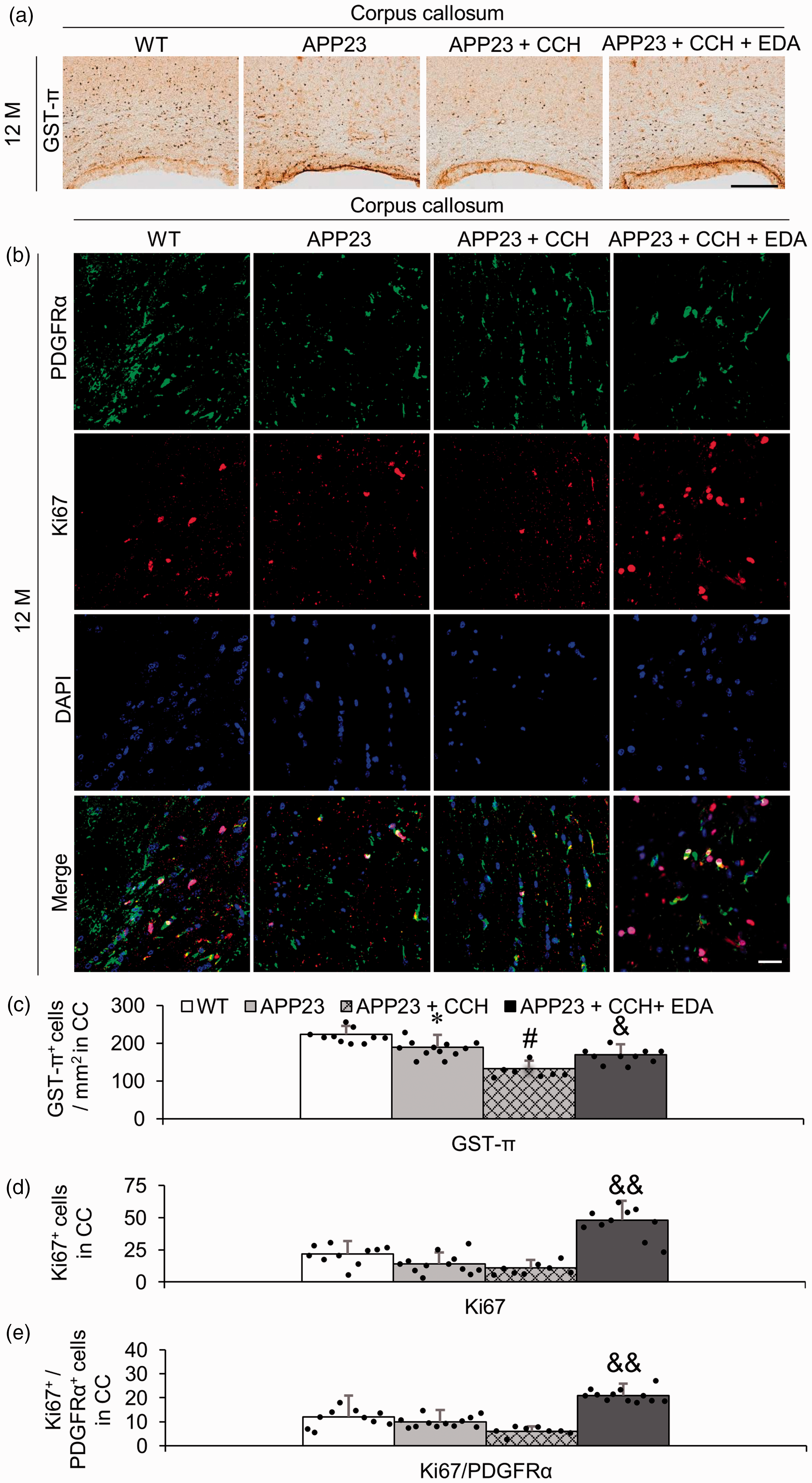
Representative immunohistochemical photomicrographs of oligodendrocyte (GST-π) in the corpus callosum (CC) (a) and associated quantitative histogram (c). Note the significant increase in the number of GST-π-positive oligodendrocytes in the CC of mice in the APP23 + CCH + EDA group compared to the CC of mice in the APP23 + CCH group (c). Representative double immunofluorescent photomicrographs of oligodendrocyte progenitor cells (OPCs, PDGFRα, green signals) and proliferation marker (Ki67, red signals) positive cells (b) and associated quantitative histograms (d and e) in the CC. Note the significant increase of total proliferating cells (d) and proliferating OPCs (e) in the CC of mice in the APP23 + CCH + EDA group compared to the CC of mice in the APP23 + CCH group. Values are means ± S.D. Scale bar = 100 um. (*p < 0.05 vs WT, **p < 0.01 vs WT; #p < 0.05 vs APP23; &p < 0.05 vs APP23 + CCH, &&p < 0.01 vs APP23 + CCH).
