
Abstract
In patients who are successfully resuscitated after initial cardiac arrest (CA), mortality and morbidity rates are high, due to ischemia/reperfusion injury to the whole body including the nervous and immune systems. How the interactions between these two critical systems contribute to post-CA outcome remains largely unknown. Using a mouse model of CA and cardiopulmonary resuscitation (CA/CPR), we demonstrate that CA/CPR induced neuroinflammation in the brain, in particular, a marked increase in pro-inflammatory cytokines, which subsequently activated the hypothalamic-pituitary-adrenal (HPA) axis. Importantly, this activation was associated with a severe immunosuppression phenotype after CA. The phenotype was characterized by a striking reduction in size of lymphoid organs accompanied by a massive loss of immune cells and reduced immune function of splenic lymphocytes. The mechanistic link between post-CA immunosuppression and the HPA axis was substantiated, as we discovered that glucocorticoid treatment, which mimics effects of the activated HPA axis, exacerbated post-CA immunosuppression, while RU486 treatment, which suppresses its effects, significantly mitigated lymphopenia and lymphoid organ atrophy and improved CA outcome. Taken together, targeting the HPA axis could be a viable immunomodulatory therapeutic to preserve immune homeostasis after CA/CPR and thus improve prognosis of post-resuscitation CA patients.
Introduction
Cardiac arrest (CA) affects more than 500,000 people in the US every year. 1 Although considerable advances in resuscitation have increased the number of CA patients who survive the initial arrest event and are admitted to the intensive care unit (ICU), the rates of survival-to-discharge and of good neurologic recovery remain extremely poor. In fact, for out-of-hospital CA (OHCA) adult patients who receive EMS treatment of full resuscitative efforts on-scene, survival to hospital discharge is only 10.4%, and survival with good functional status is 8.2%. 1 The high mortality and morbidity in this patient population is attributed primarily to a complex set of pathophysiologic processes collectively called post-CA syndrome.2,3 One key element of this syndrome is the post-CA immune response.2,3
We know that in CA patients with re-established return of spontaneous circulation (ROSC), the immune system is rapidly activated to induce a “sepsis-like” condition in the acute phase, even in the absence of infection. This is characterized by massive release of pro-inflammatory cytokines such as interleukin (IL)-6 and tumor necrosis factor (TNF)-α. 3 There are also clinical reports showing that blood leukocytes from CA patients exhibit an impaired capacity to produce cytokines,3,4 and lymphopenia is common in CA patients. 5 Collectively, clinical evidence clearly indicates post-CA immune dysfunction. However, we still do not have an adequate understanding of how systemic immunity is dysregulated after CA and how this immune dysfunction is linked to post-CA poor prognosis.
CA followed by resuscitation represents an extreme stress to the body, as the blood flow abruptly stops during CA, and upon resuscitation, ischemia/reperfusion injury is imposed on all organ systems including the nervous and immune systems, the two major regulators of homeostasis in the body. Importantly, these two systems exhibit intense bidirectional interactions, and compelling evidence has shown that these interactions can lead to imbalanced immune responses, i.e. hyperinflammation or immunosuppression.6–8 Therefore, in the present study, we investigated whether this interaction is involved in post-CA immune dysfunction using a murine model of CA and cardiopulmonary resuscitation (CPR). Our data indicate that after CA/CPR, pro-inflammatory cytokines were induced in the brain, which then activated the hypothalamic-pituitary-adrenal (HPA) axis. Importantly, we provide the first experimental evidence that activation of the HPA axis critically contributes to post-CA immunosuppression, which was characterized by drastic atrophy of lymphoid organs accompanied by a massive loss of immune cells and reduced immune function of splenic lymphocytes. Further, our data identified the HPA axis not only as a key mechanism for CA-induced immunosuppression but also as a potential therapeutic target to preserve immune homeostasis and improve functional outcome after CA/CPR.
Material and methods
The protocols for all experiments conducted in this study were approved by the Institutional Animal Care and Use Committee at Duke University. All studies were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (NIH). Experiments have been reported in compliance with the ARRIVE guidelines.
Animals and surgeries
Male C57Bl/6J mice (3–6 months old) were purchased from The Jackson Laboratory. The online tool Quickcalcs was used to randomize animals to experimental groups. Evaluations were performed in a blind fashion whenever possible. Mouse models of CA/CPR were used. 9
Cytokine enzyme-linked immunosorbent assay and measurements of corticosterone and adrenocorticotropic hormone
Quantification of IL-6, TNF-α, and IL-1β was performed using enzyme-linked immunosorbent assay (ELISA) MAX Sets (BioLegend, San Diego, CA, USA). Levels of corticosterone and adrenocorticotropic hormone (ACTH) in the blood were analyzed using competitive-enzymatic reaction kits from Cayman Chemical (Ann Arbor, MI, USA) and MD Biosciences (Cambridge, MA, USA), respectively.
Flow cytometry analysis
Single-cell suspensions of spleen, blood, Peyer’s patches (PPs), and the whole brain were prepared using established methods. Immunophenotyping was performed using a combination of surface antibodies. For intracellular staining, splenocytes were stimulated and stained anti-IFN-γ and anti-TNFα antibodies. Flow cytometry data were acquired on FACS Canto (BD Biosciences, San Jose, CA, USA) and analyzed with FlowJo software.
Blood leukocyte count and TdT-mediated dUTP nick-end labeling staining
Precision count beads (BioLegend) were used to obtain absolute counts of leukocytes. TdT-mediated dUTP nick-end labeling (TUNEL) staining was performed using the DeadEnd TUNEL System (Promega, Madison, WI, USA).
Assessments of intestinal permeability
Intestinal permeability was examined using a FITC-dextran method.
Drug administration
Corticosterone was injected via the tail vein twice daily for two days. Mice were dosed with RU486 by intraperitoneal injection at 30 min post CA/CPR, followed by a once-daily injection for two days.
Immunohistochemistry, histology, and microscopy
Immunofluorescence staining was performed on frozen tissues. Paraffin-embedded spleen and thymus sections were used for hematoxylin-eosin (H&E) staining. Images were captured on a Zeiss microscope.
Bacteriologic analysis and functional outcome
The evaluations were performed by observers who were blinded to group assignment. Bacterial load in the lung was quantified by counting bacterial colonies. A nine-point scoring system and the open field test were used to evaluate the functional outcome after CA.9,10
Statistical analysis
Sample sizes were determined based on previous publications and our preliminary data using StatMate (GraphPad, LaJolla, CA, USA). Data were analyzed using Prism 8 software (GraphPad). Data are presented as mean ± SD or median. The unpaired Student’s t-test or Mann–Whitney U test was used to compare the two groups. To compare more than two groups, one-way or two-way ANOVA with post hoc Holm–Sidak correction for multiple comparisons was performed. The level of significance was set at p < 0.05.
