
Abstract
White matter lesions are closely associated with cognitive impairment and motor dysfunction in the aged. To explore the pathophysiology of these lesions, the authors examined the expression of matrix metalloproteinase-2 (MMP-2) and MMP-9 in the white matter in a rat model of chronic cerebral hypoperfusion. After bilateral clipping of the common carotid arteries, myelin staining revealed demyelinating changes in the optic tract and the corpus callosum on day 7. Zymographic analyses indicated an increase in the level of MMP-2, but not MMP-9, after the hypoperfusion. Immunohistochemical analyses revealed the presence (most abundantly on day 3) of MMP-2–expressing activated microglia in the optic tract and corpus callosum. In contrast, the capillary endothelial cells expressed MMP-2 later. IgM-immunoreactive glial cells were absent in the sham-operated animals, but were present in the hypoperfused animals by day 3, reflecting the disrupted blood–brain barrier. These findings suggest that the main sources of the elevated MMP-2 were the microglia and the endothelium, and that these cells may contribute to the remodeling of the white matter myelin and microvascular beds in chronic cerebral hypoperfusion.
Keywords
Increasing attention has been focused on the white matter lesions in cerebrovascular diseases, because such abnormalities have become readily detectable by magnetic resonance imaging (Pantoni and Garcia, 1997a). These lesions constitute the core pathology in several dementing disorders, such as Binswanger's disease, a form of subcortical vascular dementia, and cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. A positive correlation between white matter lesions and cognitive dysfunction (Pantoni and Garcia, 1997b) has been demonstrated, although it remains controversial (Sabri et al., 1999; Hurley et al., 2000). For example, recent evidence indicates that parameters such as white matter volume (Smith et al., 2000) and lesion distribution (de Groot et al., 2000) may determine the development of cognitive dysfunction. Therefore, the elucidation of the pathomechanisms underlying these white matter lesions is important for designing the prevention and treatment of vascular dementia.
White matter lesions can be induced in rats by clipping the bilateral carotid arteries (Wakita et al., 1994). These animals exhibit long-standing cerebral hypoperfusion and behavioral disturbances, hence, they can serve as a good experimental model for studying vascular dementia (Tanaka et al., 1996), although heterogeneity in vascular dementia must be taken into account. A previous study on this system and human material implicated a dysfunction of the blood–brain barrier (BBB), perivascular edema and microglial activation as the mechanisms underlying the white matter lesions (Wakita et al., 1994). During this process, microglia may play a pivotal role because their activation and the white matter lesions occur concurrently, and both are suppressed by the administration of immunosuppressants such as cyclosporin A or FK 506 (Wakita et al., 1995, 1998).
Recent reports have suggested that macrophage/microglial cells may play a role in the pathogenesis of inflammatory demyelination, such as that observed in multiple sclerosis and Guillain-Barré syndrome, by secreting matrix metalloproteinases (MMPs) (Cuzner et al., 1996; Kieseier et al., 1999). Matrix metalloproteinases influence cell–matrix interactions in the human brain (Romanic and Madri, 1994a) and may also be involved in the disintegration of the basement membranes of microvessels during cerebral ischemia (Hamann et al., 1995). In the current study, the authors investigated the relation between white matter lesions and the expression of MMP-2 and MMP-9 in chronic cerebral hypoperfusion to investigate the role of these proteases in the degradation of BBB and myelin.
MATERIALS AND METHODS
Animals
Chronic cerebral hypoperfusion was induced in male Wistar rats (weighing 150 to 200 g; Shimizu Experimental Supply, Kyoto, Japan) as previously described (Wakita et al, 1994). Briefly, animals were anesthetized with sodium pentobarbital (25 mg/kg, intraperitoneal) and were allowed spontaneous respiration throughout the surgical procedure. Through a midline cervical incision, the common carotid arteries were exposed bilaterally and were double-ligated with silk sutures. After the operation, rats were kept in animal quarters with food and water ad libitum. At 1, 3, 7, 14, and 30 days after the ligation, animals were subjected to the experiments detailed below. The sham-operated animals were treated similarly to the operated ones, except that the common carotid arteries were not occluded.
