
Abstract
Incidences of repetitive mild TBI (r-mTBI), like those sustained by contact sports athletes and military personnel, are thought to be a risk factor for development of neurodegenerative disorders. Those suffering from chronic TBI-related illness demonstrate deficits in cerebrovascular reactivity (CVR), the ability of the cerebral vasculature to respond to a vasoactive stimulus. CVR is thus an important measure of traumatic cerebral vascular injury (TCVI), and a possible in vivo endophenotype of TBI-related neuropathogenesis. We combined laser speckle imaging of CVR in response to hypercapnic challenge with neurobehavioral assessment of learning and memory, to investigate if decreased cerebrovascular responsiveness underlies impaired cognitive function in our mouse model of chronic r-mTBI. We demonstrate a profile of blunted hypercapnia-evoked CVR in the cortices of r-mTBI mice like that of human TBI, alongside sustained memory and learning impairment, without biochemical or immunohistopathological signs of cerebral vessel laminar or endothelium constituent loss. Transient decreased expression of alpha smooth muscle actin and platelet-derived growth factor receptor β, indicative of TCVI, is obvious only at the time of the most pronounced CVR deficit. These findings implicate CVR as a valid preclinical measure of TCVI, perhaps useful for developing therapies targeting TCVI after recurrent mild head trauma.
Keywords
Introduction
There is evolving concern over repetitive mild traumatic brain injuries (r-mTBI) acquired in contact sports and military duty, which have been associated with short- and long-term functional and neuropsychological impairments,1–5 and constitute the most frequent service-related injuries seen in veterans and military personnel. 6 Traumatic cerebral vascular injury (TCVI) of varying degree is seen in almost all reported cases of severe TBI post-mortem, 7 and in the brains of patients dying in the acute phase of hours to days following a mTBI. 8 The early appearance of aberrant cerebral blood flow (CBF) in American football players9,10 and the finding that global and regional CBF is decreased in retired military personnel having sustained an injury, 11 implicate TCVI as an early occurring and enduring pathology in r-mTBI. However, resting CBF is not by itself a reliable indicator of cerebrovascular health, as CBF is tightly coupled to the metabolic demands of neural tissue and, therefore, does not discriminate between a primary vascular injury and secondary drop in CBF due to decreased neuronal metabolic demand. Veterans with a history of r-mTBI show a decreased metabolic rate of glucose in the cerebellum, vermis, pons and medial temporal lobe, compared to controls, 12 and plasma levels of brain-derived creatine kinase B are decreased in professional boxers, 13 indicating central nervous system (CNS) metabolic dysregulation following r-mTBI, which will alter resting state CBF even when the vasculature is intact.
Alternatively, cerebrovascular reactivity (CVR), which measures changes in cerebral blood vessel dilation in response to a vasoactive stimulus, and is a measure of cerebral vascular health and reserve, 14 is a more direct measure of TCVI. 7 The vasoactive stimulus most typically employed in research settings is hypercapnia – an increase in partial pressure of CO2 (paCO2), produced by voluntary breath holding, or, more reproducibly, by briefly increasing the fractional inspired concentration of CO2 (FiCO2) from atmospheric levels (approximately 0.04%) to 5–10% CO2 via a facemask. 7 CVR can be measured non-invasively, by a variety of modalities, including arterial spin labelling (ASL) and blood oxygenation level dependent (BOLD) MRI. Len et al. 15 have recently demonstrated an impairment of CVR during breath hold, in the absence of impairment of resting medial cerebral artery blood velocity (MCAv) 15 in American football athletes. Similarly, Mutch et al. 16 have reported alterations in MRI BOLD CVR in response to hypercapnic challenge, in the absence of global resting state CBF or cerebrovascular anatomical differences, in 15 adolescents diagnosed with post concussive symptoms, compared to 17 healthy controls. 16 In addition to the above studies describing impaired CVR at chronic and acute times post mild TBI,15,16 perturbed CVR has also been seen at a median of 25 months post-injury using hypercapnic challenge and BOLD/ASL MRI in human patients following a moderate to severe TBI, with poor discrimination of patient from control via the CBF metric alone. 17 Given the apparent conformity of CVR impairment across human mTBI, we studied the degree of TCVI using parallel hypercapnia-evoked CVR, and neurobehavioral profiles at two chronic time-points post-injury in our previously characterized animal model of chronic repetitive head trauma.18,19 This injury paradigm, consisting of one hit administered twice weekly, approximately 72 h apart, for 12 weeks, has been shown to induce resting CBF impairment, neuroinflammatory insult of the white matter and memory associated behavioral deficit in both hTau and wild type adult mice, at three months 19 and seven months 18 post last hit, respectively. In these studies, we report a chronic and enduring decrease in CVR, and biochemical evidence of underlying cerebrovascular damage alongside pronounced and persistent neurobehavioral impairment.
Materials and methods
Animals
Male C57BL/6 and transgenic EFAD mice were housed under standard laboratory conditions (23 ± 1°C, 50 ± 5% humidity, and 12-h light/dark cycle) with free access to food and water throughout the study. In addition to the head-injured animals, we also incorporated age-matched nine-month-old male transgenic Apolipoprotein E3 Familial Alzheimer’s Disease (EFAD) mice for use as a positive control for cerebrovascular reactivity deficit in response to hypercapnic challenge. The EFAD colony was established by crossing mice expressing five familial AD mutations (5xFAD) to homozygous apoE targeted replacement mice (apoE-TR) expressing the human apoE3 isoform. 20 The apoE-TR mice were created by gene targeting and carry one of the three human alleles (APOE2, APOE3, or APOE4) in place of the endogenous murine apoE gene. 21 These mice retain the endogenous regulatory sequences required for apoE production and express the human apoE protein at physiological levels. All mice are on a C57BL/6 background and additional details on the production and genetic background of these mice are described in the sources cited above. All experimental protocols involving animals were approved by the Institutional Animal Care and Use Committee of the Roskamp Institute. All procedures were carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals.
Injury schedule
Three-month-old male mice were randomly assigned to one of two groups: chronic repetitive mild traumatic brain injury (r-mTBI, delivered twice each calendar week i.e. 1 hit approximately every 72 h, over a period of 3 months) or repetitive Sham (r-sham; animals underwent the same duration and frequency of anesthesia as r-mTBI animals). An electromagnetic impactor (Leica Instruments) was used to generate a midline mTBI, using a 5.0 mm diameter flat face tip, 5 m/s strike velocity, 1.0 mm strike depth, and a 200 ms dwell time, as previously characterized. 22 The three-month post last injury (r-sham, n = 7; r-mTBI, n = 7) and nine-month post last injury (r-sham, n = 14; r-mTBI, n = 14) cohorts studied here were separate groups of animals, given an identical regime and intensity of r-mTBI/sham. The mice were euthanatized at either three months or nine months after the final injury/anesthesia (9 and 15 months of age, respectively), with euthanasia preceded by both behavioral testing and evaluation of cerebrovascular reactivity in the nine-month post last injury cohort, and cerebrovascular reactivity alone in the three-month post-injury cohort.
Assessment of cognitive function
In the mice assigned to euthanasia at nine months post-last injury, cognitive function was assessed at three and six months after the final injury/anesthesia (9 months of age and 12 months of age, respectively) by use of the Barnes maze (Figure 1), as described previously by our group. 22 Researchers conducting the experiments were blind to the grouping, and the Ethovision XT System (Noldus) was used to track and record the movement of each animal. Mice were given 90 s to locate and enter the target box and required to remain in the target box 30 s prior to retrieval, regardless of success. For a series of six consecutive days, four trials were given per day, with mice starting from one of four cardinal points on each trial. The inter-trial interval for each mouse on any given day of acquisition was approximately 40 min. The maze platform and retrieval box were both cleaned thoroughly between trials to limit the confounding effects of scent on performance of the mice during each trial. On the seventh day, a single probe trial lasting 60 s was performed with the mouse starting from the center of the maze and the target box removed. Escape latency measured the time taken for the mouse to enter the box. For behavioral assessment at both three months and six months post-injury, 14 r-sham and 14 r-mTBI animals were assessed. All animals were euthanized at 9 months post-injury (3 months following the second behavioral assessment, and 15 months of age), immediately following assessment of cerebrovascular reactivity.