Results
CA/CPR induces substantial immune responses in the brain
It is known that shortly after CA and resuscitation, a systemic inflammatory response occurs.3,11 To confirm and also expand these critical observations in our mouse CA/CPR model, we first measured the levels of three pro-inflammatory cytokines (IL-6, IL-1β, and TNF-α) in the blood by ELISA (Figure S1). In agreement with the previous findings, levels of all three cytokines in the serum were almost undetectable in sham-operated mice, but were markedly elevated on day 1 after CA/CPR. Further, levels of these cytokines were significantly higher in the proximal and middle small intestine of CA versus sham animals (Figure S1). Additionally, we examined intestinal permeability and found significantly higher serum levels of FITC-dextran in the CA mice at 6 h reperfusion, compared to sham-operated mice (Figure S2), indicating an increase in intestinal permeability. This increase was transient, however, as no significant difference was found between sham and CA groups at 24 h reperfusion. Together, using our CA/CPR model, we confirmed that CA/CPR leads to a systemic inflammatory response in the acute phase.
Considering that there is the critical reciprocal communication between the immune system and the central nervous system (CNS) and that both systems are impacted by the whole-body ischemia/reperfusion insult after CA/CPR, we conducted a detailed analysis on the immune response in the brain. The ELISA data showed that levels of all three pro-inflammatory cytokines (IL-6, IL-1β, and TNF-α) were significantly increased on day 1 in the post-CA brain (Figure 1(a)). Then, we analyzed the effects of CA/CPR on resident and infiltrating immune cells in the brain. The immune staining analysis using anti-Iba1 (phagocytes) and anti-GFAP (astrocytes) indicated marked activation of resident immune cells in post-CA brains (Figure 1(b) and (c)). Peripheral leukocytes in the brain can be identified based on high expression levels of CD45 (CD45+hi), 12 compared to intermediate expression of CD45 in resident immune cells (Figure 1(d)). We found that the number of CD45+hi cells was about three times higher in post-CA brains than in sham-operated brains. Among these CD45+hi cells, approximately 80% were CD11b+ cells, indicating that most of the infiltrating immune cells were monocytes/macrophages and neutrophils (Figure 1(e)), while no significant change in T and B cells was detected in post-CA brains on day 3 after CA/CPR (Figure S3).
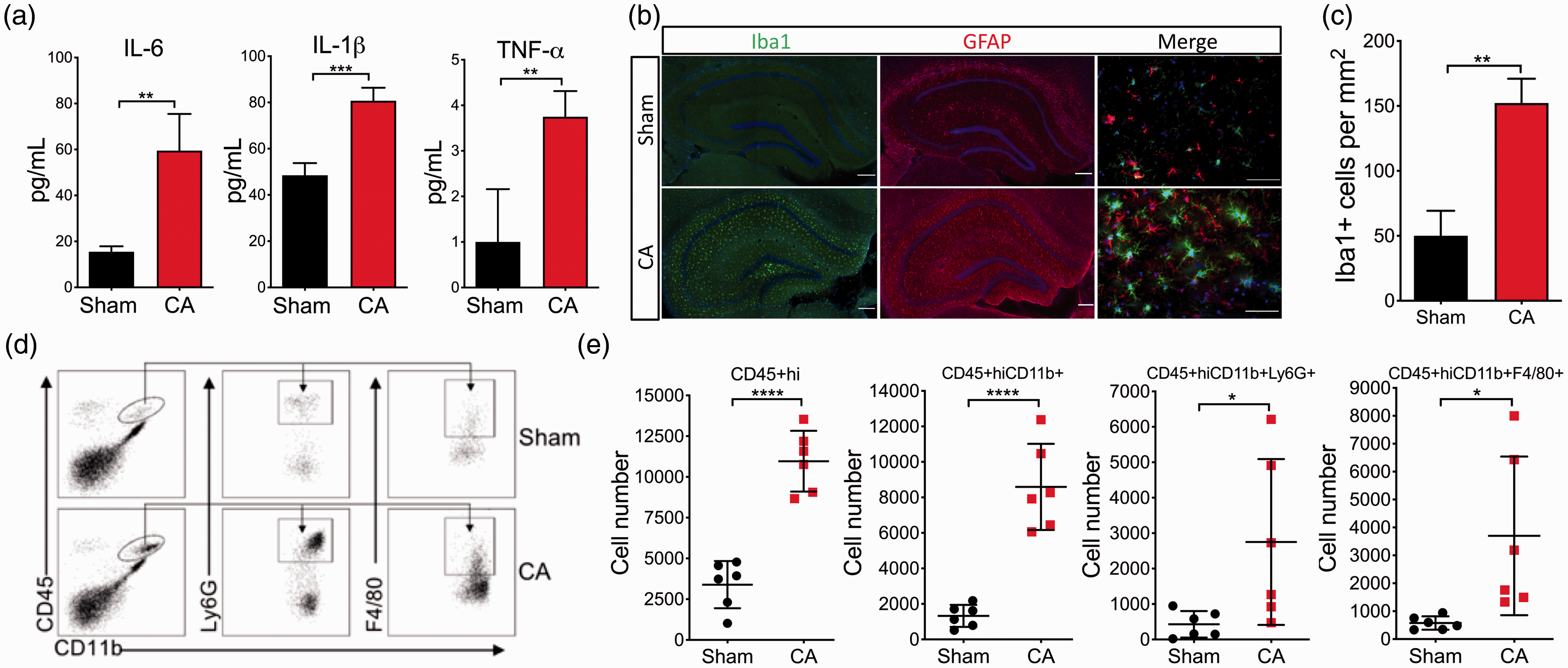
Immune responses in the brain after CA/CPR. Mice were subjected to 8.5 min CA or sham surgery: (a) levels of pro-inflammatory cytokines in the brain on day 1 after CA/CPR. The levels of three pro-inflammatory cytokines, including IL-6, IL-1β, and TNF-α, were measured with ELISA; (b–e) the cellular immune response in the brain was evaluated on day 3 after CA/CPR; (b) representative images of immunofluorescence staining for Iba1 (green; microglia/phagocytes) and GFAP (red; astrocytes); (c) quantification of densities of Iba1 positive cells in the brain (n = 3/group). (d) representative gating strategy for flow cytometric analysis of infiltrating leukocytes in the brain after CA/CPR; (e) quantification of infiltrating leukocytes (CD45+hi) and CD45+hiCD11b+ cells with their subsets, neutrophils (CD45+hiCD11b+Ly6G+), and monocytes/macrophages (CD45+hiCD11b+F4/80+), in the whole brain after CA/CPR (n = 6/group). Data are presented as mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
CA/CPR activates the HPA axis
The CA-induced neuroinflammation in the brain, as shown above, is expected to modulate the immune system. A major component in the communication between the CNS and the immune system is the HPA axis (a neuroendocrine pathway; Figure 2(a)). Notably, the HPA axis can be activated by neuroinflammatory responses in the brain. 13 Therefore, we further examined expression of pro-inflammatory cytokines locally in the hypothalamus (a key element of the HPA axis) after CA/CPR and found a significant increase in the mRNA levels of TNF-α and IL-1β (Figure 2(b)), two cytokines capable of activating the HPA axis by stimulating the hypothalamus. 14 Indeed, the levels of serum corticosterone after CA/CPR gradually increased, and the fold of this increase reached almost seven on day 3 of reperfusion (Figure 2(c)). Finally, plasma ACTH levels on day 3 after CA/CPR were found to be significantly elevated (Figure 2(d)). These findings together prove activation of the HPA axis after CA/CPR.
