
Abstract
Stroke-induced cerebral ischemia is a major cause of death and disability. The disruption of blood flow results in neuronal and glial cell death leading to brain injury. Reperfusion restores oxygen to the affected tissue, but can also cause damage through an enhanced oxidative stress and inflammatory response. This study examines mitochondrial transfer from MSC to neurons and the role it plays in neuronal preservation after oxidant injury. We observed the transfer of mitochondria from MSC to mouse neurons in vitro following hydrogen peroxide exposure. The observed transfer was dependent on cell-to-cell contact and led to increased neuronal survival and improved metabolism. A number of pro-inflammatory and mitochondrial motility genes were upregulated in neurons after hydrogen peroxide exposure. This included Miro1 and TNFAIP2, linking inflammation and mitochondrial transfer to oxidant injury. Increasing Miro1 expression in MSC improved the metabolic benefit of mitochondrial transfer after neuronal oxidant injury. Decreasing Miro1 expression had the opposite effect, decreasing the metabolic benefit of MSC co-culture. MSC transfer of mitochondria to oxidant-damaged neurons may help improve neuronal preservation and functional recovery after stroke.
Introduction
Ischemic stroke is a leading cause of death and disability and accounts for approximately 1 in 20 deaths in the United States. 1 These events disrupt blood flow and can result in neuronal and glial cell death in many brain regions.2,3 While reperfusion is important to spare viable brain tissue, it also causes tissue damage due to an enhanced inflammatory response. 4 Many studies have investigated the use of MSC transplant to improve neurologic outcome following cerebral ischemia, but this work has been limited to studying the effects of MSC on inflammation or as potential replacements for neurons.5–10 MSC can impact inflammation and cell death via several mechanisms, including the release of trophic factors, exosomes, etc. An additional mechanism recently reported includes the potential of MSC to donate mitochondria via intercellular transport mechanisms to recipient cells.11–24 In many cases, the donated mitochondria have been shown to improve the health of cells damaged by inflammation or ischemia.11,12,22
Mitochondria play a unique and important role in the central nervous system. The majority of cerebral ATP consumption is used for the electrogenic activity of neurons, and mitochondria provide over 90% of total cellular ATP via oxidative phosphorylation.25,26 The localization of mitochondria within a neuron is important for local ATP availability and the dynamics of intracellular Ca2+ signaling. 26 Given the highly polarized structure and length of the axons of neurons, mitochondrial transport and signaling are crucial for the life and function of these cells, and as such, are highly regulated.27–32 Intracellular mitochondrial transport is mediated via energy-dependent molecular motors that transport mitochondria along cytoskeletal filaments. 33 Miro1, a mitochondrial outer membrane GTPase, is a key motor protein adaptor and regulator of mitochondrial trafficking.26,34 Miro1 knockout is lethal, with animals dying shortly after birth, while its conditional knockout in neurons results in defects in axonal mitochondrial transport that is lethal in affected neurons, even while mitochondrial respiratory function and Ca2+ buffering remain unchanged (showing that defects in mitochondrial motility result in neuronal death). 35 Miro1 also interacts with HUMMR (Hypoxia Up-regulated Mitochondrial Movement Regulator), a mitochondrial trafficking factor upregulated in neuronal ischemia and with Parkinson’s disease (PD) protein PINK1, linking Miro1 function to pathological changes in mitochondrial function in stroke and PD.36,37
Intercellular mitochondrial transport from human stem cells is a process that requires cell-to-cell contact either via gap junctions or tunneling nanotubes (TNT).11–14 It has been shown to rescue cells from hypoxic injury,11,12,22 and there is evidence to suggest that this transfer occurs between neurons and other cells and may be under the direction of Miro1.11,14,38 For example, when Miro1 expression is increased in donor stem cells, there is an increase in mitochondrial transport and an improved rescue of alveolar epithelial cells after lung injury. 11 Another important protein, TNFAIP2 (tumor necrosis factor-alpha induced protein 2), has been shown to be required for TNT formation. 39 This protein is stimulated via the inflammatory response and is under the control of nuclear factor kappa B (NF-κB). 40 In cells where expression of TNFAIP2 is reduced, there is an abolishment of intercellular mitochondrial transport.11,40 Intercellular mitochondrial transport has not been well studied in neurons, and while the underlying mechanisms of this process require further elucidation, it may prove to be an important mechanism of neuronal preservation after brain ischemia. In this study, we examined the role of mitochondrial transfer to oxidant-injured neurons from MSC and the metabolic benefit conferred.