Edaravone attenuated endothelium/astrocyte remodeling
Single immunofluorescent data of an endothelium marker (CD31) showed that CCH significantly increased the area of CD31-positive vessels in the CC of AD mice at 12 M, while the administration of EDA significantly recovered these changes (Figure 3(a) and (c), ##p < 0.01 vs APP23; &&p < 0.01 vs APP23 + CCH). Double immunofluorescent data of CD31 and an astrocyte marker (GFAP) showed that the inner diameter of partial parenchymal small vessels in the CC increased significantly in the APP23 + CCH group relative to the APP23 group (Figure 3(a) and (d), ##p < 0.01 vs APP23). Additionally, partial damage of CD31-positive endothelia and dissociation of CD31-positive endothelia and GFAP-positive astrocytes were also observed in small vessels in the CC of AD mice with CCH at 12 M (Figure 3(a)). The administration of EDA strongly alleviated the pathological alterations of NVU in the CC of AD mice with CCH at 12 M (Figure 3(a) and (d), &p < 0.05 vs APP23 + CCH). No significant differences in vessels and NVU were observed between the WT and APP23 groups (Figure 3(a), (c), and (d)).
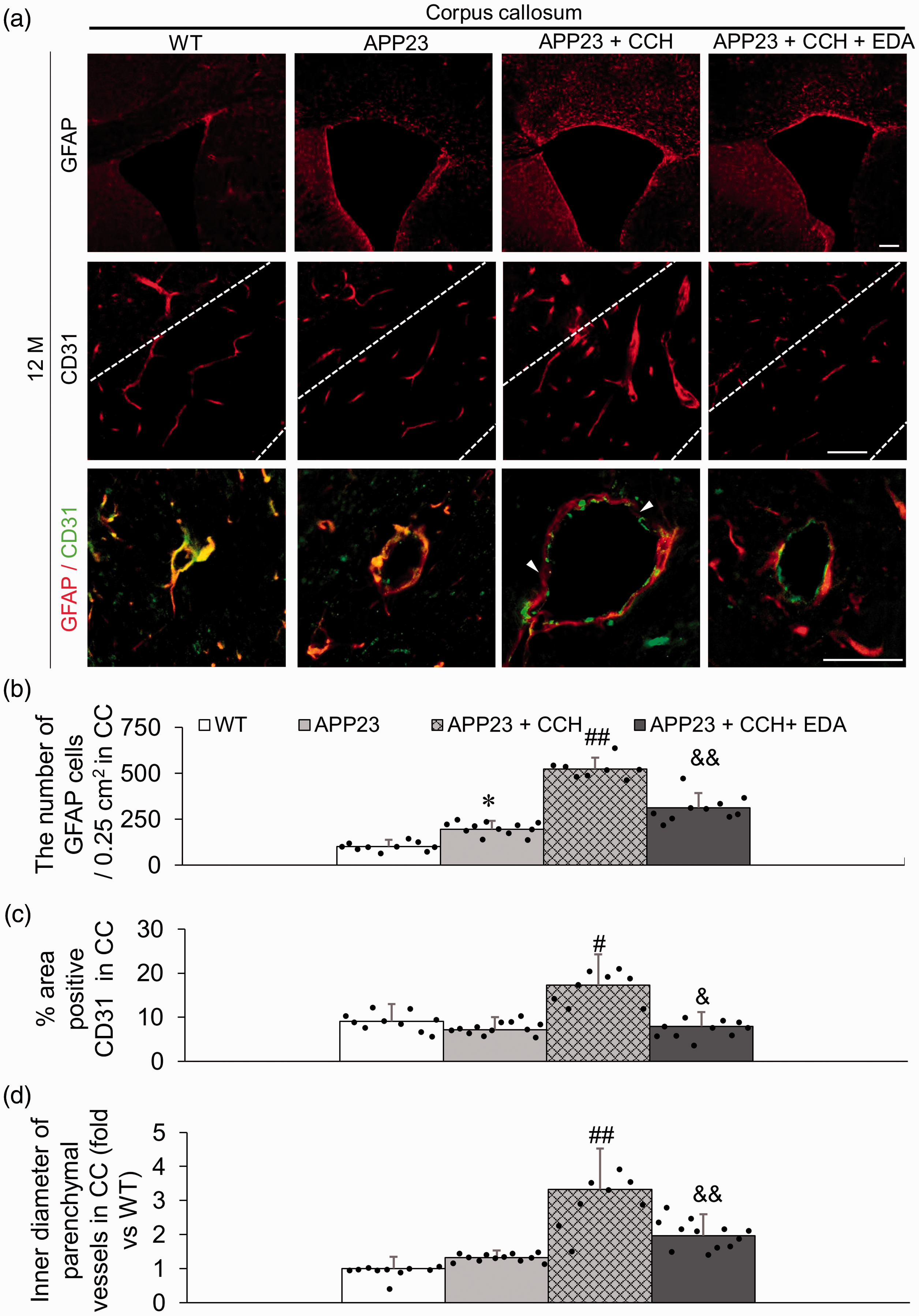
Representative immunofluorescent photomicrographs of astrocyte marker (GFAP, red signals) and endothelium marker (CD31, red signals) in the corpus callosum (CC) (a) and associated quantitative histogram (b–d). Note the significant increase in the number of GFAP-positive astrocytes and the decrease of CD31-positive areas in the CC of the APP23 + CCH + EDA group compared to the CC of mice in the APP23 + CCH group (b and c). Representative double immunofluorescent photomicrographs of GFAP (red signals) and CD31 (green signals) in the CC (a) and associated quantitative histogram (d). Note the significant decrease of inner diameter of parenchymal vessels in the CC of mice in the APP23 + CCH + EDA group compared to the CC of mice in the APP23 + CCH group. Arrowheads represent the obvious damage and dissociation of CD31-positive endothelium/GFAP-positive astrocyte unit (a). Values are means ± S.D. Scale bar = 100 um. (*p < 0.05 vs WT; #p < 0.05 vs APP23, ##p < 0.01 vs APP23; &p < 0.05 vs APP23 + CCH, &&p < 0.01 vs APP23 + CCH).