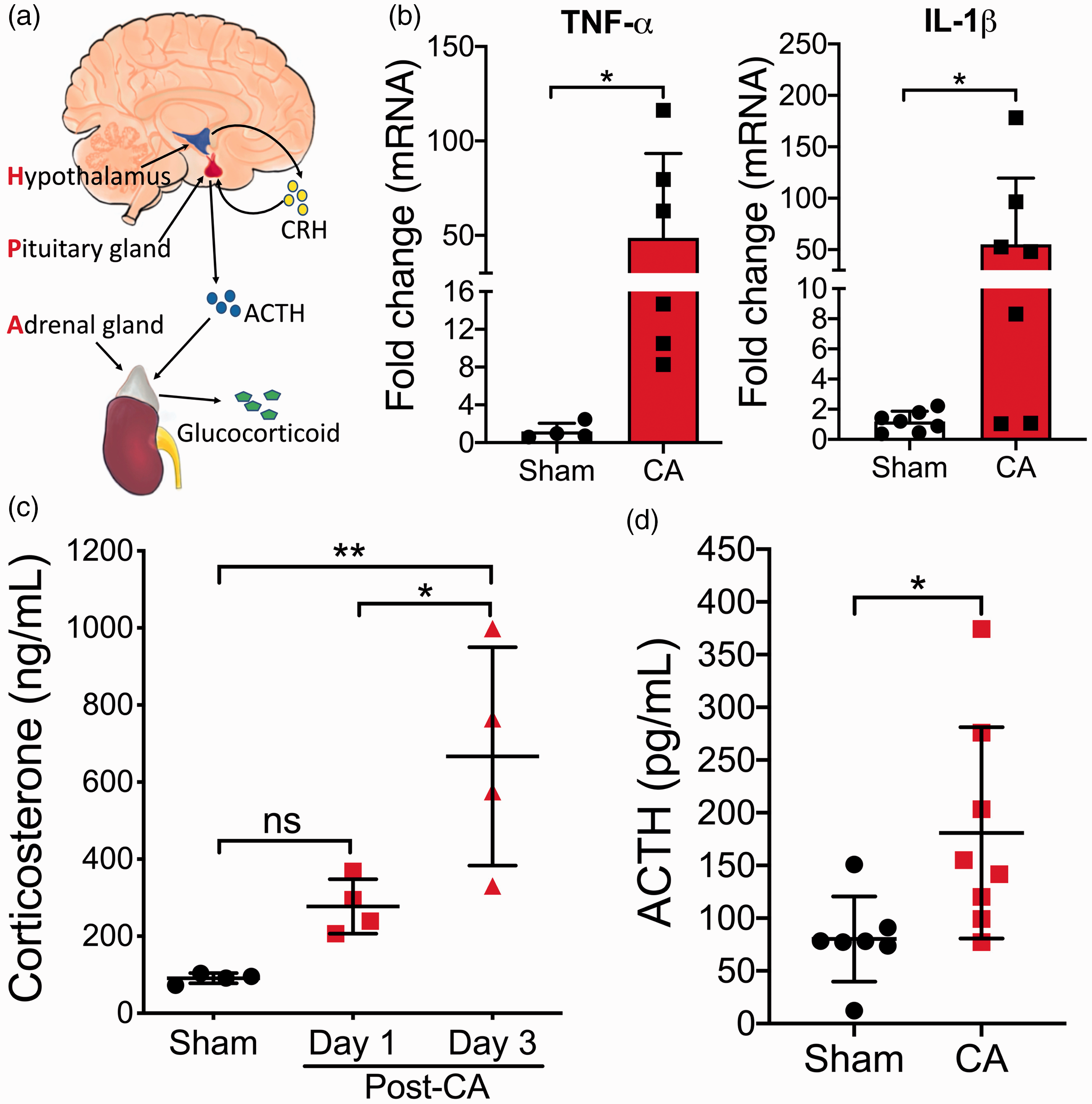
CA/CPR activates the HPA axis: (a) the HPA axis; (b–d) mice were subjected to 8.5 min CA or sham surgery; (b) the mRNA levels of TNF-α and IL-1β in the hypothalamus were analyzed (n = 4–7/group) on day 3 after CA/CPR; (c) serum levels of corticosterone were measured on days 1 and 3 after CA/CPR (n = 4/group); (d) plasma levels of ACTH were measured on day 3 after CA/CPR (n = 7–8/group). Data are presented as mean ± SD. *, p < 0.05; **, p < 0.01; ns, not significant.
CA/CPR leads to severe immunosuppression
Since glucocorticoid (e.g. corticosterone in mice) can exert potent immunosuppressive effects, 15 we speculated that CA/CPR-activated HPA axis may cause systemic immunosuppression. To test this, we analyzed the cellularity, morphology, and function of major immune organs including the spleen, thymus, blood, and PPs (a gut-associated lymphoid tissue) after CA/CPR.
First, we isolated spleens and thymi from sham and CA mice at 6, 24, and 72 h after CA/CPR and found that both spleens and thymi displayed an obvious reduction in size on day 3 after CA/CPR (Figure 3(a)). The thymus showed the most marked shrinkage, with more than 50% loss in tissue weight on day 3 after CA/CPR (Figure 3(b)). Compared to the extent of atrophy, cellularity in both the spleen and thymus appeared even more severely reduced after CA/CPR, with cell counts down to less than 20% of controls (Figure 3(c)). Histologic analysis showed marked depletion of lymphoid tissue, especially white pulp, in the spleen and structure disruption in the thymus, including reduced cortex and vanishing boundary between cortex and medulla, after CA/CPR (Figure 3(d)). These dramatic changes were, at least in part, caused by apoptotic cell death, since TUNEL-positive cells were significantly increased in both the spleen and thymus after CA/CPR (Figure 3(e,f)). Consistently, we also discovered that on day 3 after CA/CPR, the leukocyte count in the blood was significantly lower (sham: 3.03 ± 0.34 vs. CA: 1.16 ± 0.22 × 106 cells/mL; p = 0.002; Figure 3(g)), and a dramatic reduction in the number of cells in PPs (mostly immune cells) was noted (Figure 3(h)).