In vivo assessment of cerebrovascular reactivity
Nine- or 15-month old male C57BL6 mice were evaluated for CVR in vivo, at three months (r-sham, n = 7; r-sham, n = 7) or nine months (r-sham, n = 12; r-mTBI, n = 12) post-injury, respectively, using Laser Speckle Contrast Imaging (FLPI2, Moor Instruments; see Figure 2). As a positive, age-matched control for the in vivo experiments, we used nine-month-old male transgenic Apolipoprotein E3 Familial Alzheimer’s Disease mice (EFAD, n = 3), as these mice not only recapitulate some of the neuro-vascular pathology characteristic of AD, 20 in which CVR has been reported to be compromised,14,23–26 but also as the FAD mouse has been shown to exhibit a pronounced deficit in CVR to hypercapnic challenge in vivo.27–30 24 h prior to hypercapnic challenge, a large area of the parietal bone of the left hand side of the animal’s skull was thinned to transparency, and a cranial window installed over the thinned surface, using an optimized and validated technique. 31 Mice were anesthetized under 3% isoflurane in 100% oxygen, and anesthesia was maintained at 2% isoflurane in oxygen for all surgical procedures. Prior to the in vivo experiment, animals had an external pin-port catheter (custom) introduced to the left femoral artery, in order to obtain blood draws for arterial blood gas analysis prior to, and following, hypercapnic challenge (see Supplemental Figure 1). After femoral artery cannulation, the mouse was orotracheally intubated, and connected to a ventilator (Mini-vent, Model 845, Harvard Apparatus), and Capnograph (Type 340, Harvard Apparatus), and the gas mixture was switched from 2% isoflurane in 100% oxygen to 1.5% isoflurane in medical grade room air (21% O2, balance N2) for the duration of the experiment. The animal’s body temperature was kept constant at 37°C using a body temperature regulated homeothermic infrared heat pad, and mean arterial blood pressure (MABP) was recorded continuously using a tail-cuff monitor (CODA, Kent Scientific). The tidal volume and respiratory rate analog settings on the mini-vent were individually set for each mouse to keep every animal’s baseline end-tidal CO2 (ETCO2) readings between 35 and 38 mmHg, and MABP, body temperature, respiratory rate, tracheal pressure, ET CO2, and airflow recordings were monitored and recorded throughout the experiment via LabChart (AD Instruments). Upon establishment of steady baseline physiological readings, a 50–60 µL blood draw was made from the femoral artery for baseline arterial blood gas (ABG) analysis. Two minutes following the blood draw, the laser speckle camera was placed directly perpendicular to the cranial window and individual laser speckle flux images were compiled from 25 frames acquired over every 1s of time elapsed, giving one flux image every second for the duration of the experiment. Following 60 s of steady baseline readings, the gas mixture was switched from 1.5% isoflurane in medical grade room air to 1.5% isoflurane in a custom mix of 21% O2, 5% CO2, and balance N2 for 60 s. Following the 60 s of hypercapnic challenge, the inhalation gas was switched back to 1.5% isoflurane in medical grade room air, and a further 4 min of 1 s recordings were collected, after which another 50–60 µL blood draw was made from the femoral artery, for ABG analysis, using the iSTAT®1 Vetscan (Abaxis, CA, USA). Any animal displaying an ABG base excess of extracellular fluid (BeECF) outside of −4 to +4, a pH outside of 3.5 to 4.5, a saturated O2 (sO2) level of less than 90% before or after hypercapnic challenge, irregular breathing during the experiment, or average MABP below 60 mmHg was excluded from analysis a priori, giving five r-sham and five r-mTBI animals for the three-month post-injury CVR experiment, and five r-sham and six r-mTBI animals for the nine-month post-injury CVR experiment. Following in vivo recording at either three months or nine months post-injury, the isoflurane was increased to 3% for approximately 60 s and the animal was removed from the experimental setup and sacrificed under heavy isoflurane anesthesia.
Analysis of cerebral cortical vasculature
Following in vivo recordings, the animals were anesthetized with isoflurane and perfused transcardially with phosphate-buffered saline (PBS), pH 7.4, the brain was removed from the skull, and one hemisphere was dissected to separate the brainstem, cerebellum, and ophthalmic bulbs from the rest of the hemisphere for biochemical analysis. The contralateral hemisphere was post-fixed in 4% paraformaldehyde solution at 4°C for 48 h, and paraffin-embedded for immunohistochemistry (see Supplementary Materials and Methods). Western blot analysis was carried out on vascular-enriched fractions from whole cortical brain tissue dissected from sacrificed r-sham and r-mTBI mice. The cerebrovasculature from mouse brain tissue was isolated using a methodology previously established by our group. 32 Our cerebrovascular preparation consists of capillary micro-vessels and small cerebral vessels, as we are interested in studying the role of both brain vascular pericytes and smooth muscle cells in r-mTBI. Equivalent total protein amounts were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis under denatured and reduced conditions using 4–20% TGX Stain-free precast Gels (Biorad), and electro-blotted on to a low fluorescence polyvinylidene difluoride (PVDF) membrane for 2 h at 90 mA constant current. Membranes were then washed in de-ionized water and blocked for 1 h at room temperature with 5% non-fat milk in tris-buffered saline (TBS). Membranes were then incubated with primary antibodies overnight at 4°C. The following primary antibodies were used at the given concentrations; laminin (Sigma, rabbit anti-mouse laminin, L9393, 1:1000 Dilution), platelet-derived growth factor receptor β (PDGFRβ; Abcam, rabbit anti-PDGFRβ monoclonal, ab32570, 1:1000 Dilution), alpha smooth muscle actin (αSMA; Millipore, mouse anti-αSMA monoclonal, ASM-1, 1:1000 dilution), rabbit ant-mouse β-Actin (Cell Signaling Technology (CST), 13E5, 1:1000 dilution), goat anti-mouse Aminopeptidase-N (CD13; R&D Systems, AF2335, 1:1000 dilution), rabbit anti-CD31 (PECAM-1, R&D Systems, D8V9E, 1:1000 dilution). Membranes were washed with deionized water, incubated with the appropriate secondary antibody for 1 h at 4°C, washed once more, and developed using ECL chemiluminescent detection reagent (GE Life Sciences). Membranes were imaged using a Biorad ChemiDoc Western Blot Imager, and densitometry results of individual bands were collected using ImageLab 5.2 (Biorad) software. Target protein values for each lane were normalized against densitometry values for its respective β-actin value. Analysis of alpha-smooth muscle actin (αSMA) concentration in vascular-enriched fractions from 9 months and 15-months-old wild type mice was carried out by means of an enzyme-linked immunosorbent assay (ELISA; LSBio, LifeSpan BioSciences, Mouse ACTA2, LS-F21849, Seattle, WA) as per manufacturer’s direction, and data expressed as nanograms (ng) per milliliter (mL).
In vivo imaging analysis
The Moor FLPI2 image files (each 6 min in length), comprising 360 images taken at 1 s intervals, under the high resolution, low speed, spatial setting on the camera setup menu, were analyzed using the dedicated Moor Image Review software. A region of interest (ROI) measuring 0.3 mm2 was placed at a location within the area of thinned skull of the animal proximal to, and approximately 1.5 mm from, the midline suture, and devoid of any large vessels, which may be meningeal or pial in nature, and so may not correspond to the brain parenchyma from which measurements were intended to be made. A second 0.3 mm2 ROI was placed at a similarly parenchyma-like region approximately 3.0 mm distal to the midline, within the area of thinned skull (see Figure 2). All image files were scrolled through to correct for any head or camera movement during the experiment. Both proximal and distal ROIs were exported as separate graph Excel files, the flux data normalized to the 60 s of baseline immediately prior to hypercapnic challenge, and so expressed as percentage change from baseline. Every image file’s correlative Labchart data for that experiment were plotted parallel to the flux data (see Supplementary Figure 2).
Statistical analysis
Study design and reporting followed ARRIVE guidelines. For Barnes Maze, normality of data was assessed using Shapiro–Wilk test. If a given dataset was normally distributed and variances were equal, one-way ANOVA (for probe) or repeated measures ANOVA (for acquisition) were used to assess significant changes due to injury. Two-way ANOVA was used to assess significant differences between groups on only day 6 of acquisition trials.
Normality of CVR, biochemical, and immunohistochemical data sets was tested using Shapiro–Wilk. Biochemical and immunohistochemical data were analyzed using non-parametric Mann–Whitney U test. CVR data were analyzed using an unpaired, one-tailed, non-parametric Mann–Whitney U test, and the size of the effect of r-mTBI or EFAD genotype analyzed between groups at either proximal or distal ROIs. A given effect was considered significant at P < 0.05 and indicated by asterisks in the Figures. Error bars represent the standard deviation from the mean. Statistical analysis was performed, and graphs created using GraphPad Prism 8.0.
Results
Barnes maze acquisition and probe
At both three and six months following the final injury/anesthesia, r-mTBI mice travelled a significantly greater mean cumulative distance than their r-sham controls across the six days of acquisition (Figure 1(a) and (b), P < 0.001). Additionally, we observed a distinct effect of injury on cumulative distance travelled over time, reflected by a progressive separation of cumulative distance travelled by r-mTBI mice, compared to r-sham controls, across the six consecutive days of acquisition at three months post-injury (Figure 1(a), r-Sham vs. r-mTBI; P = 0.0118), and also a significant effect of injury on cumulative distance with time at six months (Figure 1(b), r-Sham vs. r-mTBI; P < 0.001). The mean velocity of r-mTBI mice was statistically significantly greater at both three and six months post-injury (Figure 1(c) and (d), P = 0.0046 and P = 0.0119, respectively), with the average velocity of r-mTBI mice being statistically greater than that of r-Sham controls on day 6 of acquisition at both three months (Figure 1(c), P < 0.05) and six months (Figure 1(d), P < 0.05) post-injury.