Edaravone attenuated inflammation and oxidative stress
The effects of EDA on inflammation of CC in APP23 mice with CCH were assessed by immunostaining using an astrocyte marker (GFAP) and a phagocytes marker (Iba-1). A few GFAP- (Figure 3(a)) and Iba-1 (Figure 4(a))-positive cells were observed in the CC of WT mice and APP23 mice at 12 M. In addition, GFAP-positive astrocytes were obvious in the CC of APP23 mice with CCH at 12 M, which was attenuated by the administration of EDA (Figure 3(a)). Compared with the APP23 + CCH group, pixel intensity of Iba-1-positive phagocytes and the number of GFAP-positive astrocytes in the CC were significantly lower after the administration of EDA at 12 M (Figures 3(b) and 4(b), &p < 0.05 vs APP23 + CCH, &&p < 0.01 vs APP23 + CCH). Moreover, to investigate the effect of EDA on oxidative stress of the CC in APP23 mice after CCH, lipid peroxidation marker (4-HNE) and nucleic acid peroxidation marker (8-OHdG) were assessed by immunostaining (Figure 4(a)). That data showed that the pixel intensities of 4-HNE and 8-OHdG in the CC of mice at 12 M in the APP23 + CCH + EDA group were much lower than in the CC of mice in the APP23 + CCH group (Figure 4(b), &p < 0.05 vs APP23 + CCH). Moreover, CCH strongly promoted the expression of GFAP, Iba-1, 4-HNE and 8-OHdG in the CC of APP23 mice at 12 M (Figures 3(a) and (b) and 4(a) and (b), #p < 0.05 vs APP23, ##p < 0.01 vs APP23). No significant differences in pixel intensity of Iba-1, 4-HNE, and 8-OHdG were observed between the WT and APP23 groups (Figure 4(b)). In contrast, a significant difference in the number of GFAP-positive astrocytes was observed between the WT and APP23 groups (Figure 3(b)).
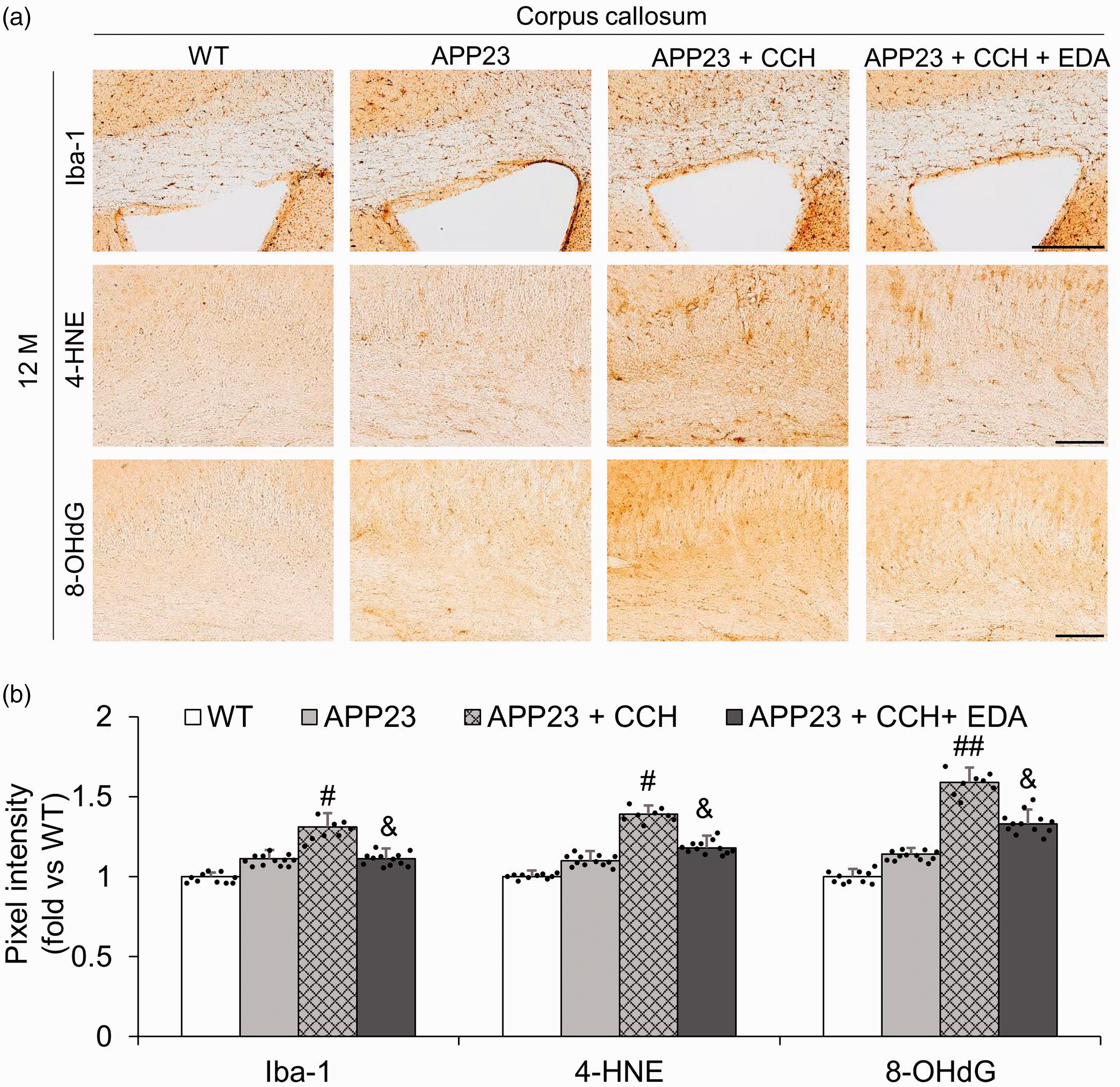
Representative immunohistochemical photomicrographs of phagocyte marker (Iba-1) and oxidative stress markers (4-HNE and 8-OHdG) in the corpus callosum (CC) (a) and associated quantitative histogram (b). Note the significant relative decreases in pixel intensities of Iba-1, 4-HNE, and 8-OHdG in the CC of mice in the APP23 + CCH + EDA group compared to the CC of mice in the APP23 + CCH group (b). Values are means ± S.D. Scale bar = 100 um. (#p < 0.05 vs APP23, ##p < 0.01 vs APP23; &p < 0.05 vs APP23 + CCH).
Discussion
The present study is the first to report the beneficial effects of EDA and related mechanisms against WMLs in an AD mouse model under CCH. The principal findings of the present study were that EDA significantly improved damaged myelin (Figure 1), attenuated the disruption of WM integrity at internodal, paranodal, and Ranvier’s nodal sites (Figure 1), enhanced the number of oligodendrocytes and the proliferation of oligodendrocyte progenitor cells (Figure 2), inhibited damage and remodeling of endothelium/astrocyte units (Figure 3), and attenuated inflammation and oxidative stress in CC of AD + CCH mice at 12 M (Figure 4).