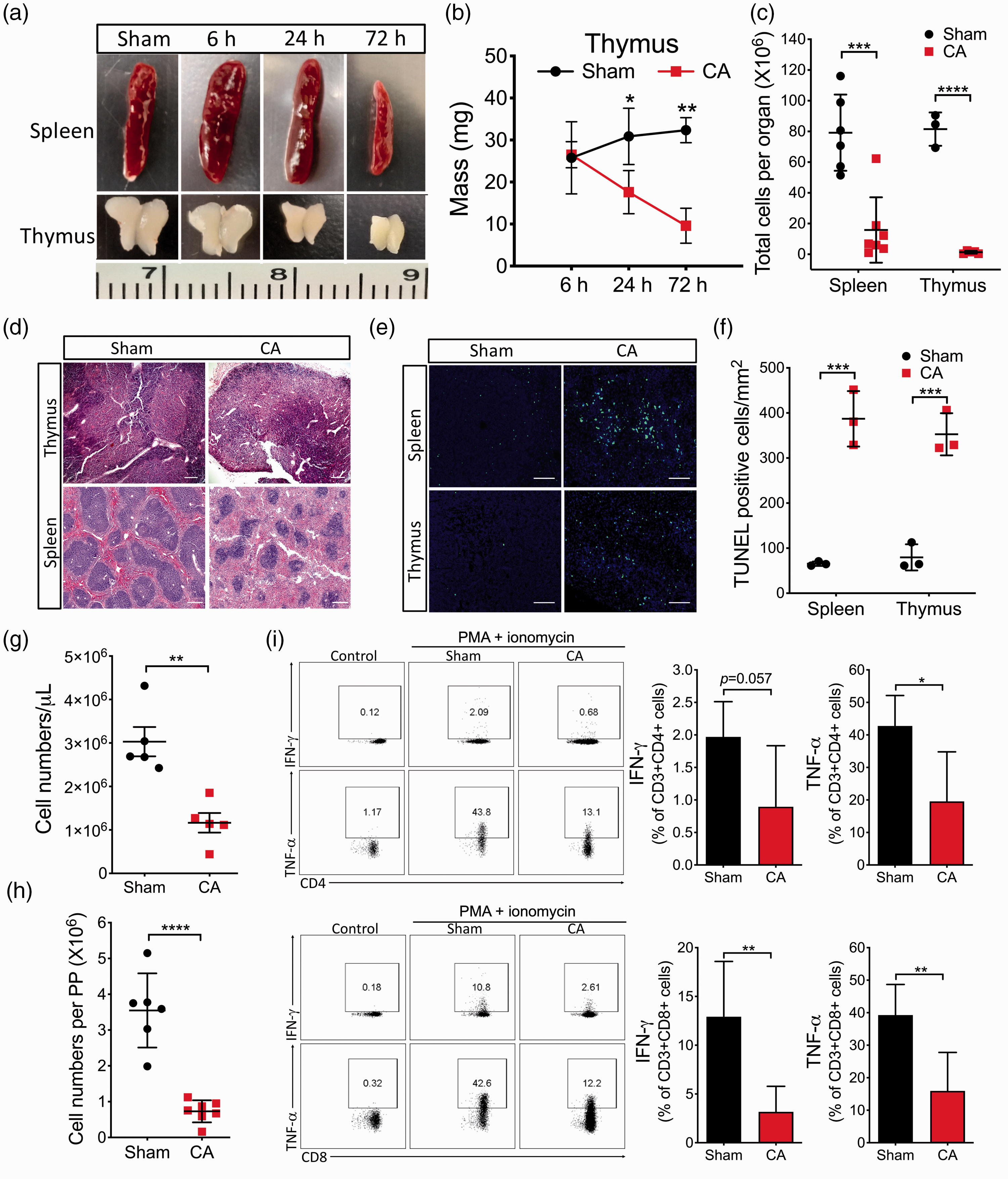
CA/CPR results in immunosuppression. Mice were subjected to 8.5 min CA or sham surgery: (a–c) atrophy of the spleen and thymus after CA/CPR; (a) representative images of spleens and thymi at different time points after CA/CPR; (b) tissue weight of thymi after CA/CPR (n = 3–4/group); (c) total cell numbers in spleens and thymi on day 3 after CA/CPR (n = 3–7/group); (d) representative images of H&E-stained sections of spleens and thymi on day 3 after CA/CPR; (e) representative fluorescence images of TUNEL staining sections of spleens and thymi on day 3 after CA/CPR; (f) quantification of TUNEL-positive cell density in spleens and thymi (n = 3/group); (g,h) leukocyte counts in the blood and cell numbers in Peyer’s patches (PPs) on day 3 after CA/CPR (n = 5–7/group); (i) cytokine production in T splenocytes by intracellular cytokine staining. On day 3 after CA/CPR, splenocytes were collected and stimulated with PMA and ionomycin (n = 5/group). Data are presented as mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
It is critical to assert that these striking changes in the morphology and cellularity occurred in response to CA/CPR, rather than to the reagents used in our CA model. Thus, we first measured blood KCl concentration before and after CA/CPR and found its rapid return to baseline (4.47 ± 0.31 vs. 4.93 ± 0.59 mmol/L: before CA/CPR vs. 15 min after resuscitation). Then, we treated healthy mice with either KCl or epinephrine at the same dose used in the CA model, but via slow infusion, and analyzed the sizes of spleens and thymi and immune cell compositions with flow cytometry (the main gating strategy outlined in Figure S4). There was no obvious effect observed by either reagent treatment (Figure S5). Last, we examined another murine CA/CPR model, the asphyxial CA model in which no reagent was used to induce CA. Supportively, massive cell loss in the spleen and, particularly, in the thymus was observed after asphyxia CA/CPR (Figure S6). Collectively, these results strongly corroborate the cause and effect relationship between CA/CPR and lymphopenia and atrophy of immune organs.
Next, to determine the extent to which cellular functions of the remaining immune cells in lymphoid organs were also negatively affected after CA/CPR, we used a common ex vivo approach, that is, evaluating the ability of T splenocytes to produce cytokines in response to PMA/ionomycin stimulation by intracellular cytokine flow cytometry. Our data demonstrated that production of both IFN-γ and TNF-α was significantly less in T cells collected from CA animals than from sham animals (Figure 3(i)).
Finally, since this study is focused on the cellular changes in the immune system after CA/CPR and there is the lack of a systemic characterization of how CA/CPR affects the frequency of immune cell subtypes, we decided to comprehensively examine the effects of CA/CPR on major immune cell populations. CA-induced temporal composition changes were first evaluated in the spleen (Figure S7). In this experiment, we included a naive group of mice without any surgery to establish baselines. As shown in Figure S7A, there was no significant difference between naive and sham groups, indicating negligible effect of surgical stress on the immune response. After CA/CPR, however, the percentage of B cells in the spleen significantly decreased over the first three days (from baseline ∼70% to ∼50%) and returned to baseline on day 5 (Figure S7B), while the percentages of CD3+ T, CD69+ T (activated T cells), and neutrophil cells increased on day 3 (Figure S7B). Since it appears that these changes in the spleen peaked on day 3 after CA/CPR, we chose this time point for the following detailed analysis (Figures 4 and S8). Consistent with a profound overall decrease in leukocyte counts in the immune organs examined (Figure 3), the absolute numbers of all major immune cell populations investigated were markedly reduced in the spleen after CA/CPR (Figure 4(a)). However, changes in the frequency of each population were different. The percentages of T cells in leukocytes were significantly increased in the spleen and PPs on day 3 after CA/CPR (Figures 4(b) and S8). In contrast, the percentages of B cells decreased in all three organs analyzed, particularly in the blood where the percentage markedly declined from 64.50% ± 2.02% (sham) to 13.69% ± 5.63% (CA) (Figure S8A). Notably, the percentages of neutrophils increased in all CA samples (Figures 4(b) and S8). Thus, our data indicated marked alterations in the composition of immune cells in immune organs after CA/CPR.