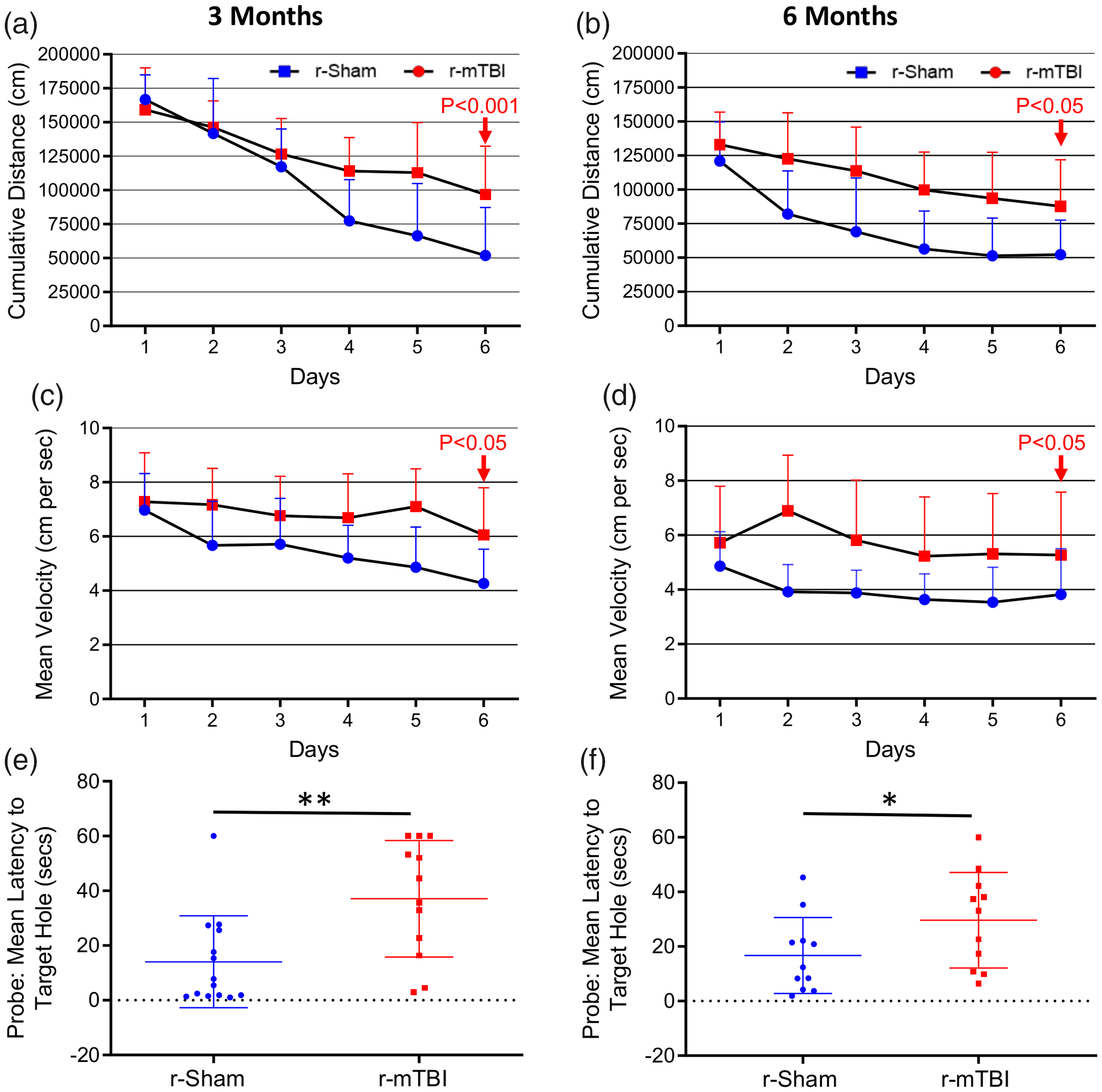
Evaluation of learning (acquisition) and retention of spatial memory of wild type mice using the Barnes maze at three and six months following repetitive mild traumatic brain injury. Mice were tested in the Barnes maze for their ability to locate a black box at the target hole. During the course of the six days of acquisition at both the three months and six months chronic time-points post-injury, the r-mTBI mice travelled a greater mean cumulative distance to reach the target hole, compared to sham controls (a and b, P < 0.001, repeated measures ANOVA). In cumulative distance data, the injury by time interaction term was statistically significant across the six days of acquisition at the three months post-injury time-point (P < 0.0001, Repeated Measures ANOVA); however, this was not seen at the six-month time-point (P = 0.08, repeated measures ANOVA). There was also a significant effect of injury on mean velocity between groups across all six days of acquisition at both the three month and the six-month time-points (c and d; P < 0.05, repeated measures ANOVA) with a significant increase in velocity of r-mTBI mice vs. r-Sham controls on day 6 of acquisition at both three and six months post-injury (d; P < 0.05, two-way ANOVA). Evaluation of spatial memory of wild type mice using the Barnes maze at three and six months following repetitive mild traumatic brain injury. Mice were tested in the Barnes maze for their ability to locate a black box at the target hole. During the course of the six days of acquisition at both the three months and six-month chronic time-points post-injury, the r-mTBI-treated mice travelled a greater mean cumulative distance to reach the target hole, compared to sham controls (a and b, P < 0.001, repeated measures two-way ANOVA). Evaluation of spatial memory retention (Probe) of wild type mice using the Barnes maze, at three months and six months following r-mTBI (e and f, respectively). For the probe trial (the day immediately following the 6 consecutive days of acquisition testing), the target box was removed, and mice were placed in the middle of the table for a single, 60-s trial. Probe test performance was significantly impaired in the r-mTBI mice at three months (e, P < 0.01, Student t-test) and six months (f, P < 0.01, Student t-test), compared to r-Sham controls. Data are presented as mean ± standard deviation (SD); 12 r-Sham, and 12 r-mTBI at three months; 11 r-Sham, and 11 r-mTBI at six months.
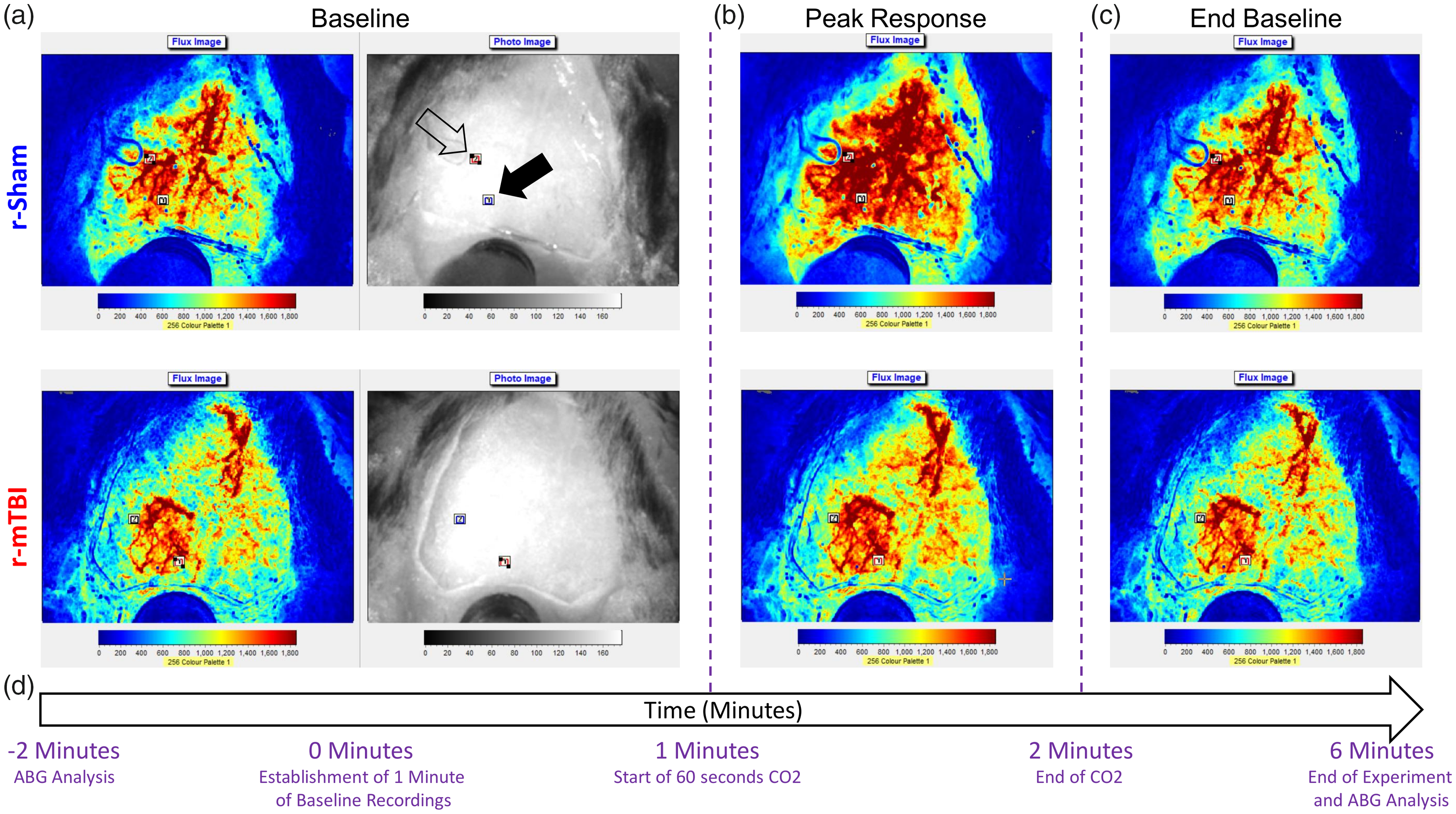
Time course of experimental assessment of cerebrovascular reactivity in male C57BL/6 mice. Representative laser speckle contrast flux and bright field images taken from the dorsal view by the Moor FLPI2 Laser Speckle Contrast Imager of fixed heads of a r-sham and r-mTBI mouse as viewed during the 60-s baseline immediately prior to hypercapnic challenge (a, first and second images from left, respectively, of top and bottom panels), during the peak response to hypercapnia (b, third image, top and bottom panel), and during the final return to baseline flux (c, fourth image, top and bottom). The time course of the experiment with relation to the representative images is shown in (d). Two 0.3 mm squared rectangular regions of interest (ROIs) were positioned over areas of the cranial window with no obvious large arteries or transparency occlusion, with one ROI being positioned proximal to the midline site of injury (a, top panel, second image, closed arrowhead) and the second being positioned distally to the midline site of injury (a, top panel, second image, open arrowhead). The flux data for the entirety of the experiment duration for each mouse were normalized to the 60-s baseline of the animal, and ABG analysis was evaluated immediately before and after the experiment (d).