RNA blot analysis
The rats were deeply anesthetized with sodium pentobarbital (50 mg/kg, intraperitoneal) and were perfused transcardially with 0.01 mol/L phosphate-buffered saline (PBS). Three animals each were examined at four different time points after carotid occlusion and for the sham operation. Total RNA was extracted using Isogen (Nippon Gene, Tokyo, Japan). Poly (A) RNA (10 μg) was purified by oligo-dT-cellulose chromatography, resolved on 1.2% agarose/2.2 mol/L formaldehyde gels, and then transferred to a nylon membrane. The membrane was hybridized with 32P-labeled MMP-2 or MMP-9 probes at 65°C in RapidHyb (Amersham, Arlington Heights, IL, U.S.A.) for 3 hours. Each probe was prepared by the random priming method using a polymerase chain reaction product derived from the human MMP-2 cDNA (primers: 5′-GCCTACACTGGGACCTGTCACT and 5′-ACGACTGCATCCAGGTTATCAG) and the human MMP-9 cDNA fragment (BamHI-HindIII, 1.3 kb) as templates. The filter then was washed at 60°C in standard sodium citrate containing 0.1% sodium dodecyl sulfate. After exposure on Imaging Plate, densitometry was performed with a BAS2000 Image Analyzer (Fuji Photo Film, Tokyo, Japan). The same membranes were reprobed with rat GAPDH cDNA to assess the quality and quantity of each RNA sample.
Immunohistochemistry
Under deep anesthesia, rats were perfused with 0.01 mol/L PBS and then with a fixative containing 4% paraformaldehyde and 0.2% picric acid in 0.1 mol/L phosphate buffer (PB, pH 7.4). The brains were removed and postfixed for 24 hours in 4% paraformaldehyde in 0.1 mol/L PB (pH 7.4), and then stored in 15% sucrose in 0.1 mol/L PB (pH 7.4). Five animals were examined for each time point after carotid occlusion and for the sham operation.
Serial sections (20-μm-thick) were cut on a cryostat and incubated overnight with a mouse anti–MMP-2 antibody (diluted 1:10,000; Fuji Chemical Industries, Toyama, Japan), a goat anti–MMP-9 antibody (diluted 1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.), a biotinylated goat anti-rat IgM (μ) antibody (diluted 1:1000; Kirkegaad & Perry Laboratories, Gaithersburg, MD, U.S.A.), and biotinylated Ricinus communis agglutinin-1 (RCA-1; diluted 1:1000; Vector Laboratories, Burlingame, CA, U.S.A.). According to the manufacturer's data sheet, the anti–MMP-2 antibody recognizes both the latent and active form of MMP-2 in their common COOH-terminal region. Sections incubated with the anti-MMP-2 or anti-MMP-9 antibodies then were treated with appropriate biotinylated secondary antibodies (diluted 1:200; Vector Laboratories). All these sections were subsequently treated with an avidin-biotin complex (diluted 1:200; Vector Laboratories), and were visualized with 0.01% diaminobenzidine tetrahydrochloride and 0.005% H2O2 in 50 mmol/L Tris HCl (pH 7.6). MMP-9 immunoreactivity also was examined in the brain sections of a rat suture model of middle cerebral artery occlusion (MCAO; n = 4), which had been prepared for another experiment.
To test the specificity of the immunoreaction, control sections were incubated with normal mouse IgG instead of the primary antibody. The number of nuclei with MMP-2–immunoreactive cytoplasm was counted in 10 representative fields (per 0.25 mm2) of the optic tract and corpus callosum immunostained with anti–MMP-2 antibody (n = 5). To confirm the cellular source, sections were labeled by biotinylated RCA-1, a marker for microglia, followed by fluorescein isothiocyanate-labeled avidin (diluted 1:40; Dako, Carpinteria, CA, U.S.A.). Alternatively, sections were labeled by rabbit anti-glial fibrillary acidic protein antibody (1:2000; Dako), a marker for astroglia, and subsequently rhodamine-labeled anti-rabbit antibody (1:40; Dako). After taking a photograph, the same sections were treated with anti-MMP-2 antibody, and finally visualized with diaminobenzidine tetrahydrochloride.
Zymography
Minced forebrain tissues were incubated with gentle rotation at 4°C for 20 hours in an extraction buffer consisting of 0.5% Triton-X 100, 0.5 U/mL aprotinin, and 0.01% sodium azide in 0.01 mol/L PBS. Samples then were centrifuged at 14,000 g for 15 minutes at 4°C, and the supernatants were collected. The protein content was adjusted to 10 mg/mL, and the samples were stored at −20°C until use. For each time point after carotid occlusion and for the sham operation, four animals were studied.
The gelatinolytic activity of these samples was detected by sodium dodecyl sulfate-polyacrylamide gel electrophoresis zymography (Kleiner and Stetler-Stevenson, 1994). Briefly, polyacrylamide gels (10%) were copolymerized with gelatin (1 mg/mL). Equal amounts of tissue extract (50 μg) then were subjected to electrophoresis. Sample gels were agitated in 10 mmol/L Tris-HCl (pH 8.0) containing 2.5% Triton X-100 (30 minutes × 2) to restore the activity of the proteins. The gels were rinsed in 50 mmol/L Tris-HCl (pH 8.0) for 30 minutes, and then incubated overnight twice at 37°C in 50 mL of 50 mmol/L Tris-HCl (pH 8.0) containing 0.5 mmol/L CaCl2 and 1.0 μmol/L ZnCl2. After incubation, the gels were stained with Coomassie blue R-250 and then destained with 10% methanol containing 7% acetic acid.