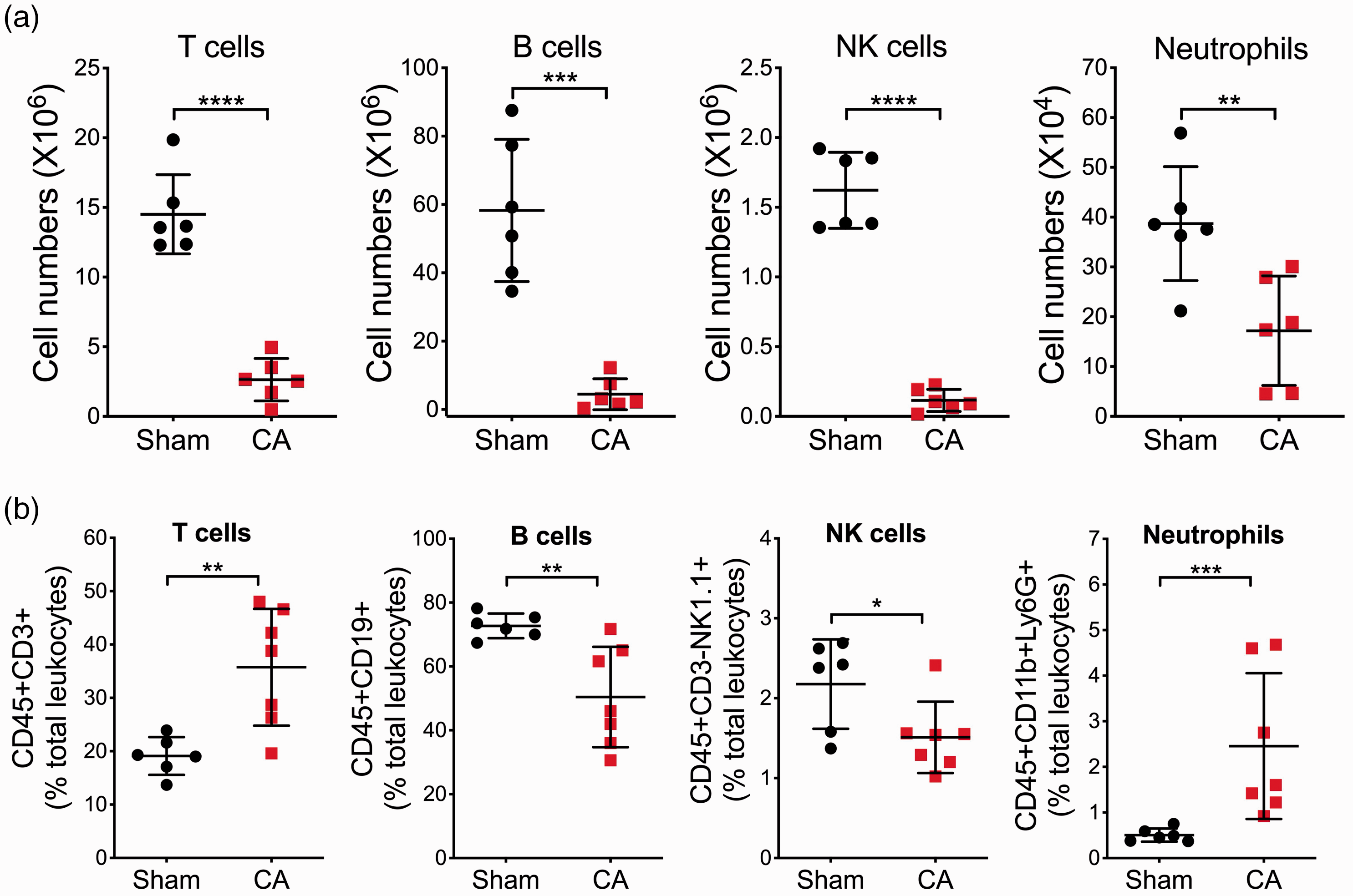
CA/CPR induces marked changes in the number and frequency of major splenocyte subsets. Mice were subjected to sham or 8.5 min CA. On day 3 post-CA, single-cell suspensions were prepared from the spleen and then subjected to flow cytometric analysis. The following leukocyte subpopulations were shown here: T cells (CD45+CD3+), B cells (CD45+CD3–CD19+), natural killer cells (NK; CD45+CD3–NK1.1+), and neutrophils (CD3–CD11b+Ly6G+): (a) cell numbers; (b) relative percentage changes of immune cell populations. Data are presented as mean ± SD (n = 6-7/group). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Taken together, we provide strong evidence that CA/CPR caused leukopenia in the circulation, drastic atrophy and profound cell loss in lymphoid organs including PP, spleen, and thymus, and impaired function of the remaining immune cells, which collectively manifests an immunosuppression phenotype in the subacute phase after CA/CPR.
Post-CA immunosuppression is mediated by activation of the HPA axis
We then set out to determine the mechanistic link between the post-CA immunosuppression and the HPA axis. First, we hypothesized that if elevated glucocorticoid concentrations directly contribute to post-CA immunosuppression, post-CA treatment with corticosterone would exacerbate lymphopenia. To test this hypothesis, we used a subthreshold CA duration (7 min CA) after which serum levels of corticosterone were not markedly elevated compared to our standard CA duration (8.5 min) (Figure 5(a)). Further, unlike 8.5 min CA, 7 min CA did not lead to obvious leukocyte depletion and lymphoid organ atrophy on day 3 post-surgery (Figure 5(b) and (c)), suggesting a potential correlation between the extent of post-CA immunosuppression and the severity of CA injury. Then, we treated 7-min CA mice with corticosterone or vehicle for two days starting on postoperative day 1. Remarkably, the corticosterone-treated mice almost phenocopied the immunosuppression observed in 8.5-min CA mice (Figure 5(b) and (c)). By contrast, no obvious effect was observed in sham mice treated with corticosterone, indicating that corticosterone at this dose alone is not sufficient to lead to a noticeable immunosuppression phenotype in mice without CA/CPR.