The free radical scavenger EDA has been shown to improve motor and cognitive deficits in the present mouse model of AD with CCH partly by targeting multiple key pathways of the disease’s pathogenesis including Aβ deposition, oxidative stress, Tau hyperphosphorylation, glial activation, neuroinflammation, and neuronal loss in grey matter. 22 , 23 In the present paper, the LDF result showed that the value of CBF in APP23 + CCH + EDA mice was relatively higher than that in APP23 + CCH mice at 12 M indicating that EDA significantly recovered CBF in APP23 + CCH mice at 12 M (Supplemental Figure 1). This could be one of the mechanisms related to the protective effects of EDA on WM pathology in the present mouse model. However, the precise effects of EDA in the WM of the present mouse model remains unknown. As reported, WMLs caused by CCH accelerated the progression of AD and vascular cognitive impairment. The major characteristic of WMLs was the damage and loss of myelin and the disruption of WM integrity in the present AD + CCH mouse model, which was reported in our previous studies. 11 , 12 MBP is a pivotal protein composing the compact myelin sheath and MAG is a key myelin protein involved in the maintenance of axon-glial integrity. First, we observed that EDA therapy significantly improved WM atrophy or loss in the CC of APP23 mice with CCH, indicating the overall impact of EDA therapy on CCH-induced WMLs in APP23 mice (Supplemental Figure 2). Additionally, histochemical staining showed that EDA significantly ameliorated CCH-induced WM degradation not only in the MBP but also in the axon-glial MAG in the CC of APP23 mice at 12 M (Figure 1). LFB staining also demonstrated fewer vacuoles, disorganized myelinated fibers, and a higher density of myelinated fibers in the CC of EDA-administered mice relative to APP23 + CCH mice at 12 M (Figure 1). However, in the present study, the density of total axonal neurofilaments did not significantly decrease in both APP23 and APP23 + CCH mice (Figure 1(b)). Only some axon debris was found as a bulk pattern in APP23 + CCH mice, which was improved by EDA administration to some degree (Figure 1(a)), indicating that the loss of WM intensity could result from severe demyelination, but not as a consequence of the severe loss of axons. The pivotal molecular components of the node and paranode are the NF186 complex and Caspr, respectively. 12 , 28 , 29 Double immunofluorescent analysis showed that the administration of EDA significantly increased the length of NF186 and decreased Caspr gap length in the CC compared with APP23 + CCH mice, suggesting that disruption of the node of Ranvier and the breakdown of paranodal septate-like junctions were greatly ameliorated by the administration of EDA in APP23 + CCH mice at 12 M (Figure 1).