These gels were optically scanned by a flatbed scanner (8 bit, 600 dpi). The image was digitally inverted by Adobe Photoshop (Adobe Systems, Mountain View, CA, U.S.A.) so that an integration of the bands would be reported as a positive value. Using NIH Image 1.62b7, the integrated density of each band was calculated by summing the pixel values within a selected area and subtracting the background density. The digestion areas then were quantified as volume units of pixel intensity × mm2. Results for each time point were presented as a fold-value versus the sham-operated animals.
Statistical analysis
Differences between groups were determined by a one-factor analysis of variance followed by Fisher's protected least significant difference procedure between each group. The bars in the figures represent standard deviation. P < 0.05 was considered to be statistically significant.
RESULTS
The authors examined the expression of MMP-2 and MMP-9 in a rat model of chronic cerebral hypoperfusion induced by bilateral clipping of the common carotid arteries. The operation was successful in all animals (n = 56) except 1, which fell in a moribund condition within 24 hours after the operation. This animal was excluded from the statistical analysis, but its brain with a massive infarct was analyzed further by zymography to examine the effects of a severe ischemic insult to the brain.
Klüver–Barrera staining revealed demyelinating changes in the optic tract and the corpus callosum on day 7. These changes consisted of vacuoles, tortuosity of the myelinated fibers, and a numeric decrease of nuclei of oligodendroglia, most of which were aligned in interfascicular distribution and had the morphology of oligodendroglia (Fig. 1A and 1B). Immunohistochemical analyses showed that MMP-2–positive glial cells appeared in white matter regions such as the corpus callosum and optic tract, but not in the gray matter. The number of MMP-2–immunoreactive cells was largest on day 3 after the hypoperfusion (Fig. 1C to 1F). In contrast, the endothelial cells of the microvessels were more heavily immunostained with anti–MMP-2 on day 7 in both the white and gray matter (Fig. 1G). IgM-immunoreactive glia, which were representative of those cells taking up extravasated serum proteins because of the disrupted BBB, were absent in the sham-operated animals (Fig. 1H), but emerged after chronic cerebral hypoperfusion for 3 days (Fig. 1I). The number of IgM-immunoreactive glia decreased after day 7 (data not shown). In the double-labeling experiment, approximately 90% of MMP-2–immunoreactive cells were identified as microglia