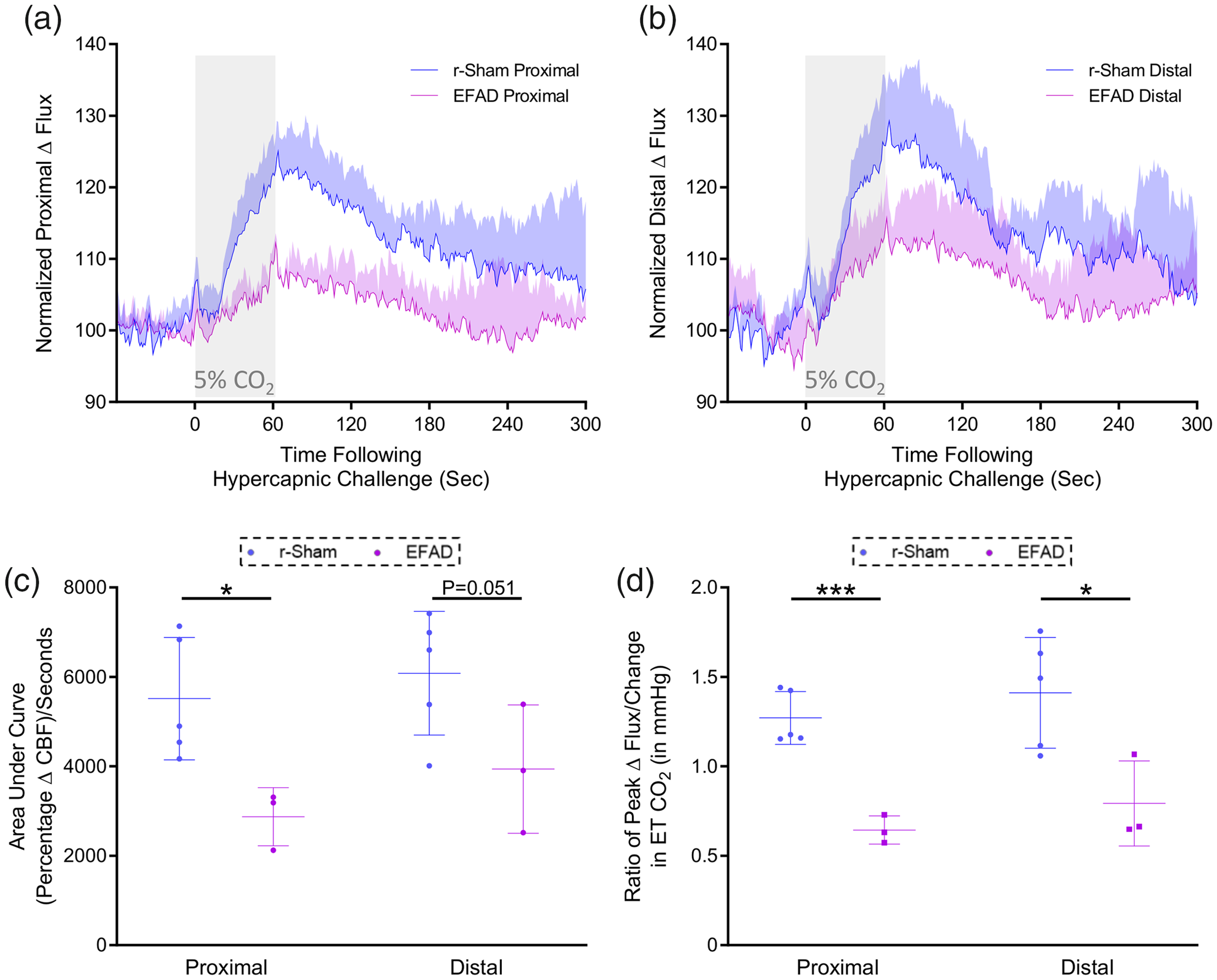
Cerebrovascular reactivity at nine months of age. CO2 evoked responses of the cerebrovasculature of positive control ApoE3FAD mutant mice are diminished, as compared to the responses of C57BL/6 mice, at nine months post-injury. Representative traces of CBF flux, as measured by laser speckle contrast imaging, show a reduced mean response in CBF at ROIs both proximal (a) and distal (b) to the site of injury, in the brains of age-matched positive control ApoE3FAD mice, versus r-sham controls, following 60 s of 5% CO2 hypercapnic challenge. The area under the curve (c, AUC) and peak response of each individual mouse, normalized to the animal’s peak change in ETCO2 (d, CVR ratio) were both analyzed using a non-parametric unpaired Wilcoxon Mann–Whitney U test, within ROI and between groups. The AUC of the proximal ROI CBF of EFAD mice was significantly less than that or age-matched r-sham animals (c, P < 0.05, Mann–Whitney U test). The AUC of the distal ROI CBF was not significantly different between EFAD and r-sham mice; however, there was a trend towards significance (c, P = 0.051, Mann–Whitney). There was a statistically significant difference in CVR ratio between groups at nine months of age (d, P < 0.05, Mann–Whitney), with a significantly lower value in CVR of EFAD mice of 0.64 ± 0.07, compared to a value of 1.27 ± 0.14 for r-sham controls, and a significantly lower value in CVR of EFAD mice of 0.79 ± 0.23, compared to a value of 1.41 ± 0.31 for r-sham controls, at the proximal and distal ROIs, respectively (d, P = 0.05, Mann-Whitney; r-Sham vs. EFAD, *P < 0.05, ***P < 0.001). R-sham mice; n = 5 per group. EFAD; n = 3 per group. CVR ratio; % Peak Response in CBF/Change in ETCO2 (mmHg).
Spatial memory was also analyzed at 3 and 6 months following final injury/anesthesia (9 and 12 months of age, respectively). Probe test performance was profoundly impaired in r-mTBI mice, compared to r-sham controls, at both three months (Figure 1(e), P < 0.01) and six months (Figure 1(f), P < 0.05) post-injury.
CVR at three months post-injury in nine months old wild type mice
CVR of mice in response to inhalation of 5% CO2 in medical grade room air for 60 s, and concomitant transient increase in ETCO2 to 55–60 mmHg, was markedly decreased in nine-month-old EFAD transgenic mice, compared to age matched wild type r-sham controls. The mean response of CBF from ROIs both proximal and distal to the midline site of injury of nine-month old r-sham and EFAD mice (Figure 3(a) and (b), respectively) was analyzed using both area under the curve (Figure 3(c)) and the CVR ratio, a more clinically relevant index of CVR compared to the AUC, expressed by further normalizing the peak response percentage change in CBF from baseline, to the peak change in ETCO2 from baseline for each animal. AUC analysis demonstrates a significant decrease in response of EFAD transgenic mice of approximately 51.87% and 37.82% mice, at the proximal and distal ROIs, respectively, compared to r-sham controls (Figure 3(c); Mann–Whitney, P < 0.05), and CVR ratio analysis showed a decrease in peak CBF response, normalized to peak change in ETCO2, of EFAD transgenic mice, of approximately 49% and 44%, at the proximal and distal ROIs, respectively, relative to r-sham controls (Figure 3(d), Mann–Whitney, P < 0.05).
The cerebrovascular reactivity at three months post-injury in response to inhalation of 5% CO2 in medical grade room air for 60 s, and concomitant transient increase in ETCO2 to 55–60 mmHg were markedly decreased in r-mTBI mice, compared to r-sham controls, at three months post-last mTBI. The mean response of CBF from ROIs both proximal and distal to the midline site of injury of nine-month-old r-sham and r-mTBI mice (Figure 4(a) and (b), respectively) was analyzed using both area under the curve (Figure 4(c)) and the CVR ratio. The AUC analysis demonstrates a significant decrease in response of r-mTBI mice of approximately 47% and 44% at the proximal and distal ROI, respectively, compared to r-sham controls (Figure 4(c); Mann–Whitney, P < 0.05), and CVR ratio analysis showed a decrease in ETCO2-normalized peak CBF response of r-mTBI mice, of 30% and 46%, at the proximal and distal ROIs, respectively, relative to r-sham controls (Figure 4(d), Mann–Whitney, P < 0.05).