Materials and methods
Experimental animals
All experimental protocols were approved by the University of Colorado Institutional Animal Care and Use Committee (IACUC) and conform to the National Institutes of Health guidelines for the care and use of animals in research. Experiments were performed using P0-P3 C57Bl/6 mice purchased from Charles River Laboratories (Wilmington, MA). Animals were used to obtain primary neuronal cultures, and prior to euthanasia were housed with a standard 12-h light-dark cycle with free access to food and water. There were no in vivo experiments performed for this study.
Cell cultures
Newborn mice (P0–P3) were anesthetized by hypothermia and euthanized by decapitation. Cerebral cortical tissue was dissected and digested with papain (LS003126, Worthington Biochemical Corporation, Lakewood, NJ) followed by trituration. Neurons were plated in culture vessels coated with Poly-D-Lysine (Sigma) and Laminin (Sigma) and grown in Neurobasal-A Medium (Gibco, Life Technologies #10888022) supplemented with B27 (Gibco #17504044), L-glutamine, pen/strep, 10 µM uridine (Sigma U3750) and 10 µM 5-fluoro-2ʹ-deoxyuridine (Sigma F0503) at 37°C in a humidified incubator under standard mammalian cell culture conditions (21% oxygen, 5% CO2 and 74% nitrogen). Neurons were cultured for 10–14 days prior to experiments.
Human MSC (Human Marrow Stromal Cells, adults; Cell Application, Inc.; 492-05 F) were cultured in T-75 cm2 flasks or well plates with media changed every other day using DMEM/F12 1:1 (Gibco #11330-032) supplemented with FGHb at 2 ng/ml (Recombinant Human FGFbasic; PeproTech, NJ, USA), 10% FBS (Gibco), 2.5 mM Glutamine and 1% Pen/Strep. Cells were maintained in a humidified atmosphere of 5% CO2 at 37°C. Passage numbers used for experiments ranged from P5 to P9.
H2O2 oxidant injury
Primary cortical neurons were oxidant damaged with H2O2 (RICCA CAT# 3819-16; 3% (W/W)) at 150 µM for 2 h. Cells were then plated with or without co-culture with MSCs in a ratio of 1:10 MSC:neurons for 24 h. The expression levels of Miro1 and TNFAIP2 were assessed via Western blot. All experiments were repeated at least three times for reproducibility.
Flow cytometry
Mitochondria were labelled with MitoTracker dyes (100 nM) for 45 min before treatment. Cells were trypsinized and suspended in sorting buffer (25 mM HEPES, 1% BSA in PBS), then immediately analyzed on a Gallios analyzer. Cells were gated based on FSC-A versus SSC-A scatter, and DAPI staining was used to exclude dead cells. Data were analyzed using the FlowJo V10 software.
MTT assay
Neuronal viability was evaluated by the colorimetric MTT assay based on the measurement of the reducing yellow tetrazolium dye (Acros Organics; AC 158992500, MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) to its insoluble purple formazan crystal under mitochondrial dehydrogenases within viable cells. MTT is prepared by dissolving MTT reagent (5 mg/mL) with 1× PBS and then 10-fold diluting it in serum-free media. Cells were incubated for 3 h, followed by adding acidified ethanol (absolute isopropanol/0.04M HCL) to dissolve the purple crystals. The absorbance of solubilized MTT formazan is measured at a wavelength of 570 nm.