based on their RCA-1 immunoreactivity (Fig. 2), the rest were astroglia with glial fibrillary acidic protein immunoreactivity. RCA-1 immunoreactive cells had enlarged cell bodies with thick processes, which were characteristic of activated microglia. A quantitative study revealed that the number of MMP-2–immunoreactive glia began to increase on day 1, peaked on day 3, and decreased by day 7 in both the optic tract and the corpus callosum of the hypoperfused animals (Fig. 3).
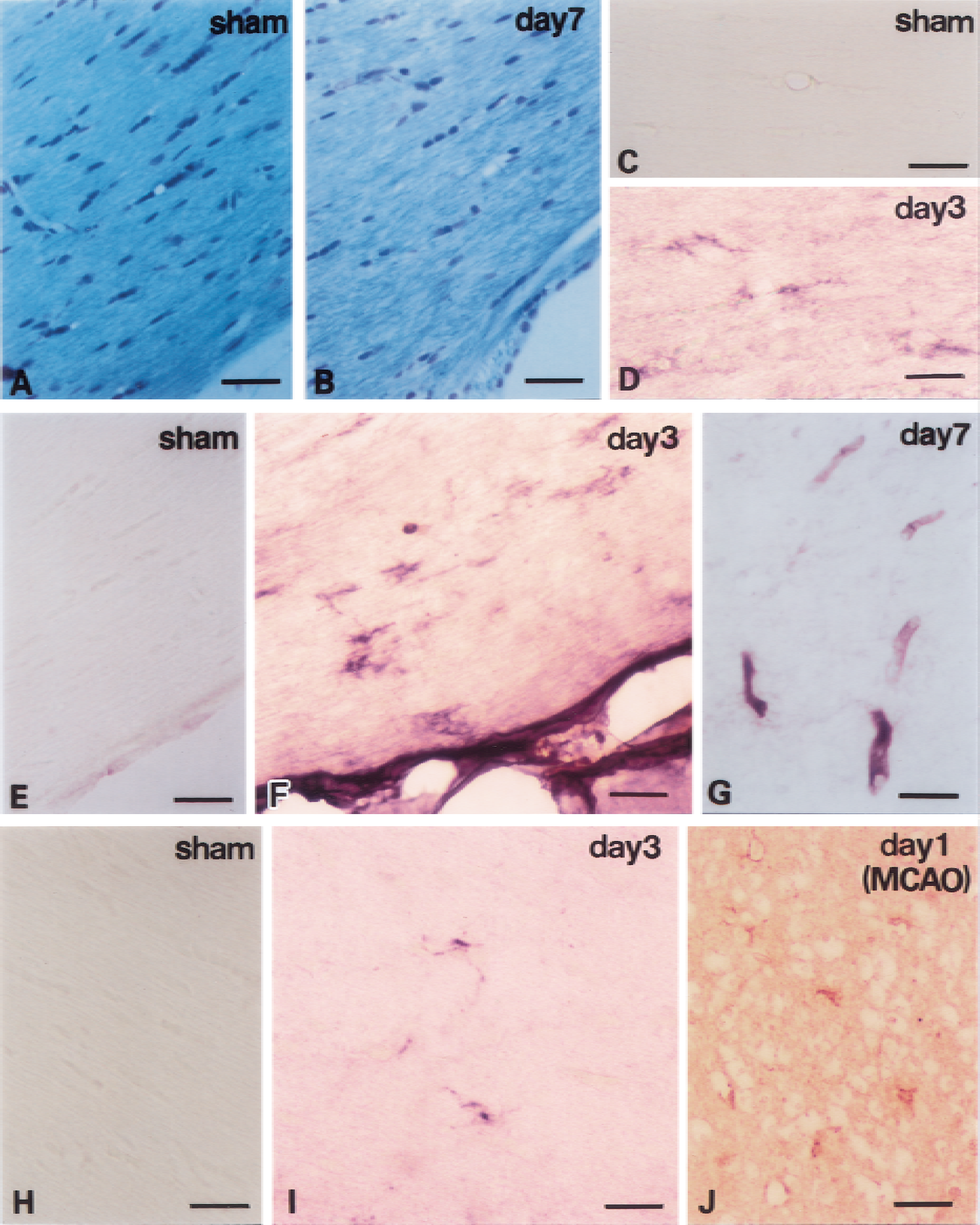
Histologic changes induced in the corpus callosum
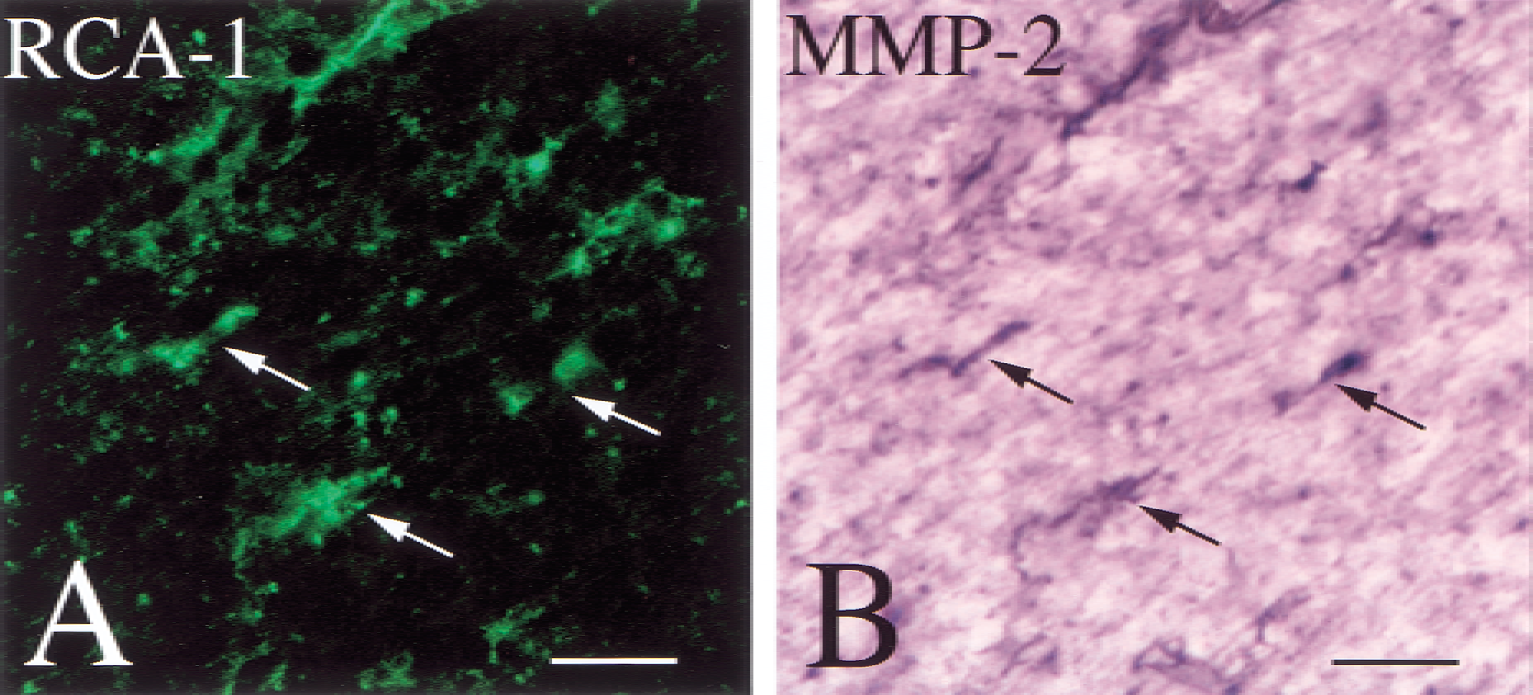
Histologic changes induced in the optic tract after chronic cerebral hypoperfusion for 3 days. Sections were subjected to double labeling for RCA-1
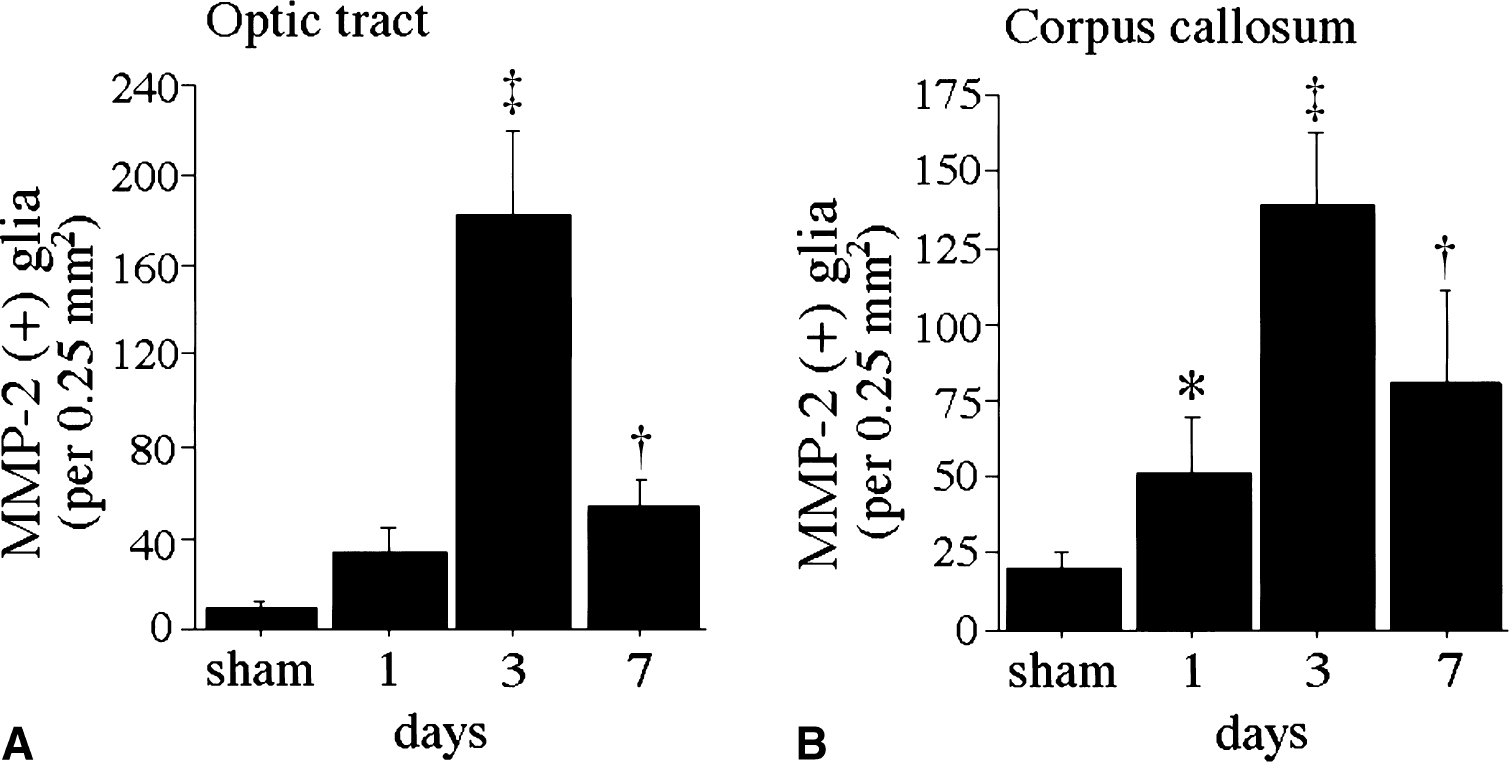
Histograms of the numerical density of MMP-2 immunoreactive glia in the optic tract
Northern blot analysis showed that the level of MMP-2 mRNA was up-regulated by 1.8-fold on day 3 (Fig. 4A and 4B) and remained greater than the basal level for up to 30 days. On zymography, a 72-kDa band of pro–MMP-2 was observed in the sham-operated animals (Fig. 5A). Seven days after the chronic cerebral hypoperfusion, the level of pro–MMP-2 had increased significantly as compared with the controls (Fig. 5B;P < 0.001).