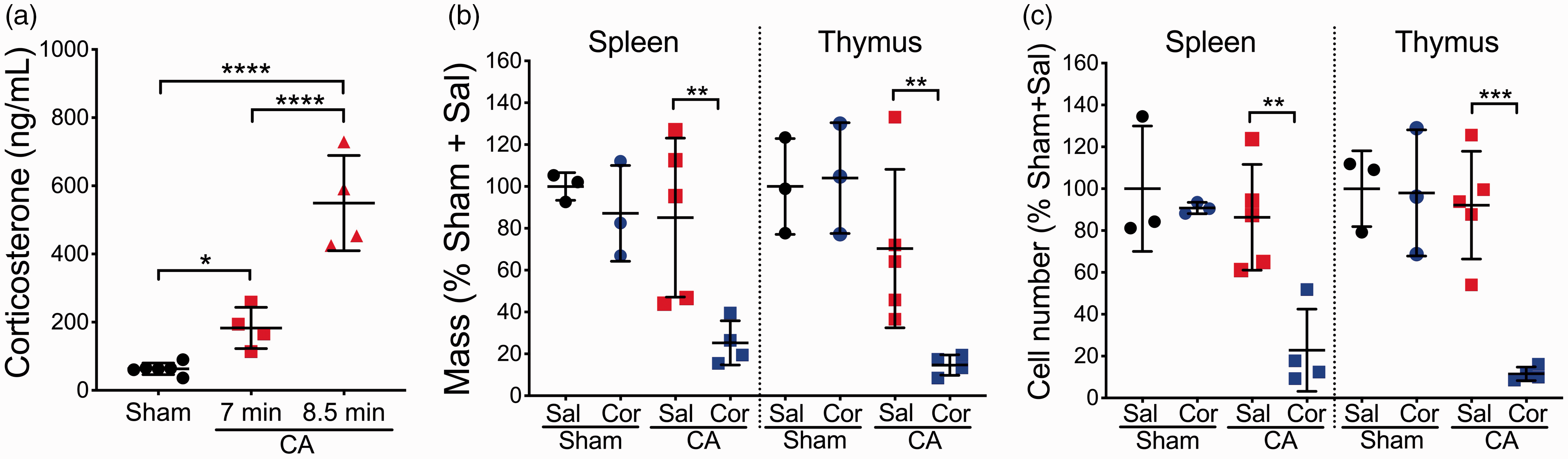
Glucocorticoid contributes to CA/CPR-induced immunosuppression: (a) mice were subjected to 7 min CA, 8.5 min CA, or sham surgery. Serum levels of corticosterone were measured on day 3 after CA/CPR (n = 4–6/group); (b,c) effects of corticosterone treatment on immunosuppression. Mice (n = 3–6/group) were subjected to 7 min CA or sham surgery. Mice were dosed with saline (Sal) or corticosterone (Cor). On day 3 after CA/CPR, organ weight (b) and the total cell numbers of spleen and thymus (c) were assessed. Data are presented as mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
To further substantiate the link, we used the glucocorticoid receptor antagonist RU486 to suppress the effects of the activated HPA axis and examined its effect on the post-CA immunosuppression (Figure 6). In line with our reasoning, splenic atrophy and immune cell depletion were significantly mitigated in the RU486-treated mice after CA/CPR (Figure 6(a) and (b)). Consistently, the numbers of all examined immune cells were dramatically decreased on day 3 after CA/CPR in the vehicle group; by contrast, RU486 treatment significantly prevented the massive declines (Figures 6(c) to (f) and S9). Of note, RU486 treatment had no obvious effect on the composition and numbers of immune cells in sham mice (Figure S10).
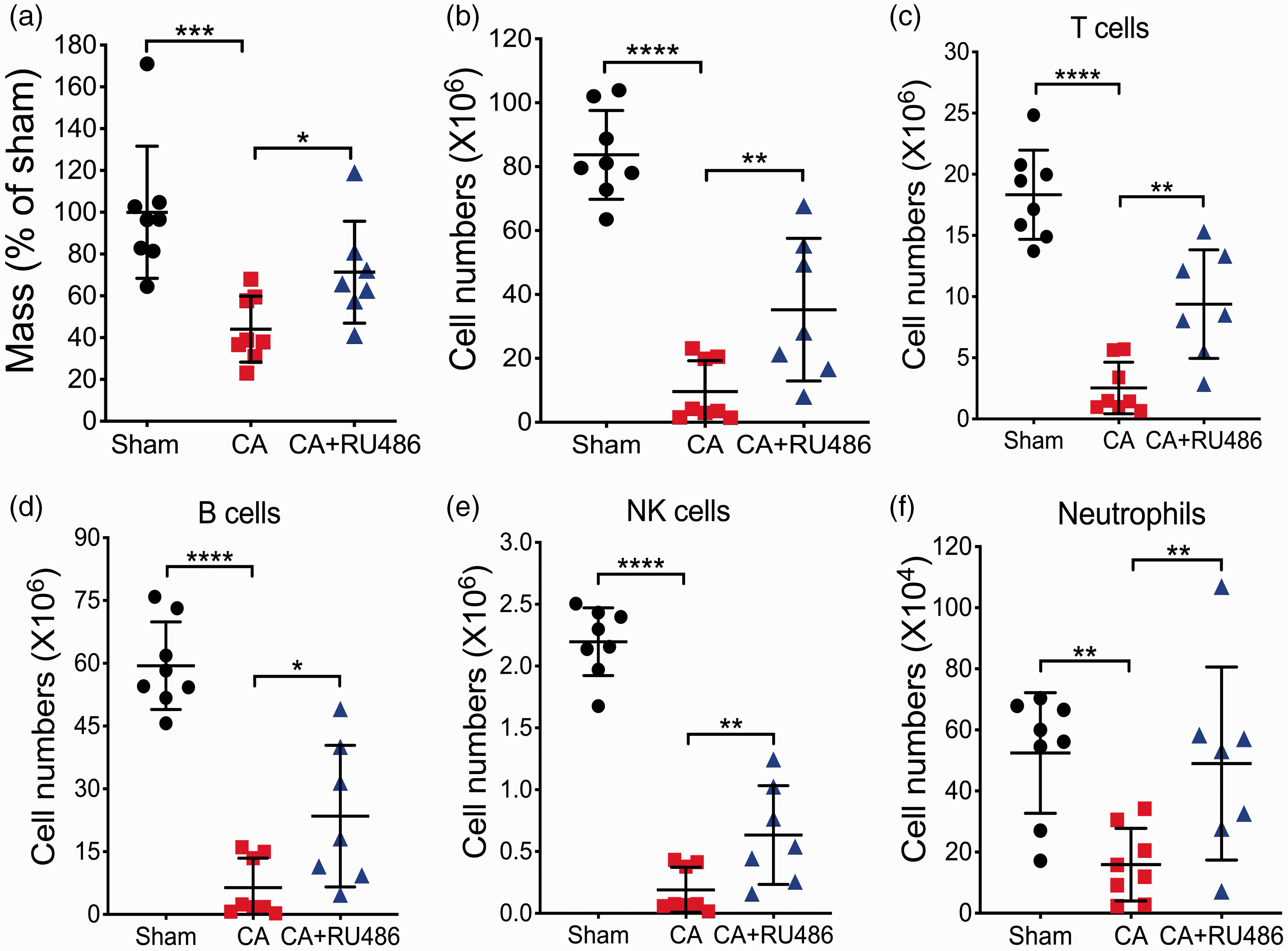
Suppression of the HPA axis alleviates CA/CPR-induced immunosuppression. Mice were subjected to 8.5 min CA or sham surgery and then treated with vehicle or RU486. On day 3 after CA/CPR, spleen tissues were weighed (a) and the numbers of total cells (b) and major immune cell populations (c–f) in spleens were counted or analyzed by flow cytometry. Data are presented as mean ± SD (n = 7–8/group). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Finally, we asked whether RU486 treatment can improve CA outcome and restore impaired immunity (Figure 7). Interestingly, RU486- versus vehicle-treated CA mice performed significantly better in both functional scoring and the open field test (Figure 7(a) and (b)). Our pilot study indicated that no bacterial colonization was normally found in the lung in both vehicle- and RU486-treated sham mice. However, we found that most vehicle-treated CA mice displayed increased bacterial numbers in the lung (Figure 7(c)), suggesting that CA mice were highly susceptible to spontaneous pneumonia. In contrast, RU486-treated CA mice showed significantly reduced pulmonary bacterial counts (Figure 7(c)), thus suggesting a link between HPA axis-mediated immune dysregulation and increased susceptibility to bacterial infection.