Western blot analysis
Ten-day old primary neurons were treated with H2O2 (Ricca #3819-16; 3% Stock) (150 mM) for 2 h followed by co-culture with human MSC (Cell Application, Inc.; 492-05 F). Protein was extracted after 24 h in co-culture from these cells using RIPA buffer (pH 7.4) with a protein inhibitor cocktail. Lysates were resolved in 10% SDS-PAGE and transferred onto a nitrocellulose membrane. Blots were probed with antibodies for Miro 1 (Rho GTPase 1 or RHOT1, ThermoFisher, PA5-42646) and TNFAIP2 (Novus, NBP1-33480) followed by secondary (Goat anti-Rabbit – HRP antibody, Jackson #111-035-1440) to evaluate expression levels. β-actin (abcam, ab49900) served as a loading control.
Measurement of metabolic activity
Metabolic activity was measured using the Seahorse XF-96 Extracellular Flux Analyzer (Seahorse Biosciences, North Billeric, MA, USA) to measure oxygen concentration rate (OCR) and extracellular acidification rate (ECAR) for our samples; neurons without and after H2O2 treatment, H2O2 treated neurons co-cultured with MSC and MSC alone, similar to other experiments described above. These measurements after conversion to OCR and ECAR then enabled a direct quantification of mitochondrial respiration, assessing basal mitochondrial function and stress for these samples. This protocol has been described previously.41–43 Briefly, neurons were plated in XF 96-well Seahorse plates (V3-PS; 101085-004) (30,000–60,000/well) and maintained for up to 10 days. Prior to analysis, the culture medium was replaced with unbuffered XF assay medium containing sodium pyruvate, L-glutamine and glucose at pH 7.4 and cells were then allowed to equilibrate in a non-CO2 incubator immediately before metabolic flux analysis using the Seahorse XF to allow for precise measurements of Milli-pH unit changes. OCR and ECAR were measured under both basal conditions and after injection of compounds through drug injection ports. With the described protocol, we assessed the basic energy metabolism profiles of these samples as well as key parameters of mitochondrial function along with sequential injection of four mitochondria perturbing agents oligomycin, FCCP and rotenone/antimycin A. Results were reviewed using the Seahorse XF data viewer which automatically saves data in a Microsoft Excel (.xls) file.
Knockdown and overexpression of Miro 1
Knockdown
Small interfering RNA (siRNA, Silencer Pre-Designed SiRNA; AM16708) was purchased from ThermoFisher to knock down the Miro1 gene (ThermoFisher, Waltham, MA, USA). RNA interference was performed on MSC cells by using Lipofectamine RNAiMax (Invitrogen, ThermoFisher, Waltham, MA, USA) transfection reagent following the manufacturer’s instructions. MSC were seeded on 6-well plates with 60% to 80% confluency the day before transfection was performed. Cells were transfected with the final concentration of 50 nM siRNA for one to three days before co-culture with H2O2-treated neurons.
Overexpression
The human Miro1 gene cloned into a pcDNA3.1 vector was purchased from Alta Biotech (CO, USA). MSC at 60% to 80% confluency were transfected with 5 μg/ul of cDNA using Lipofectamine 3000 (Invitrogen ThermoFisher, Waltham, MA, USA) following the manufacturer’s instructions. MSC were seeded on 6-well plates with 60% to 80% confluency the day before transfection was performed. Incubation of transfected MSC was carried out for two to four days, followed by co-culture with H2O2 treated neurons.
Transcriptomic analysis
RNA was extracted from each sample and hybridized to the Mouse Clariom S Transcriptome Array (Affymetrix) according the manufacturer’s instructions. Data analysis was performed in R (http://www.r-project.org), using publicly available packages through Bioconductor (http://www.bioconductor.org). The .CEL microarray datafiles were background corrected and normalized using the RMA algorithm, resulting in log2 gene expression values. Multiple probe sets for a gene were then collapsed to one entry per gene based on the mean best-expressed probe set for that gene. Genes with significant differential expression (using a repeated measures of ANOVA in R for evaluation) were defined as those in H202-treated samples compared to untreated samples by pair-wise comparisons with significance for each gene set to a left of p < 1.76e-6 (e.g. p < 0.05 with strict Bonferroni correction for 28,463 genes).