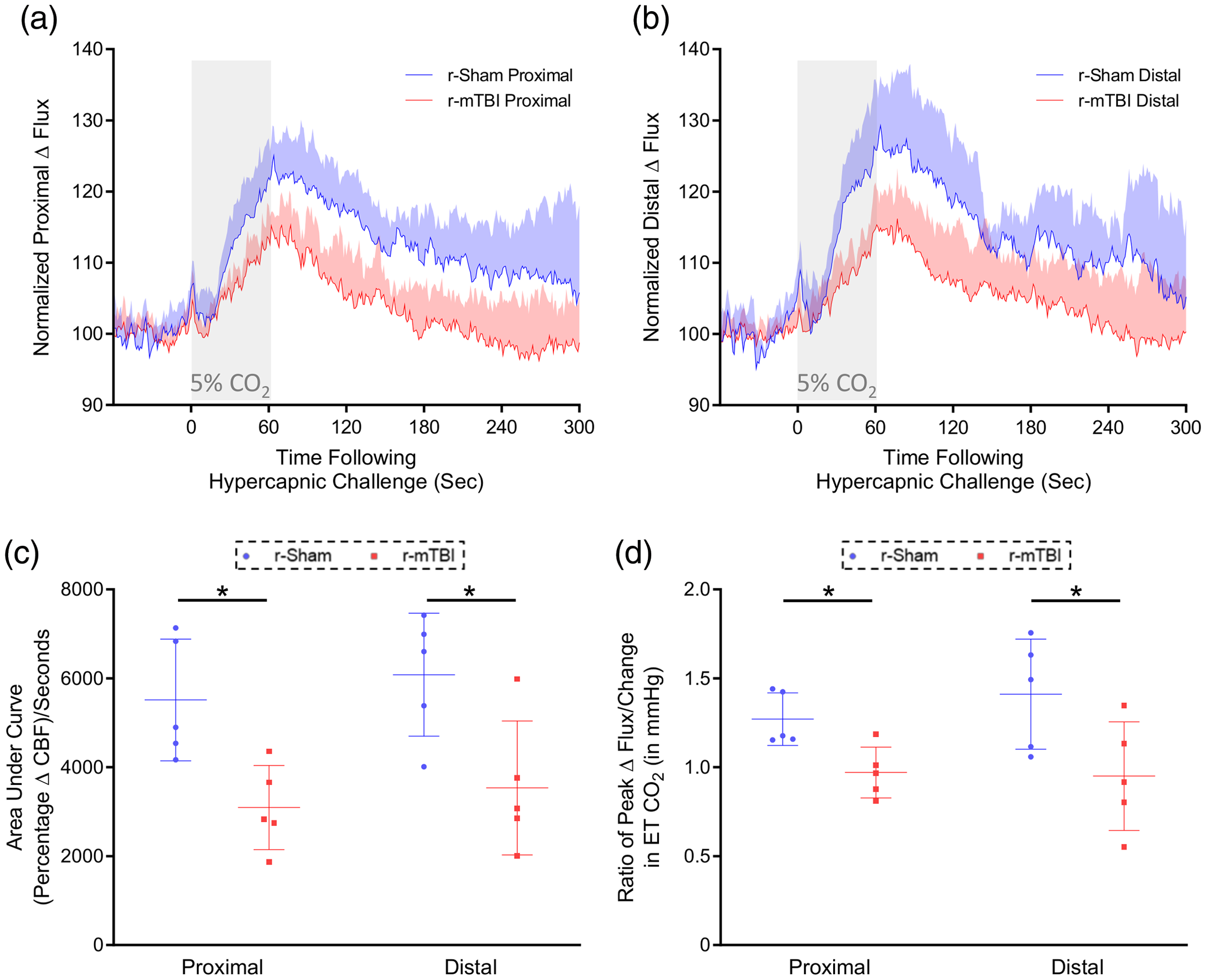
Cerebrovascular reactivity at three months post-injury. CO2 evoked responses of the cerebrovasculature of C57BL/6 mice are diminished at three months post-injury. Representative traces of CBF flux, as measured by laser speckle contrast imaging, show a reduced mean response in CBF at ROIs both proximal (a) and distal (b) to the site of injury, in the brains of both r-mTBI mice, versus r-sham controls, following 60 s of 5% CO2 hypercapnic challenge. The area under the curve (c, AUC) and peak response of each individual mouse, normalized to the animal’s peak change in ETCO2 (d, CVR ratio), were both analyzed using a non-parametric, unpaired Wilcoxon Mann–Whitney U test. The AUC of both the proximal and distal ROI CBF of r-mTBI mice was significantly less than that of r-sham controls (c, P < 0.05, Mann–Whitney). There was a statistically significant difference in CVR ratio between groups at both the proximal and distal ROIs at three months post-injury (d, P < 0.05, Mann–Whitney), with a significantly lower value in proximal CVR ratio of r-mTBI mice of 0.97 ± 0.14, compared to a value of 1.27 ± 0.16 for r-sham controls (d, Mann–Whitney; r-sham vs. r-mTBI, P < 0.05), and a significantly lower value of distal CVR ratio of r-mTBI mice of 0.95 ± 0.30, compared to a value of 1.41 ± 0.31 (d, Mann–Whitney, P < 0.05). R-sham and r-mTBI mice; n = 5 per group. CVR ratio; % Peak Response in CBF/Change in ETCO2 (mmHg).
CVR at 9 months post-injury in 15 months old wild type mice
The cerebrovascular reactivity at 9 months post-injury in response to inhalation of 5% CO2 for 60 s, and concomitant transient increase in ETCO2 to 55–60 mmHg, was markedly decreased in 14–15-months-old r-mTBI mice, compared to r-sham controls, at nine months post-last mTBI. The mean response of CBF from ROIs both proximal and distal to the midline site of injury of 15-months old r-sham and r-mTBI mice (Figure 5(a) and (b), respectively) was analyzed using both area of curve (AUC, Figure 5(c)) and the CVR ratio. AUC analysis demonstrates a statistically significant decrease in AUC of r-mTBI mice of approximately 36% at the proximal ROI, compared to r-sham controls (Figure 5(c); Mann–Whitney, P < 0.05). There was a decrease in AUC value of r-mTBI mice of approximately 20% at the distal ROI, compared to r-sham controls; however, this was not significant (Figure 5(c); Mann–Whitney, P > 0.05). CVR ratio analysis shows no correlate decrease in ETCO2-normalized peak CBF response of r-mTBI animals, relative to r-sham controls, at the proximal or distal ROI (Figure 5(d), Mann–Whitney, P > 0.05), but a trend towards a decrease at the proximal ROI (Figure 5(d), Wilcoxon, P = 0.14).
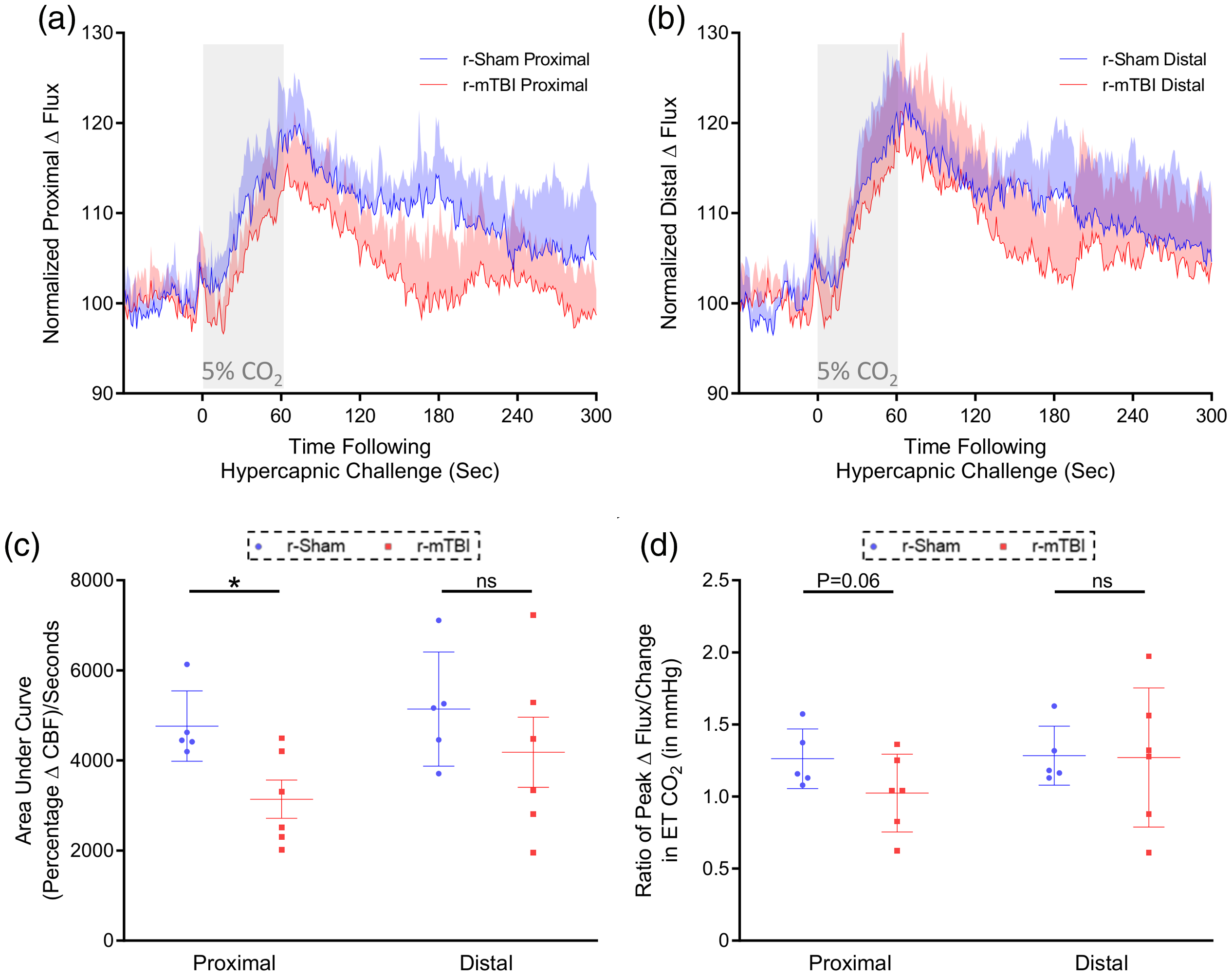
Cerebrovascular reactivity at nine months post-injury. CO2 evoked responses of the cerebrovasculature of C57BL/6 mice at nine months post-injury. Representative traces of CBF flux, as measured by laser speckle contrast imaging, showed the response in CBF at ROIs both proximal (a) and distal (b) to the site of injury, in the brains of r-mTBI mice, versus r-sham controls, following 60 s of 5% CO2 hypercapnic challenge, at nine months post-injury. The area under curve (c, AUC) and peak response of each individual mouse, normalized to the animal’s peak change in ETCO2 (d, CVR ratio), were both analyzed within ROI using a non-parametric, unpaired Wilcoxon Mann–Whitney U test. There was a statistically significant decrease in AUC of r-mTBI mice, compared to r-sham controls, at the proximal ROI at nine months post-injury (c, Mann–Whitney, P < 0.01). There was no statistically significant difference in CVR ratio between groups at either the proximal or distal ROI at nine months post-injury (d, Mann–Whitney, P > 0.05); however, the CVR ratio at the proximal ROI showed a trend toward significant decrease (d, Mann–Whitney, P = 0.06). Neither the AUC, nor the CVR ratio analysis of the distal ROI reported statistically significant difference between r-sham and r-mTBI at nine months post-injury (d, Mann–Whitney, P > 0.05). R-sham, n = 5, r-mTBI, n = 6. CVR ratio; % Peak Response in CBF/Change in ETCO2 (mmHg).