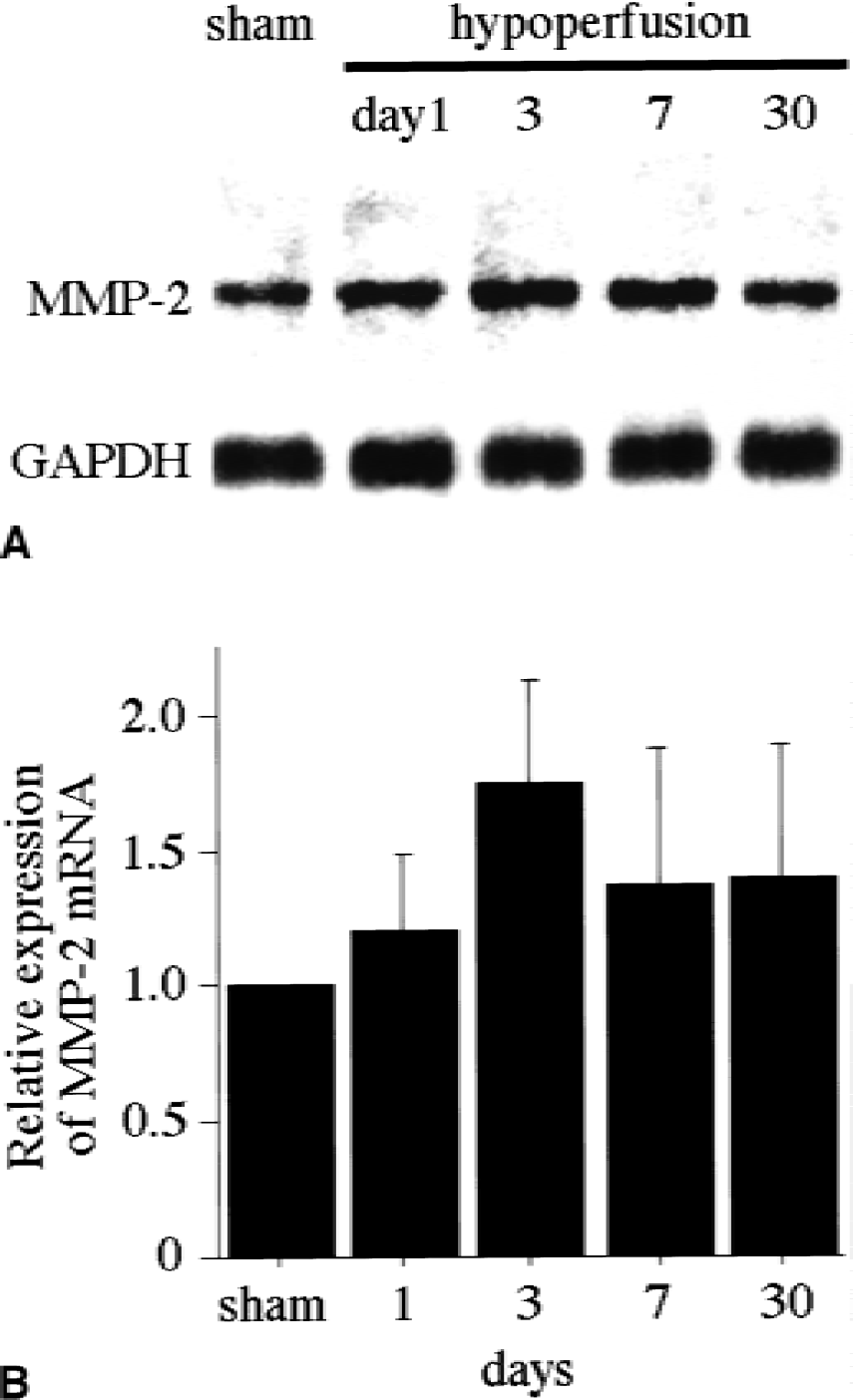
Effects of chronic cerebral hypoperfusion on the levels of MMP-2 mRNA.