Statistical analysis
All experiments were repeated in at least triplicate for reproducibility, with results reported as a mean ± standard deviation based on normalized values, or values as shown in each individual figure. Continuous variables were assessed using Student t test with p < 0.05 for significance. Transcriptomic analysis used a repeated measure of ANOVA in R for evaluation with p < 0.05 with strict Bonferroni correction for 28,463 genes (details above).
Results
Mitochondrial transfer to neurons from MSC
To determine if MSC transfer mitochondria to neurons, we used a co-culture system. Our data demonstrated mitochondrial transfer from MSC to neurons after oxidative injury in vitro (Figure 1). These experiments were performed with primary cortical neurons stained with the fluorescent probe MitoTracker Green FM and MSC whose mitochondria were stained with MitoTracker Deep Red FM. Following oxidant injury, MSC were added to the neurons and co-cultured for 2 h with direct contact or separated with a porous trans-well membrane before being analyzed using a Gallios flow cytometer (Figure 1(b) to (e)). We observed cells containing mitochondria labelled with both MitoTracker dyes in the direct co-culture samples, indicating that mitochondrial transfer occurred between neurons and MSC (Figure 1). Confocal images confirmed oxidant-damaged neurons containing green and red mitochondria existed in proximity to MSC after co-culture (Figure 1(a)). This phenomenon was not observed when a trans-well membrane was placed between neurons and MSC, indicating that cell-to-cell contact was necessary for mitochondrial transfer to occur (Figure 1(e)).
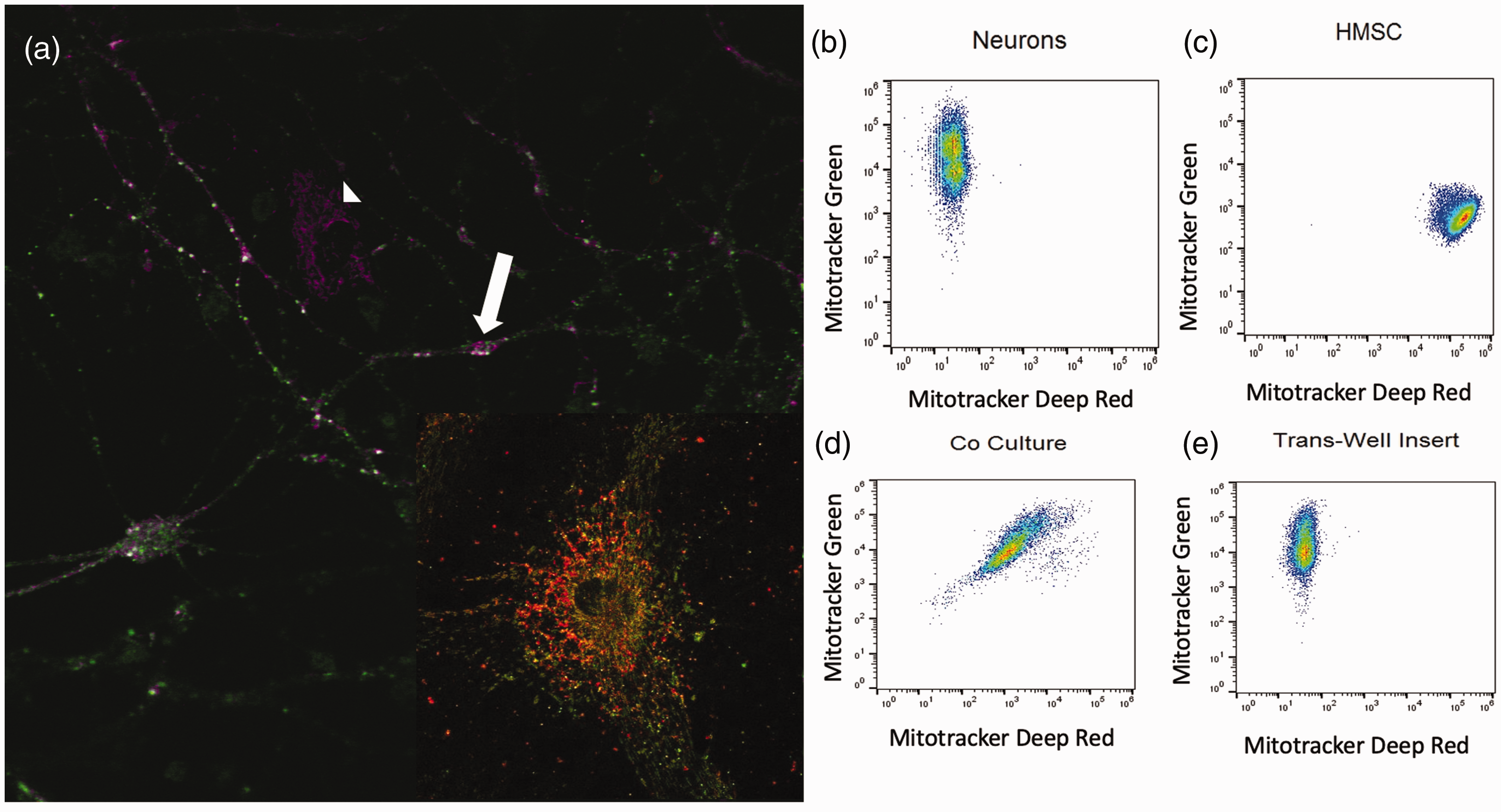