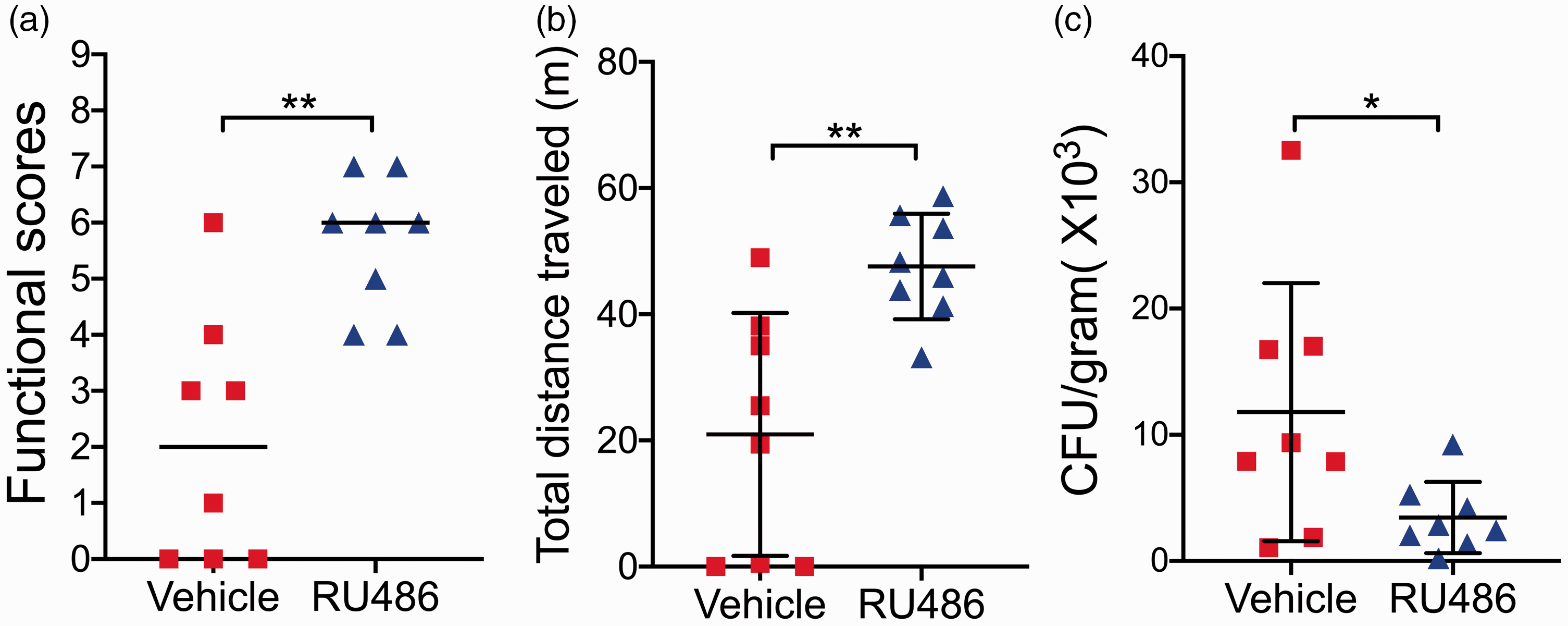
Suppression of the HPA axis improves CA outcome. A cohort of mice were subjected to 8.5 min CA or sham surgery and then treated with vehicle or RU486. Functional scoring (a) and the open field test (b) were performed on day 3 after CA/CPR. After behavioral tests, bacterial load in the lung was assessed (c). Data are presented as mean ± SD or median (n = 8/group). *p < 0.05; **p < 0.01.
Discussion
Understanding how the immune system responds to CA/CPR is essential to improving post-resuscitation care. Here, we provide, to our knowledge, the first direct experimental evidence that CA/CPR results in immunosuppression, as demonstrated by a massive loss of immune cells in the circulation and the immune organs, and functional impairment in the remaining immune cells. Further, our data indicated that this impaired immunity was mediated, at least in part, by the HPA axis, which is activated by inflammatory response in the post-CA brain, and alleviating immunosuppression via targeting the HPA axis improved functional outcome after CA/CPR.
Critically, the immune responses in our CA/CPR mouse model, as revealed in this study, recapitulate central aspects of the established clinical immune phenotype. Most notably, we found that after CA/CPR, pro-inflammatory cytokine levels were significantly increased in the blood in the acute phase, followed by a marked decrease in the total number of blood leukocytes together with increased serum levels of corticosterone in the late phase. We also observed that gut permeability was increased after CA/CPR. These findings align well with clinical reports. It has been reported that serum levels of IL-6 and TNF-α are significantly increased in CA patients on admission; 3 lymphopenia was diagnosed in 40% of a cohort of mixed in-hospital and OHCA patients; 5 cortisol concentrations in the blood of CA patients roughly double within 24 h after ROSC; 16 and urinary concentration of intestinal fatty acid-binding protein is much higher in CA patients on admission, indicating impaired intestinal permeability. 17 Taken together, the data presented here are expected to have important clinical implications.
By using an experimental CA model, we were able to expand on previous clinical findings by characterizing post-CA changes in cytokines and immune cell populations in various organs, including the brain. To date, only few studies have reported on the immune response in the post-CA brain.18,19 These studies found a flux of peripheral immune cells into the brain after CA/CPR, but it remains to be clarified as to which populations of immune cells infiltrate the brain. For example, Deng et al. showed that most of the infiltrating lymphocytes are CD4+ T cells, while Zhang et al. found no increase in CD4+ T cells.18,19 In the present study, we confirmed infiltration of peripheral immune cells into the post-CA brain; however, on day 3 after CA/CPR, we did not find any significant change in T cells, but instead most of the infiltrating immune cells were monocytes/macrophages and neutrophils. This finding is consistent with the notion that ischemia-induced sterile inflammation is characterized by recruitment of phagocytic leukocytes. Of note, we used a well-established flow cytometry-based strategy to identify the infiltrating immune cells in the post-CA brain.12,20–22 In the very first paper describing this strategy, the authors provide strong evidence that CD45+hi cells are infiltrating peripheral leukocytes. They also demonstrated that neuroinflammation increases CD45 expression in the resident immune cells, but even with this increased expression, the population of infiltrated peripheral immune cells can still be clearly separated from the resident cells. 12 However, in future studies that are focused on neuroinflammation in the post-CA brain, other approaches, such as fluorescent labeling of peripheral cells, should be exploited to further clarify and virtualize what immune cells infiltrate into the brain after CA/CPR.
The key finding in this study is that CA/CPR resulted in immunosuppression after CA/CPR. Previous clinical research has provided some evidence suggesting a dysfunctional immune system in CA patients, which may predispose them to post-CA infections.4,5,23 Our current study constitutes a crucial step toward a better understanding of CA-induced impairment of the immune system. First, we found that total numbers of immune cells in the blood and in major immune organs (spleen, thymus, and PPs) were dramatically decreased after CA/CPR. This loss of immune cells is expected to negatively impact the capacity of both innate, and in the long run, adaptive immune responses and thus account for increased susceptibility to infections after CA/CPR. Indeed, in a retrospective, single-center study, Villois et al. 5 reported that CA patients with lymphopenia more often developed infection during their hospital stay. Second, our data demonstrated that the drastic reduction in cellularity and gross morphology in the spleen and thymus was due in part to increased apoptotic cell death. The etiology of apoptosis in these immune organs is not yet known. Clarifying the responsible mechanism(s) may inform development of anti-apoptotic strategies in CA treatment.