Analysis of vascular density and mural cell coverage
Western blot analysis of isolated cerebrovascular fractions from wild type r-sham and r-mTBI mouse cortical tissue revealed a statistically significant decrease in expression of PDGFRβ at three months post-injury (Figure 6(c), unpaired, two-tailed Mann–Whitney U test, P < 0.01). There was no apparent change in the expression level of the cerebrovascular-associated basal membrane constituent Laminin, the endothelial marker CD31, or the pericyte cellular marker CD13; however, there was a non-significant decrease in levels of the smooth musculature marker αSMA between r-sham and r-mTBI mice at three months post-injury (Mann–Whitney, P = 0.3), but not at nine months post-injury, as assessed via immunoblot analysis. Enzyme-linked immunosorbent assay (ELISA) analysis of the same vascular-enriched fractions confirmed this trend towards significance of reduction in the expression of αSMA of approximately 34% at three months post-injury (Figure 7(a), Mann–Whitney, P = 0.058), alongside the approximate 40% decrease in levels of PDGFRβ demonstrated via immunoblot analysis, with no difference in either marker detected at nine months following r-mTBI (Figure 7(b), Mann–Whitney, P > 0.05). The average area per image of laminin-positive capillaries throughout layers I–II of the cortices of r-mTBI mice was not statistically significantly different to that of r-sham controls at either three months or nine months post-injury (Supplemental Figures 4 and 5(I), Mann–Whitney, P > 0.05), suggesting that there is no change in the overall density of the cerebral vasculature during the times at which both CVR, and learning and memory are impaired. In addition, there was no statistically significant difference in the average coverage of laminin-positive vessel ROIs by CD13 immuno-positive pericytes between groups at either three months or nine months following injury (Supplemental Figures 4 and 5(II), Mann–Whitney, P > 0.05).
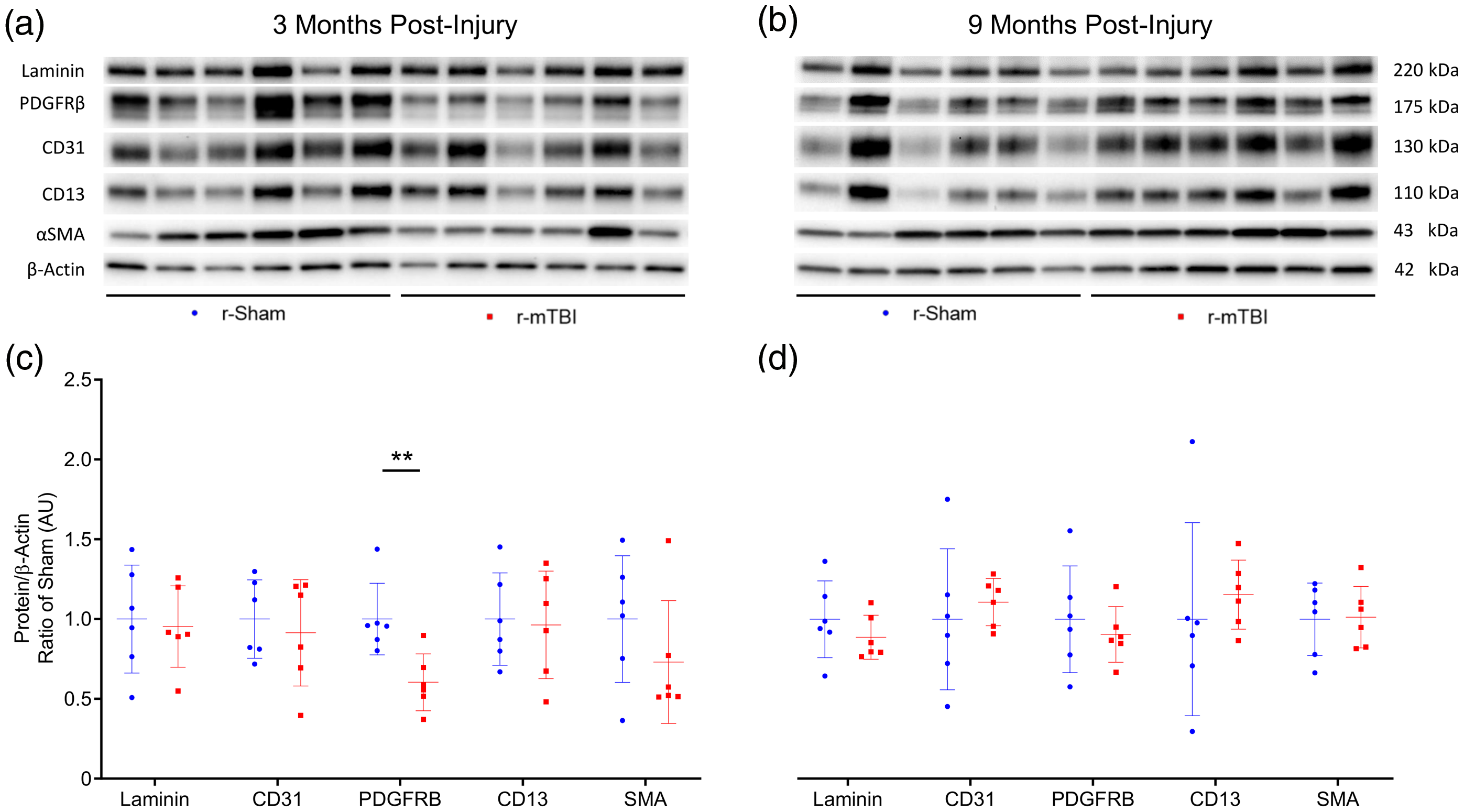
Western immunoblot analyses of cerebrovascular cellular markers in cortical vascular-enriched fractions at three months and nine months post-injury. Representative images of Western blot bands shown in (a) and (b) were analyzed via densitometry to compile the graphs shown in (c) and (d). Expression of laminin, CD31, CD13, and αSMA were not altered in cortical vascular fractions of r-mTBI mice, as compared to r-sham, at three or nine months post-injury (c and d, P > 0.05, Mann–Whitney); however, the expression of the receptor complex PDGFRβ was significantly decreased in r-mTBI cortical vasculature, compared to the vascular fractions of r-sham controls (c, P < 0.01, Mann–Whitney) at three months post-injury. This decreased expression of PDGFRβ was not significant at nine months following r-mTBI (d, P > 0.05, Mann–Whitney) wild type mice; six r-Sham and six r-mTBI. Values expressed as AU. All densitometry values for individual bands in A and B were normalized to the β-Actin value for their respective lane and this ratio further normalized to ratio of Sham values and used for statistical analysis in (c) and (d). No statistical significance between groups unless otherwise stated by horizontal bars and text. Error bars represent ± SD.
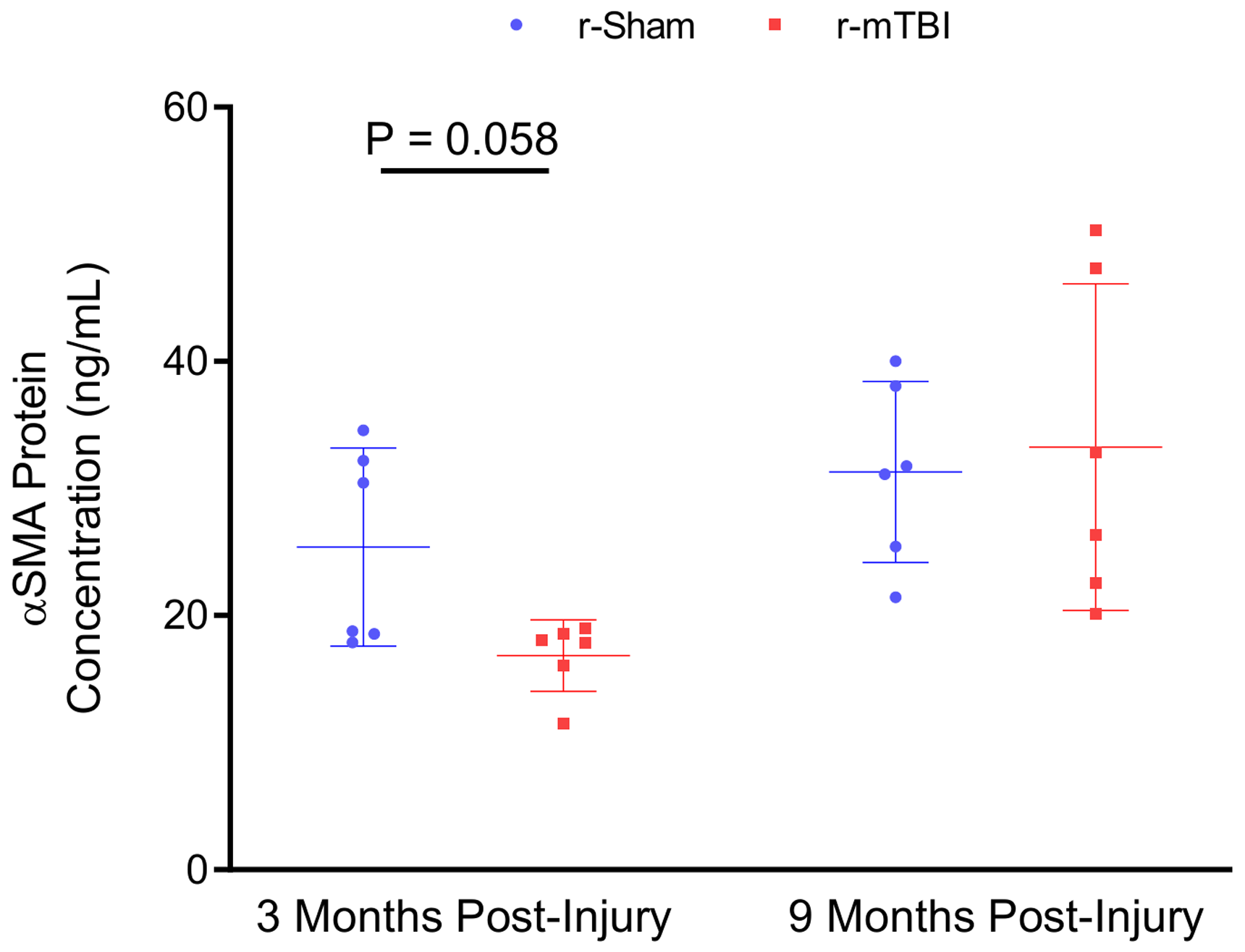
ELISA measurement of αSMA concentration in cortical vascular-enriched fractions at three months and nine months post-injury. Biochemical analysis of the vasculature revealed a trend towards a decrease in levels of αSMA in the brains of r-mTBI at three months post-injury, compared to r-sham controls (P = 0.058, Mann–Whitney). There was no statistically significant difference in levels of αSMA between r-sham and r-mTBI mice at nine months post-injury (P > 0.05, Mann–Whitney). Error bars represent ± SD.