(a) Oxidant-damaged neurons (white arrow) with mitochondria labeled with MitoTracker Green accepting mitochondria from a neighboring MSC (white arrowhead) with mitochondria labeled with Mitotracker Far Red after co-culture neuron at high power showing both red and green mitochondria. (b) Flow cytometry of primary cortical neurons labeled with MitoTracker Green. (c) Flow cytometry of MSC labeled with MitoTracker Far Red. (d) Flow cytometry of MitoTracker Green-labeled primary cortical neurons following hydrogen peroxide exposure and co-culture with MSC labeled with MitoTracker Far Red resulting in a double positive cell population. (e) Flow Cytometry of MitoTracker Green-labeled primary cortical neurons following oxidant injury and indirect co-culture with MSC labeled with MitoTracker Far Red. MSC and neurons were co-cultured with a 0.2 µm pore size membrane lying in between cells resulting in no double positive cell population of neurons.
Metabolic benefits from co-culture
After demonstrating that mitochondrial transfer occurs between MSC and neurons, we then assessed the metabolic benefits to neurons after co-culture with MSC. First, we examined cell survival after hydrogen peroxide injury and co-culture with MSC. Using the MTT assay as an indicator of cell viability, we observed lower rates of viability in cells that suffered oxidant injury when compared to untreated cells (Figure 2(a)). Furthermore, we saw an increase in MTT conversion after co-culture with MSC, indicating higher levels of cell viability. Consistent with improvements observed in ATP levels, the benefits of co-culture were not observed when MSC and neurons were separated using a trans-well insert (Figure 2(a)). Additionally, the Seahorse assay demonstrated significant recovery of metabolic function in neurons after co-culture with MSC with improvements in mitochondrial respiration (Figure 2(b)), basal metabolic rate (Figure 2(d)), spare respiratory capacity (Figure 2(d)), proton leak (Figure 2(e)) and ATP production (Figure 2(e)).