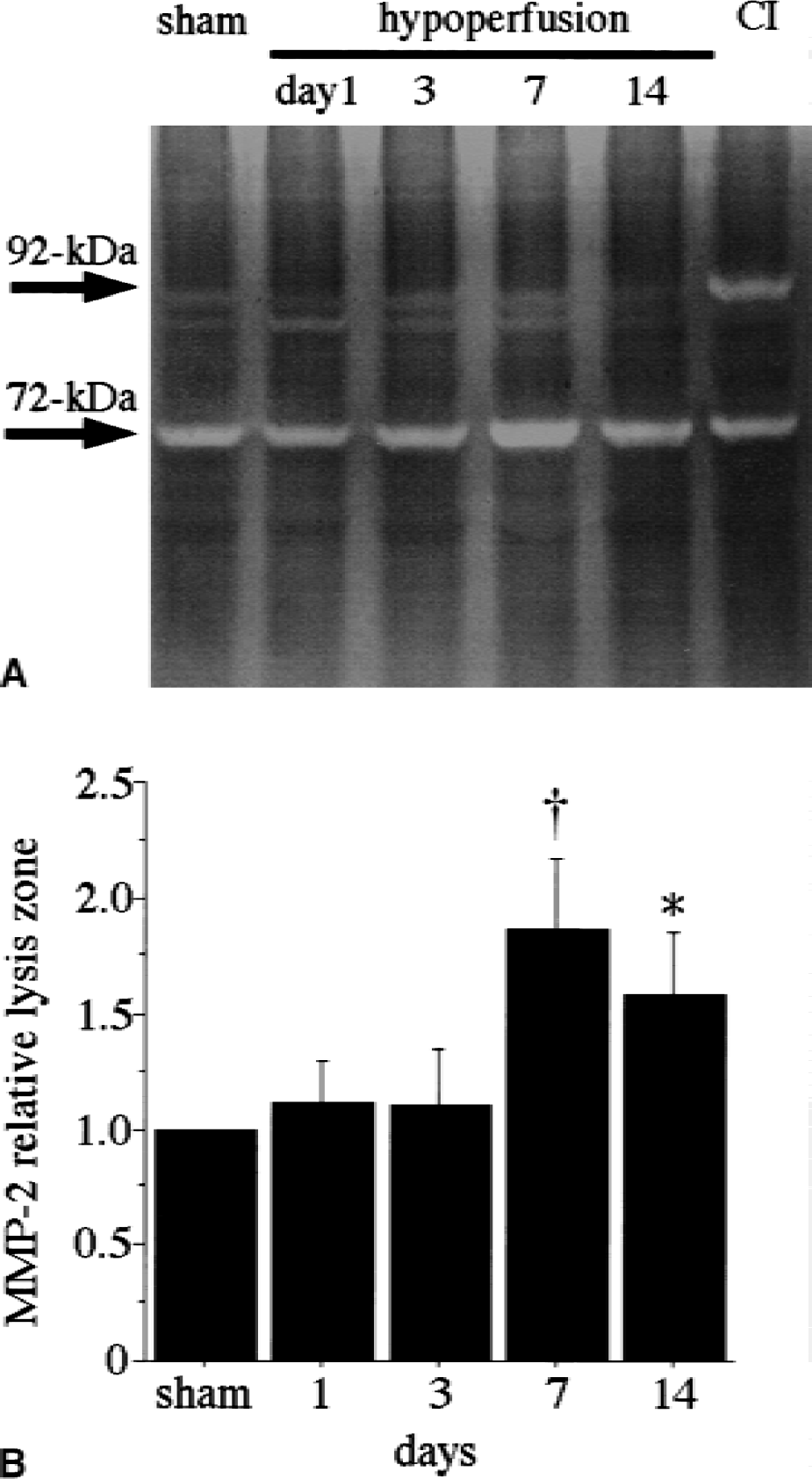
Effects of chronic cerebral hypoperfusion on the levels of MMP-2 and MMP-9.
In contrast, MMP-9 (92 kDa) was not induced either at the mRNA or the protein level in this model; immunohistochemical staining with anti–MMP-9 failed to detect any brain substructures in spite of the fact that the same antibody shows immunopositive glia in a rat MCAO model (Fig. 1J). Northern blot analysis with MMP-9 probes also failed to show any positive bands (data not shown). Zymography revealed only a faint band in the samples after hypoperfusion, in contrast with the robust band in the sample with massive cerebral infarction (Fig. 5A).
DISCUSSION
In this study, the authors demonstrated that activated microglia in the white matter produced MMP-2 maximally on day 3 of chronic cerebral hypoperfusion (Fig. 3), preceding the rarefaction of the white matter. Because extravasated IgM appeared during the same period, the MMP-2 derived from the microglia may contribute to the BBB breakdown through its activity as a type IV collagenase (Rosenberg et al., 1992). The resulting perivascular edema may further exacerbate the degradation of the white matter myelin through the actions of extravasated serum factors (Akiguchi et al., 1998). The BBB breakdown also leads to leukocyte diapedesis (Romanic and Mandri, 1994b), and the infiltrating leukocytes may cause inflammatory demyelination. MMP-2 itself has strong activity in degrading myelin basic protein, approximately 100× more potent than MMP-9 (Chandler et al., 1995). Thus, the MMP-2 released from microglia may be directly involved in the remodeling of the white matter myelin.