Previous studies reported that oligodendrogenesis spontaneously occurred once myelin was damaged after short and sublethal ischemic injury, in an acute phase of CCH, or in partial human AD patients and mouse models, thereby mediating a compensatory response for self-repairing WMLs.30–34 However, long-term CCH interfered with such endogenous repair mechanisms associated with oligodendrogenesis in damaged WM partly due to excessive deleterious oxidative stress and inflammation. 17 ,35–37 Moreover, although the mechanisms of disruption of oligodendrocyte lineage cells during the course of AD pathology are still unclear, WM inflammation and oxidative stress could be underlying reasons. The present study showed that the administration of EDA significantly increased the number of GST-π-positive OLs and promoted the proliferation of OPCs in the CC of APP23 mice within 8 months of treatment with CCH (Figure 2), contributing to the improvement of WMLs. This may be owed, in part, to the suppressed inflammation and oxidative stress after EDA administration indicated by the lower positive signal densities of Iba-1, 4-HNE, and 8-OHdG in the CC of mice in the APP23 + CCH + EDA group relative to that of mice in the APP23 + CCH group (Figures 3 and 4). On the other hand, although the number of GFAP-positive astrocytes in the APP23 + CCH + EDA group was lower than in the APP23 + CCH group, considering the complexity of subtypes and the function of astrocytes, these values might only indicate decreased inflammation in the CC of APP23 + CCH mice after EDA treatment. Further investigations to better elaborate the relationship between inflammation and astrocytes by using more advanced analytical methods and multiple astrocyte markers of different subtypes should be applied to the present models in our future study based on a previous paper. 38
Additionally, based on a previous report by Miyamoto et al., oligodendrogenesis peaked after 7-day CCH induced by 0.18 mm microcoils and returned to the sham level after 28 days in C57BL/6 mice or in C57BL/6 mice following the administration of EDA (twice per week, 3 mg/kg, i.p.) over a period of 28 days. 17 However, we observed that oligodendrogenesis was relatively enhanced in APP23 mice following EDA therapy, even after 8-month CCH, compared with APP23 + CCH mice. There are two possible reasons for this observation. First, long-term administration of EDA over a period of 8 months might restore oligodendrogenesis, probably by alleviating the pathogenesis of AD, NVU damage, and CBF as reported previously 22 , 23 and based on the present data set (Figure 3 and Supplemental Figure 1). Second, AD pathology might have influenced oligodendrogenesis in the CC of CCH mice in this experiment compared with the single CCH effects on WT C57BL/6 mice.