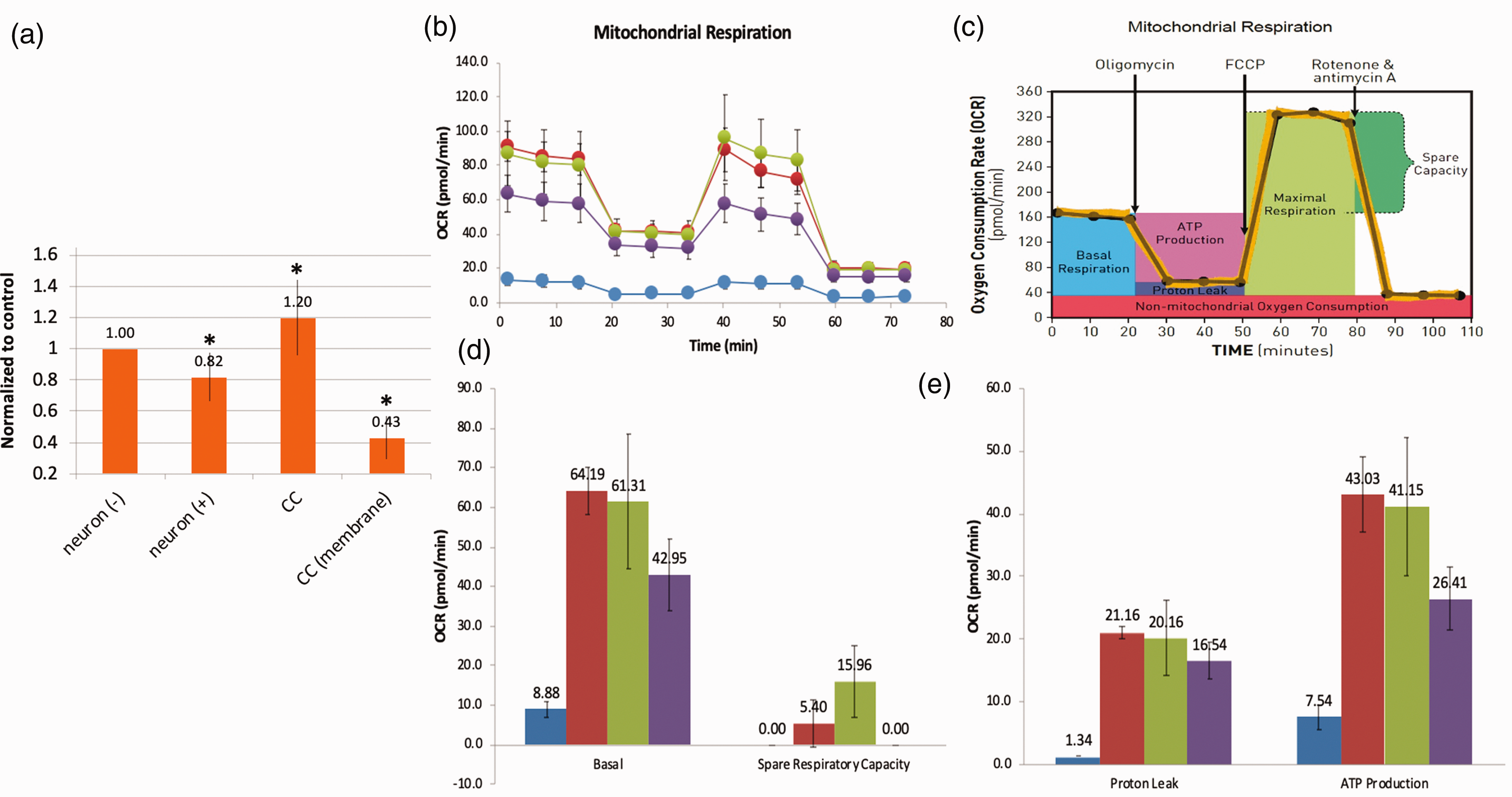
Metabolic benefit of co-culture of MSC with oxidant-damaged neurons. Murine primary cortical neurons underwent in vitro oxidant injury with hydrogen peroxide followed by co-culture with MSC. (a) MTT assay. Neuronal cell viability declined after oxidant injury, improved after co-culture, but not if MSC were separated from neurons with a semi-porous membrane (CC membrane). (b–e) Seahorse XF Assay (pmol/min/60,000 cells). From left to right (see legend): MSC controls (blue); neuronal control (red); oxidant-damaged neurons in co-culture with MSC (green); neurons after hydrogen peroxide exposure (purple). (b) There is evidence of significant mitochondrial dysfunction in neurons after oxidant injury that recovers in co-culture with MSC when assessing mitochondrial respiration in neurons. If confidence intervals do not cross, differences are significant (see C for Figure key). (d) Basal mitochondrial respiration and spare respiratory capacity all worsened with oxidant injury and then improved with co-culture with MSC. (e) Proton leak and ATP production worsened in neurons after oxidant injury and recovered after co-culture. *Significant difference from control.