Third, we investigated the functional capacity of the immune cells that remain after CA/CPR. The capacity of leukocytes to produce cytokines has been commonly used as an indicator of immune functions. This capacity appears to be impaired in CA patients, as demonstrated by reduced production of cytokines after stimulating whole blood with bacterial antigens.4,23 However, this approach did not consider the fact that lymphopenia is common in CA patients 5 and that less cytokine production may be due to fewer lymphocytes in CA patients. Thus, we used a quantitative flow cytometry approach with intracellular cytokine staining, and our data support the notion that the remaining immune cells display suppressed immune function after CA/CPR.
More importantly, our work provides evidence that post-CA immunosuppression primarily involves the HPA axis, which may play a pivotal role in spontaneous pneumonia after CA/CPR. We first confirmed activation of the HPA axis after CA/CPR, which is likely due to the increased pro-inflammatory cytokines in the post-CA brain. A massive increase in corticosterone observed on day 3 after CA/CPR led us to then speculate that this dramatic release of glucocorticoid via the HPA axis could contribute to CA-induced systemic immunosuppression. Indeed, post-CA treatment with glucocorticoid exacerbated immunosuppression, while administration of glucocorticoid receptor antagonist RU486 markedly mitigated cell loss and atrophy in the spleen and thymus. Finally, we found that RU486 treatment reduced the bacterial counts in the lungs and improved functional outcome after CA/CPR. Collectively, our data indicate that during the subacute phase, dysregulation of the HPA axis is a major mechanism responsible for post-CA leukopenia and increased susceptibility to bacterial infections. Interestingly, dysregulation of the HPA axis after CA/CPR has been reported before. 24 This previous work showed that the response ability of the HPA axis is impaired during the late stage (i.e. 15–20 days) after CA/CPR.
Our results together demonstrate that modulation of the HPA axis could be a viable therapeutic strategy to improve immune status and thus reduce risk for developing infections in post-CA patients. However, it must be noted that in the clinic, the role of the HPA axis in CA is a subject of debate. This is clearly reflected by recent critical care guidelines that suggest the use of corticosteroids in the setting of CA with very low quality of evidence. 25 However, it seems that glucocorticoid use during CPR can improve the rate of resuscitation from CA,26,27 which may be due to its favorable effects on hemodynamic stability. While this hyperacute glucocorticoid use may be beneficial, our findings indicate that stimulation of the HPA axis in the late phase may exacerbate immune dysfunction and increase risk for infection, leading to worse CA outcome. Therefore, the HPA axis plays distinct roles at different stages of CA pathophysiology, which should be harnessed accordingly for the treatment of CA patients.
The features of post-resuscitation immune dysfunction revealed here exhibit many similarities to immunosuppressive phenotypes defined in other diseases such as sepsis, stroke, and spinal cord injury.6,28–31 Since this phenotype has been closely associated with an increased risk for infection and poor prognosis in patients,6,29,30 much attention has been directed to understand the mechanisms underlying this phenotype in various diseases. Notably, despite apparently similar manifestations of immunosuppression, such as lymphopenia and lymphoid atrophy, each disease has its unique etiology and pathophysiology, and consequently, the mechanisms underlying disease-induced immunosuppression can be distinct. For example, data presented here and from others indicate that the HPA axis is critically involved in lymphopenia after CA and ischemic stroke, 28 while this mechanism is less clear for spinal cord injury and sepsis.30,31 Further, there is no strong evidence that suppressing the HPA axis with RU486 alone can improve neurologic outcome and reduce bacterial colonization in the lung after stroke, which is in contrast to our RU486 data in CA. Together, these important differences highlight that detailed mechanistic studies are required to fully understand the immunosuppressive phenotype observed in individual critical illness.
Some limitations are noted in our study. Using our animal model, we have showed that CA/CPR resulted in neuronal death in the brain and neurologic deficits.9,32 But, we did not extensively examine these outcomes in the present study because this study was aimed to dissect how the immune response is altered after CA/CPR during the acute and subacute phases. However, our data indeed uncovered a potential link between neuronal injury and immunosuppression after CA. This critical aspect warrants further investigation. More, we understand that the partial rescue of the CA-induced immunosuppression phenotype by RU486 may suggest involvement of other mechanisms. For example, the sympathetic nervous system (SNS), another neuroendocrine pathway, may be involved, as the SNS has been implicated in the immunosuppression phenotype observed in stroke and spinal cord injury.28,31 Other possible mechanisms could be related to cell-mediated regulation of the immune system. For example, regulatory T (Treg) and myeloid-derived suppressor cells (MSDC) possess strong immunosuppressive properties.30,33 Future studies could be designed to explore these potential mechanisms.
In summary, we propose that CA/CPR activates the HPA axis, which leads to profound suppression in cellular immune responses in the subacute phase. CA-induced immunosuppression could be a protective response to limit adverse effects of an overactivated immune response that occurs in the acute phase after CA/CPR. On the other hand, our data suggest that post-CA immunosuppression likely drives the development of post-CA infectious complications and poor prognosis in CA patients at the late stage. Although such a determination will require more experimental and clinical evidence, disease-induced immunosuppression has been increasingly recognized as a key contributor to poor outcomes and increased healthcare costs in ICU patients and may be used a criterion to stratify patients for personalized treatment. This has led to extensive on-going efforts to profile the immune status in critical illnesses including septic shock and severe trauma. 34 Our data clearly underscore that CA also deserves such clinical effort.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X20948612 - Supplemental material for Cardiac arrest and resuscitation activates the hypothalamic-pituitary-adrenal axis and results in severe immunosuppression
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X20948612 for Cardiac arrest and resuscitation activates the hypothalamic-pituitary-adrenal axis and results in severe immunosuppression by Qiang Zhao, Yuntian Shen, Ran Li, Jiangbo Wu, Jingjun Lyu, Maorong Jiang, Liping Lu, Minghua Zhu, Wei Wang, Zhuoran Wang, Qiang Liu, Ulrike Hoffmann, Jörn Karhausen, Huaxin Sheng, Weiguo Zhang and Wei Yang in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Author's note
Qiang Liu is now affiliated with Department of Neurology, Tianjin Medical University General Hospital, Tianjin, China.
Acknowledgements
We thank Pei Miao for her excellent technical support and Kathy Gage for her excellent editorial contributions.
Authors’ contributions
Conceived and supervised the study and wrote the manuscript: UH, JK, WZ, WY. Designed and performed the experiments: QZ, YS, JW, RL, JL, MJ, MZ, WW, LL, ZW, HS. Analyzed the data: QZ, YS, JW, RL, JL, MJ, MZ, WW, LL, LQ, UH, JK, WZ, WY.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Department of Anesthesiology (Duke University Medical Center), American Heart Association grants (16GRNT30270003, 18CSA34080277), and National Institutes of Health grant (R56HL126891).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.