Discussion
Epidemiological and neuropathologic evidence emerging over the last decade supports a causal link between a history of head trauma and the development of later life neurodegenerative co-morbidity33–36 and dementia,35–39 including, but not limited to, the tauopathies Alzheimer’s disease (AD)35,40 and chronic traumatic encephalopathy (CTE).35,41–44 All phase III clinical trials using drugs with demonstrated efficacy in multiple pre-clinical models have failed,45,46 indicating the need for better stratification of injury mechanism, severity, and endophenotype by biomarkers more sensitive than neurological assessment alone. 47 Clinical imaging of aberrant protein aggregates as a biomarker in patients with a history of TBI has been limited.48,49 However, detection of cerebrovascular pathophysiology via CBF and CVR modalities is well established, and as cerebrovascular pathology such as TCVI is not only known to be sufficient for development of neurodegenerative illness in human patients, 50 but also to precede clinical symptomology in diseases such as AD,51–53 these imaging metrics represent early occurring and easily detectable point-of-care biomarkers, and possible prognostics, for TBI-related illness and recovery, respectively.
Although CVR deficit is a symptom of many neurodegenerative diseases,23,54,55 recent work by Diaz-Arrastia et al.7,17,56,57 has demonstrated global, whole gray, and white matter CVR deficit in moderate-to-severe TBI patients over a timeframe of six months to eight years following injury, using BOLD MRI hypercapnic challenge,7,17,56,57 and a correlation of global gray matter CVR, but not resting CBF, with chronic TBI symptoms. 17 Furthermore, regions of parenchymal tissue distant from areas of encephalomalacia and gross tissue damage exhibited resting CBF comparable to healthy controls, but showed CVR deficit, suggesting that normal appearing areas of tissue with apparently adequate vessel density and resting state perfusion are still damaged enough to have a decreased response to vasoactive stimuli.17,56 In addition, whole brain CVR from TBI patients with marked encephalomalacia does not differ from that of TBI patients without detectable parenchymal lesions, 56 indicating gross tissue damage may not be necessary for the diffuse TCVI-associated pathophysiology seen here, and perhaps in lesser severities of TBI. The prevalence of CVR perturbation is not limited to the chronic phase following moderate to severe forms of injury. Mutch et al.16,58 have demonstrated alterations in global CVR in adolescents following sports-related concussion. Bailey et al. 59 have shown the magnitude of decreased CVR in adult boxers to correlate with both the number of mTBIs sustained and the degree of neuropsychological deficit incurred. Svaldi et al. 60 showed only transient decreases in CVR in asymptomatic female soccer players following a single season of play, as compared to pre-season baseline values, correlating with the number and degree of high acceleration events (HAEs). The finding that both symptomatic boxers 59 and asymptomatic soccer players60,61 exhibit decreased CVR supports its use as a biomarker of TCVI across mTBI populations. The recovery of CVR in female athletes, as compared to the ongoing deficit in boxers, is possibly due to a comparatively greater load and frequency of mTBIs attained during a boxing season, as compared with that over a season of soccer. Indeed, those soccer players sustaining the greatest load of higher force HAEs demonstrated the worst CVR, indicating there may be a threshold of mTBIs needed for prolonged CVR perturbation in humans, much as is the case with pathological phenotypes in animal models of r-mTBI, where typically the highest frequencies of closed head trauma give the most extreme r-mTBI-related sequalae, such as pathognomonic tauopathy.19,62
The injury paradigm used in this study was chosen to emulate the r-mTBI frequency to which athletes and military personnel may be exposed over the course of an entire career. To assess the disseminate nature of CVR in the brain parenchyma of our experimental animals, blood flux recordings were taken from two optimally distant, equally sized ROIs placed over areas devoid of any large vessels. We report a profound r-mTBI dependent decrease in CVR at three months post-injury recorded from separate ROIs when analyzed via either the AUC of the CBF flux, or the ETCO2-normalized peak CBF response. There is a trend towards recovery of CVR deficit at nine months post-injury, reflected by the lack of a deficit in either the AUC or ETCO2-normalized CVR values recorded from the more lateral of cortical ROIs examined, despite a conserved significant deficit in the CVR AUC, and trend towards decrease in ETCO2-normalized CVR (P = 0.06) at the ROI more proximal to the sagittal suture. The fractional microvascular volume of the cerebral cortex of C57Bl/6 mice is known to increase with distance lateral from midline, 63 perhaps masking a present but diminished injury effect at this ROI at the later time-point. Regardless, the CVR impairment can be interpreted in one of two ways; either the CVR detriment in our animal model is short-lived, and shows a propensity to resolve following reprieve from trauma, as is the case in the above human studies by Mutch16,58 and Svaldi, 61 or the effect at the more chronic nine months post-injury time-point is only detectable with a more sensitive, global imaging modality, much as with chronic human TBI CVR dysfunction.17,56,57
To elucidate possible underlying causes of our observed alterations in CVR following head trauma, we examined markers for vascular endothelial and mural cells. Leptomeningeal and precapillary arteriolar encompassing smooth muscle cells expressing the contractile protein αSMA, 64 and pericytes expressing the cellular markers PDGFRβ and CD13, have both been shown to regulate CBF in vivo.65–67 Pericytes initiate and maintain retrograde signaling upstream to larger arterioles via sensing and conductance of a hyperpolarizing potassium cation current,68,69 and contribute greatest to vascular resistance in the CNS. 70 The capillary strata of the cerebral vasculature also responds and dilates first in mouse imaging studies,71,72 and the initial BOLD MRI signal in humans emanates from localized parenchymal areas, 73 suggesting engagement of the capillaries, and perhaps, by default, pericytes, first in physio-normal neurovascular coupling. Kisler et al.74,75 have shown that modest cerebral vessel pericyte degeneration in mice heterozygous for PDGFRβ loss-of-function (PDGFRβ+/−), 76 and tamoxifen-inducible acute, global ablation of cortical pericytes in mice, 75 both result in a similar blunted capillary neurovascular coupling in absence of impaired endothelial, astroglial, or smooth muscle cell-dependent vasodilation or compromised neuronal cell viability, implicating healthy pericyte presence and function as crucial for normal CBF regulation. Indeed, studies have shown both a rapid cerebral vessel pericyte cell death, 77 and an acute rarefaction and pursuant increase in pericytes 78 following a single controlled cortical impact (CCI) in mice,77,78 and pericyte loss is seen in humans following moderate to severe TBI.79,80 These data together indicate pericyte degeneration may be incipient in TCVI pathogenesis. We report a decreased expression of PDGFRβ in tandem with αSMA, but without pericyte vessel coverage loss, as assessed by CD13 (most ubiquitously expressed by pericytes 81 ) at three months following r-mTBI, perhaps suggesting arteriolar smooth musculature damage with concomitant capillary pericyte injury. To our knowledge, there exists no documentation of arteriolar-specific loss of PDGFRβ or αSMA expression following TBI; however, human primary cell cultures of pericytes are known to express approximately four times as much of the receptor as human primary SMCs 82 and to shed soluble PDGFRβ upon hypoxic and amyloid beta-induced injury. 82 Hence, the decrease in PDGFRβ seen here is likely reflective of pericyte injury. The lack of evidence of mural cell marker loss or damage at the later of the time-points analyzed may suggest a latent recovery of TCVI following cessation of mTBIs as causative for the less pronounced CVR deficit with time following injury in our r-mTBI animals, as compared to r-sham mice, much as with the above human population of athletes, when compared to healthy controls.60,61,83 Indeed, although human post mortem studies show microvascular cell apoptosis up to 20 days following severe TBI, 84 Danaila et al. 85 have reported post-traumatic artery ECs and SMCs exhibiting mixed apoptotic, parapoptotic, and regenerative markers at four weeks following craniocerebral injury. A similar profile of mural cell regeneration with time in our r-mTBI mice could account for the earlier decrease, and later restoration, of levels of αSMA and PDGFRβ in our immunoblot analyses. As the nature of mild head trauma in the human population precludes analysis of the relatively acute vasculopathies underlying CVR impairment, emulation of a profile of CVR deficit akin to human studies further validates our animal model of r-mTBI and offers a pre-clinical platform for translational research.