The up-regulation of macrophage/microglial MMPs in white matter lesions is not exclusive to chronic cerebral hypoperfusion. Infiltrating macrophage/microglia express multiple MMP genes also in tumor necrosis factor-α transgenic mice and in mice with experimental allergic encephalomyelitis (Pagenstecher et al., 1998). Interestingly, the MMPs are differentially induced in each model—MMP-2 and MMP-9 in tumor necrosis factor-α transgenic mice, MMP-9 in experimental allergic encephalomyelitis, and MMP-2 in chronic cerebral hypoperfusion. Thus, the elevated macrophage/microglial MMP levels commonly observed within white matter lesions imply a shared pathophysiology in these models of apparently different etiology. The cytokine network, including tumor necrosis factor-α, may underlie the localized expression of microglial MMPs in the white matter.
The MMP-2 detected in the zymographic analysis represents the total expression of this enzyme in the whole brain, its major sources being the microglia and endothelial cells. The immunohistochemical data suggest that the endothelium was the major source of MMP-2 expression, which peaked on day 7 of the chronic hypoperfusion. The peak expression of MMP-2 on day 7 coincided with the recovery of blood flow in this model (Tsuchiya et al., 1992; Tomimoto et al., 1997). Endothelium-derived MMP-2 was implicated in the degradation of the vascular basement membrane and the extracellular matrix, which leads to angiogenesis by facilitating endothelial migration, proliferation, and capillary tube formation (Moses, 1997). Therefore, the endothelium-derived MMP-2 may function to restore microvascular flow. In spontaneously hypertensive rats, MMP-2 is up-regulated 5 days after MCAO (Mun-Bryce and Rosenberg, 1998; Rosenberg et al., 1998). In the human brain, elevated MMP-2 levels persist in the ischemic lesion up to several years after a stroke (Clark et al., 1997; Anthony et al., 1997). These findings suggest that the MMP-2-associated microvascular remodeling in the brain may be long lasting after a stroke.
A single band of 72 kDa that corresponds to pro–MMP-2 was observed by zymography. The failure to detect active MMP-2 may be attributable to the detection limit of zymography (Kleiner and Stetler-Stevenson, 1994), because homogenized tissues contain relatively low amounts of active MMP-2. Pro–MMP-2 is known to be activated by thrombin (Nguyen et al., 1999), plasminogen activators (Cuzner and Opdenakker, 1999), and membrane type 1-MMP (Nguyen et al., 2000), and is inhibited by tissue inhibitor of metalloproteinase-1 and −2 (TIMP-1, 2) (Romanic and Madri, 1994a). Collagen type I also is known to facilitate the activation of pro–MMP-2 by inducing MT1-MMP in the endothelium (Haas et al., 1998); collagen has been shown to accumulate in the basement membrane of the microvessels after cerebral infarction (Garcia et al., 1981; Zhang et al., 1994). Little is known, however, about the behavior of these modulators during or after cerebral ischemia or chronic cerebral hypoperfusion. Thus, the determination of the upstream mechanisms controlling MMP-2 activity under these pathologic conditions is an important subject for future studies.
The pattern of expression of MMP-9 is different depending on the severity of ischemic insult. In a rat model of MCAO, MMP-9 has been reported to be up-regulated during the early phase of ischemia (Mun-Bryce and Rosenberg, 1998; Rosenberg et al., 1998). MMP-9 may be detrimental to ischemic brain tissues, because the infarct size can be reduced by an MMP-9 inhibitor (Romanic et al., 1998). In contrast, MMP-9 expression was negligible as determined by its mRNA and protein levels in the present chronic hypoperfusion model. Hence, a different pathophysiology between these two models of cerebral ischemia may be reflected by the distinct patterns of MMP-9 expression.
In summary, the current results indicate that chronic ischemic stress in the brain induces the expression of MMP-2 at the protein levels. The main cellular sources of MMP-2 in the brain were the microglia and vascular endothelium, which were likely to play specific and cooperative roles in remodeling the white matter myelin and microvascular beds. Consistent with this, MMP-2 was reported to have a close relation with demyelination in multiple sclerosis (Maeda and Sobel, 1996) and BBB opening in cerebral hemorrhage (Rosenberg et al., 1992). Although the observed induction of MMP-2 may reflect a compensatory reaction, the excessive production of MMP-2 may also lead to white matter damage, BBB disruption, and microvascular damage.
Footnotes
Acknowledgments:
The authors are grateful to Ms. H. Nakabayashi, M. Fukuda, K. Imai, and A. Miyazaki for their technical and/or secretarial assistance.