Gene expression profile changes after H2O2 exposure
To further elucidate the underlying mechanisms surrounding the phenomena we report, we examined gene expression profile changes occurring in neurons after oxidant injury. A number of pathways involved in mitochondrial motility, cytoskeletal integrity, and inflammation were altered (Figure 3(a) and (b)). Looking more closely at pathways related to mitochondrial motility and inflammation, a number of genes were upregulated or downregulated in a consistent manner (Figure 3(c)). In order to better understand the role of some of the key members of these pathways, graphical representation of two of the top scored networks identified by ingenuity pathway analysis (IPA) was evaluated. Canonical pathways from IPA for control neurons compared to oxidant damaged neurons are shown. A number of mitochondrial motility genes related to the Miro1 (Rhot1) pathway were altered (Figure 3(d)). Additionally, a number of upstream regulators of tumor necrosis factor alpha (TNF-a) were altered (Figure 3(e)). From this information, two that have been previously reported to play a role in mitochondrial transfer in other cell types and were altered in this analysis were studied (Miro1 and TNFAIP2).11,39 Whole protein lysates were obtained from primary cortical neurons or oxidant damaged neurons and expression levels of Miro1 and TNFAIP2 were assessed via Western blot. Oxidant damaged neurons showed increased levels of protein expression for both Miro1 and TNFAIP2 in comparison to primary cortical neurons. These elevated expression levels returned to normal when oxidant damaged neurons were co-cultured with MSC (Figure 4(a) and (b)). Co-culture also improved metabolic activity of oxidant damaged neurons in comparison to oxidant-damaged controls. We then upregulated or inhibited Miro1 expression in MSC to assess the effects this had on the metabolic benefits of mitochondrial transfer after co-culture. When Miro1 was upregulated, there was an increase in cell viability above standard co-culture of MSC with oxidant-damaged neurons (Figure 4(c)). In contrast, after Miro1 inhibition, cell viability was diminished and no longer significantly different than controls (Figure 4(c)).