There are limitations of this experiment. Although we observed that the number of GST-π-positive OLs increased after EDA treatment in APP23 + CCH mice, the origin of these OLs or other OPC-derived cell types remains unknown. Additional studies are needed to understand this issue in detail.
Dysfunction of the NVU is one vascular hypothesis to explain AD, 39 and CCH significantly induced both vascular remodeling and NVU damage in grey matter in the present AD model. 11 , 23 , 26 EDA has been shown to alleviate cognitive deficits partly by improving the vascular Aβ burden, oxidative stress, neuroinflammation, and Tau hyperphosphorylation, not only in single APP/PS1 mice, 19 but also in APP23 mice with CCH (Figures 3 and 4). 22 All the above mechanisms might be related to the recovery of NVU damage and vascular remodeling. 26 ,40–43 Our present data set suggests that EDA reduced the density of CD31-positive vessels, inhibited CD31-positive endothelium/GFAP-positive astrocyte remodeling, improved the damage of CD31-positive vessels, and retarded the dissociation of CD31-positive endothelium/GFAP-positive astrocytes in the CC of APP23 mice with CCH (Figure 3). Therefore, our present data indicated that EDA improved endothelium/astrocyte unit dysfunction in WM of the present AD model with CCH (Figure 3), which might contribute to the improvement of motor and cognitive deficits with clinical and pathological benefits. 22 , 23
Recent imaging studies demonstrated hyperintensities of WM in AD patients and that the levels of structural abnormalities in WM are related to the progression of AD.44–51 In addition, CCH has been shown to contribute to the development of WMLs and cognitive impairment in AD patients.52–54 Although the mechanisms of WMLs in single AD or AD with CCH are very complicated, the damage to myelin, disruption of WM integrity, changes of oligodendrocyte lineage cells, damage and remodeling of NVUs, WM inflammation, and WM oxidative stress have been shown to be involved in the pathophysiological progression of WMLs in AD patients or in AD animal models.11,12,34,55–60 In the present study, EDA shed light on the protective effects on multiple key pathological alterations in the WM of a novel mouse model of AD with CCH. This may provide experimental proof for a large-scale clinical trial to examine its efficacy and safety in familial AD or AD with CCH.
However, on the other hand, we emphasize that the present study is only an experimental study that has used a small rodent model that contains a smaller cerebral WM content than large animal species or humans. To the best of our knowledge, basic anatomical and physiological differences between species can give rise to the different outcomes of cell therapy or traditional medication therapy for AD or cerebrovascular diseases. 61 , 62 Therefore, the benefits of EDA observed in the present mouse model need to be tested in larger animal species for a thorough understanding prior to clinical trials since large animal species appear to have a closer anatomy to humans than rodents.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X20968927 - Supplemental material for Protective effects of edaravone on white matter pathology in a novel mouse model of Alzheimer’s disease with chronic cerebral hypoperfusion
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X20968927 for Protective effects of edaravone on white matter pathology in a novel mouse model of Alzheimer’s disease with chronic cerebral hypoperfusion by Tian Feng, Toru Yamashita, Ryo Sasaki, Koh Tadokoro, Namiko Matsumoto, Nozomi Hishikawa and Koji Abe in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was partly supported by a Grant-in-Aid for Scientific Research (B) 25293202, (C) 15K09316 and Challenging Research 15K15527 and Young Research 15K21181, and by Grants-in-Aid from the Research Committees (Mizusawa H, Nakashima K, Nishizawa M, Sasaki H, and Aoki M) from the Ministry of Health, Labour and Welfare of Japan.
Acknowledgements
We specially thank Yan-Jiang Wang (Third Military Medical University, China) for excellent technical assistance with respect to the administration of edaravone. We thank Xin-Fu Zhou (University of South Australia, Australia) for excellent theoretical guidance in designing the present manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
KA, TF, and TY designed the research; TF, RS, KT, and NM performed the experiments; TF, KT, NM, and NH analyzed the data; TF and KA wrote the manuscript.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.