Compromised CVR in TBI may be due to an inability of cerebral vessels to react normally to vasoactive stimuli, rather than a decreased density of the vessels themselves. 56 Indeed, there was no difference in expression of either the basal lamina protein laminin, or the endothelial cellular marker CD31, as assessed via immunoblot, or in the average area of laminin positive vessels, as quantified by laser confocal immunofluorescence, between the r-sham and r-mTBI mice in this study, approximating the above human data, wherein CVR impairment occurs in absence of cerebral vascular anatomical differences.17,56,58 The preserved cerebral vessel density in both our r-mTBI mice and human chronic TBI patients17,56,58 could be said to conflict with reports of both early cerebral vasculature morphological changes in human patients following severe TBI, 84 and cerebrovascular cellular stress and apoptosis, 86 and aberrant morphology following TBI in animal models.86,87 Obenaus et al. 88 have catalogued an acute rarefaction of global cerebrovascular density at 1 day following mTBI in mice, 88 with a recovery to baseline levels by 14 days post-injury, and Hayward et al. 89 have shown acute cerebrovascular loss, before revascularization with persistent hypoperfusion at 14 days post injury in a rat model of TBI. 89 Steinman et al. 90 have shown blunted resting and hypercapnia-induced CBF at four weeks following CCI in mice, despite recovery of microvascular density and volume detriments recorded at one day post injury. These data not only suggest an acute loss and later return of cerebrovascular density following brain trauma in animal models, and possibly human patients, but also demonstrate that revascularization does not necessarily translate to functional recovery. The unperturbed cerebrovascular density despite decreased cerebrovascular responsiveness at chronic periods following repeat mild TBI in our animals is thus unsurprising, especially as it is known that while both moderate and severe TBI leads to similar cerebrovascular pallor acutely, only animals exposed to the lesser, moderate intensity of TBI show recovery of vascular density. 91
The unaltered expression of CD31 alongside normal laminin-positive vessel density in the brains of r-mTBI mice suggests the cerebral endothelium may be functionally impaired, as opposed to physically absent, following injury. This hypothesis is supported by clinical observations of early decreases of reactive hyperemia index, a measure of microvascular endothelial function, in patients admitted with acute brain injury, 92 and preclinical studies demonstrating early activation of the endothelium, 93 and loss of endothelial nitric oxide synthase-(eNOS) dependent vasogenic tone94,95 and eNOS-dependent improvement in CBF, 96 following TBI. Further evidence that TCVI-associated microvascular dysfunction can occur with normative, intact vessel volume can be seen in a study by Wei et al., 97 showing mTBI in rats elicits a pronounced decrease in CVR via hypercapnic challenge for up to three weeks post-injury, with the degree of deficit in response to either hypercapnia or topical application of acetylcholine quantitatively greater than that observed with application of the nitric oxide (NO) donor sodium nitroprusside (SNP), 97 indicating that while the smooth musculature retains the ability to respond to NO, production of NO itself seems impaired. 97 Concomitantly, Kenney et al. 57 demonstrated that a single oral 25 mg dose of sildenafil was sufficient to potentiate CVR in moderate to severe TBI patients exhibiting CVR deficits in lesioned and normal appearing gray matter tissue with normal resting CBF, indicating intact and functional vessel density, as is indicated in our study, where there was no change in cerebral vascular lamina or endothelial cellular markers post-injury. Patients receiving a twice daily oral dose of 25 mg sildenafil for a course of eight weeks showed improved CVR relative to baseline. In this study, there was no significant effect of prolonged treatment on neurocognitive function or self-reported symptoms with sildenafil administration, 57 despite rescued CVR. The lack of an amelioration in clinical symptoms complement to improved CVR following a short treatment 57 is neither surprising nor disparaging for prospective treatment earlier in disease progression, as the neurodegenerative effects consequent to a protracted ischemia are slowly evolving, 74 and chronic TBI-related illness is known to be accompanied by hither-to irreversible white matter tract damage and neuroinflammation98,99 alongside the TCVI seen in these patients. Indeed, early treatment of TCVI with angiogenic therapeutics has been shown to ameliorate cognitive symptoms in animal models of brain injury.100–102 Treatment with simvastatin in 18-month-old transgenic mice over-expressing transforming growth Factor-β 1 (TGF-1), considered a model of cerebrovascular disease, is shown to restore nitric oxide levels to baseline in vivo and to rescue acetylcholine-induced CVR deficit ex vivo. 103 Synergistic rescue of both cerebral artery CVR deficit ex vivo, and impaired spatial memory, in AD mice has been observed following treatment with simvastatin. 28 Most importantly, these neurocognitive and physiological improvements occurred without obvious resolution of amyloid pathology, and only in the adult treated, and not the aged, group of AD mice. 28 The reversal of both cognitive and CVR deficits by angiogenic statins at an earlier stage of neuropathology in animal models of AD, and the above success in the rescue of vascular responsiveness and behavioral phenotype in animal models of TBI, along with the proof of concept of targetability of TCVI by CVR analyses from both Kenney et al. 57 and Wei et al., 97 all suggests that treatment of TCVI may have therapeutic efficacy in the pathogenesis of TBI and CTE-like neurodegenerative disease.
In summary, we have demonstrated a chronic functional deficit of the cerebral vasculature following closed head repeat mild traumatic brain injury coinciding with cerebrovascular-specific pathology and neurobehavioral decline. These results, which resemble those across a spectrum of mTBI patients in the human population, posit a causative role for TCVI in preclinical r-mTBI-related pathogenesis, and justification for the use of CVR as a prospective therapeutic metric in pre-clinical and human TBI research.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X20954015 - Supplemental material for Impairment of cerebrovascular reactivity in response to hypercapnic challenge in a mouse model of repetitive mild traumatic brain injury
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X20954015 for Impairment of cerebrovascular reactivity in response to hypercapnic challenge in a mouse model of repetitive mild traumatic brain injury by Cillian E Lynch, Maxwell Eisenbaum, Moustafa Algamal, Matilde Balbi, Scott Ferguson, Benoit Mouzon, Nicole Saltiel, Joseph Ojo, Ramon Diaz-Arrastia, Mike Mullan, Fiona Crawford and Corbin Bachmeier in Journal of Cerebral Blood Flow & Metabolism
Supplemental Material
sj-pdf-2-jcb-10.1177_0271678X20954015 - Supplemental material for Impairment of cerebrovascular reactivity in response to hypercapnic challenge in a mouse model of repetitive mild traumatic brain injury
Supplemental material, sj-pdf-2-jcb-10.1177_0271678X20954015 for Impairment of cerebrovascular reactivity in response to hypercapnic challenge in a mouse model of repetitive mild traumatic brain injury by Cillian E Lynch, Maxwell Eisenbaum, Moustafa Algamal, Matilde Balbi, Scott Ferguson, Benoit Mouzon, Nicole Saltiel, Joseph Ojo, Ramon Diaz-Arrastia, Mike Mullan, Fiona Crawford and Corbin Bachmeier in Journal of Cerebral Blood Flow & Metabolism
Supplemental Material
sj-pdf-3-jcb-10.1177_0271678X20954015 - Supplemental material for Impairment of cerebrovascular reactivity in response to hypercapnic challenge in a mouse model of repetitive mild traumatic brain injury
Supplemental material, sj-pdf-3-jcb-10.1177_0271678X20954015 for Impairment of cerebrovascular reactivity in response to hypercapnic challenge in a mouse model of repetitive mild traumatic brain injury by Cillian E Lynch, Maxwell Eisenbaum, Moustafa Algamal, Matilde Balbi, Scott Ferguson, Benoit Mouzon, Nicole Saltiel, Joseph Ojo, Ramon Diaz-Arrastia, Mike Mullan, Fiona Crawford and Corbin Bachmeier in Journal of Cerebral Blood Flow & Metabolism
Supplemental Material
sj-pdf-4-jcb-10.1177_0271678X20954015 - Supplemental material for Impairment of cerebrovascular reactivity in response to hypercapnic challenge in a mouse model of repetitive mild traumatic brain injury
Supplemental material, sj-pdf-4-jcb-10.1177_0271678X20954015 for Impairment of cerebrovascular reactivity in response to hypercapnic challenge in a mouse model of repetitive mild traumatic brain injury by Cillian E Lynch, Maxwell Eisenbaum, Moustafa Algamal, Matilde Balbi, Scott Ferguson, Benoit Mouzon, Nicole Saltiel, Joseph Ojo, Ramon Diaz-Arrastia, Mike Mullan, Fiona Crawford and Corbin Bachmeier in Journal of Cerebral Blood Flow & Metabolism
Supplemental Material
sj-pdf-5-jcb-10.1177_0271678X20954015 - Supplemental material for Impairment of cerebrovascular reactivity in response to hypercapnic challenge in a mouse model of repetitive mild traumatic brain injury
Supplemental material, sj-pdf-5-jcb-10.1177_0271678X20954015 for Impairment of cerebrovascular reactivity in response to hypercapnic challenge in a mouse model of repetitive mild traumatic brain injury by Cillian E Lynch, Maxwell Eisenbaum, Moustafa Algamal, Matilde Balbi, Scott Ferguson, Benoit Mouzon, Nicole Saltiel, Joseph Ojo, Ramon Diaz-Arrastia, Mike Mullan, Fiona Crawford and Corbin Bachmeier in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Roskamp Institute and the National Institute on Aging of the National Institutes of Health under award number R01AG041971 (Bachmeier) and the Department of Defense PRARP award W81XWH-16-1-0724 (Bachmeier). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health nor the Department of Defense. Dr. Crawford is a Research Career Scientist at the James A. Haley Veterans Hospital, Tampa, FL. Dr. Bachmeier is a Research Scientist at the Bay Pines VA Healthcare System, Bay Pines, FL.
Acknowledgements
The authors would like to thank Dr Andy Shih (Principal Investigator, Center for Developmental Biology and Regenerative Medicine, 1900 9th Ave Seattle, WA 98101) for his help in the training of cranial window surgical technique.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
ME, MA, MB, SF, BM, NS, JO, RDA, MM, and FC contributed to the revision of the manuscript and approved its submission for publication. CL and CB conceived and designed the study, analyzed and interpreted the data, and prepared the manuscript.
Supplementary material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.