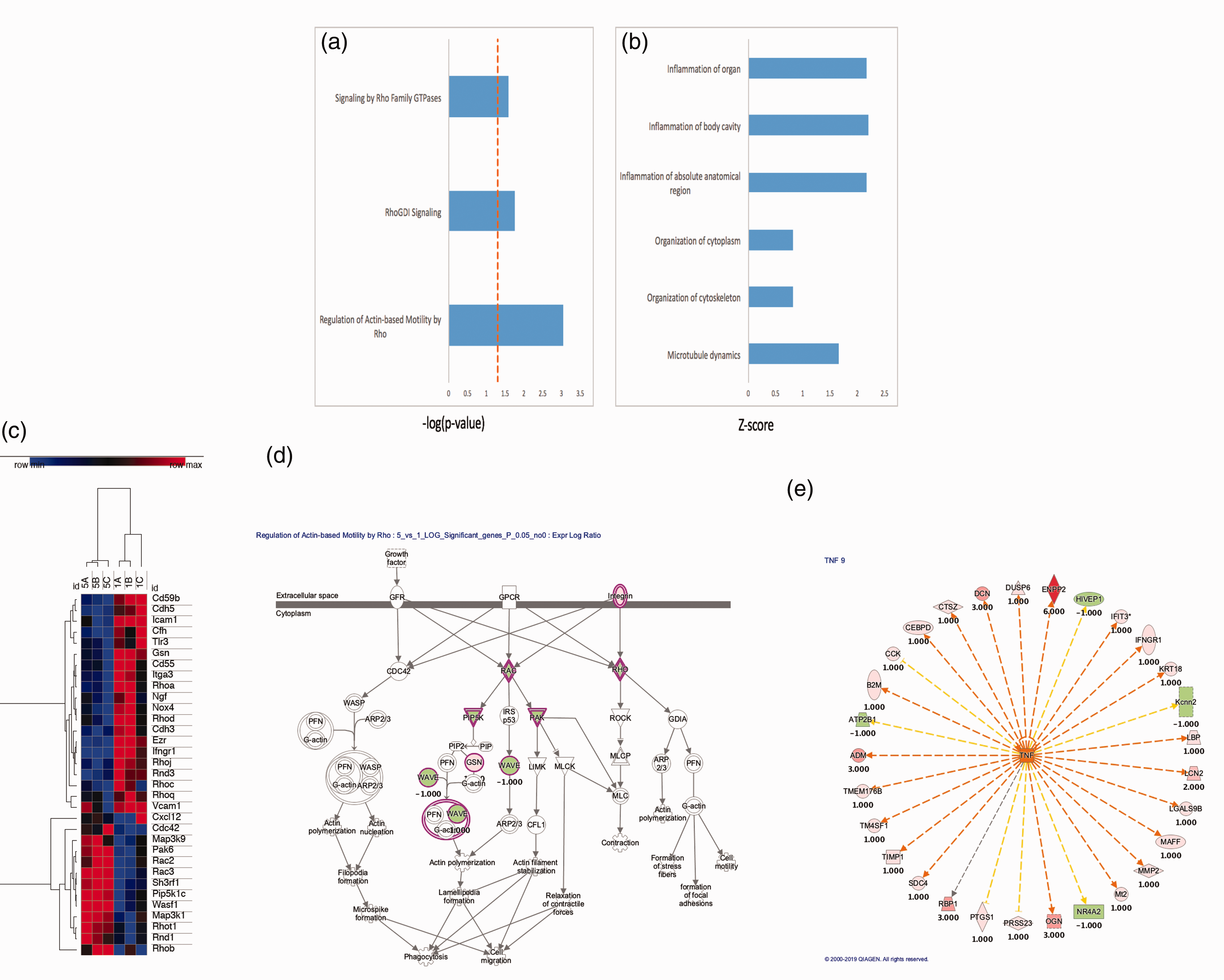
Clariom S Mouse array of murine neurons after hydrogen peroxide injury. (a) Rho Family GTPases and related pathways were upregulated. (b) Inflammatory pathways, cytoskeleton and microtubule pathways were also upregulated. (c) Gene expression microarray analysis comparing control neurons (1 A-C) with H2O2-oxidant damaged neurons (5 A-C): Clustering of the samples according to their expression profile. High expression is indicated in red. Low expression is indicated in blue. Hierarchical clustering was performed using the one minus Pearson correlation. (d and e) Graphical representation of two top scored networks identified by ingenuity pathway analysis (IPA). Canonical pathways from IPA for control neurons compared to oxidant damaged neurons. Molecular relationships between genes after treatment are shown demonstrating changes in genes related to mitochondrial motility (d) and upstream regulators of tumor necrosis factor (TNF) (e) with positive numbers representing upregulation with oxidant damage and negative numbers representing downregulation.
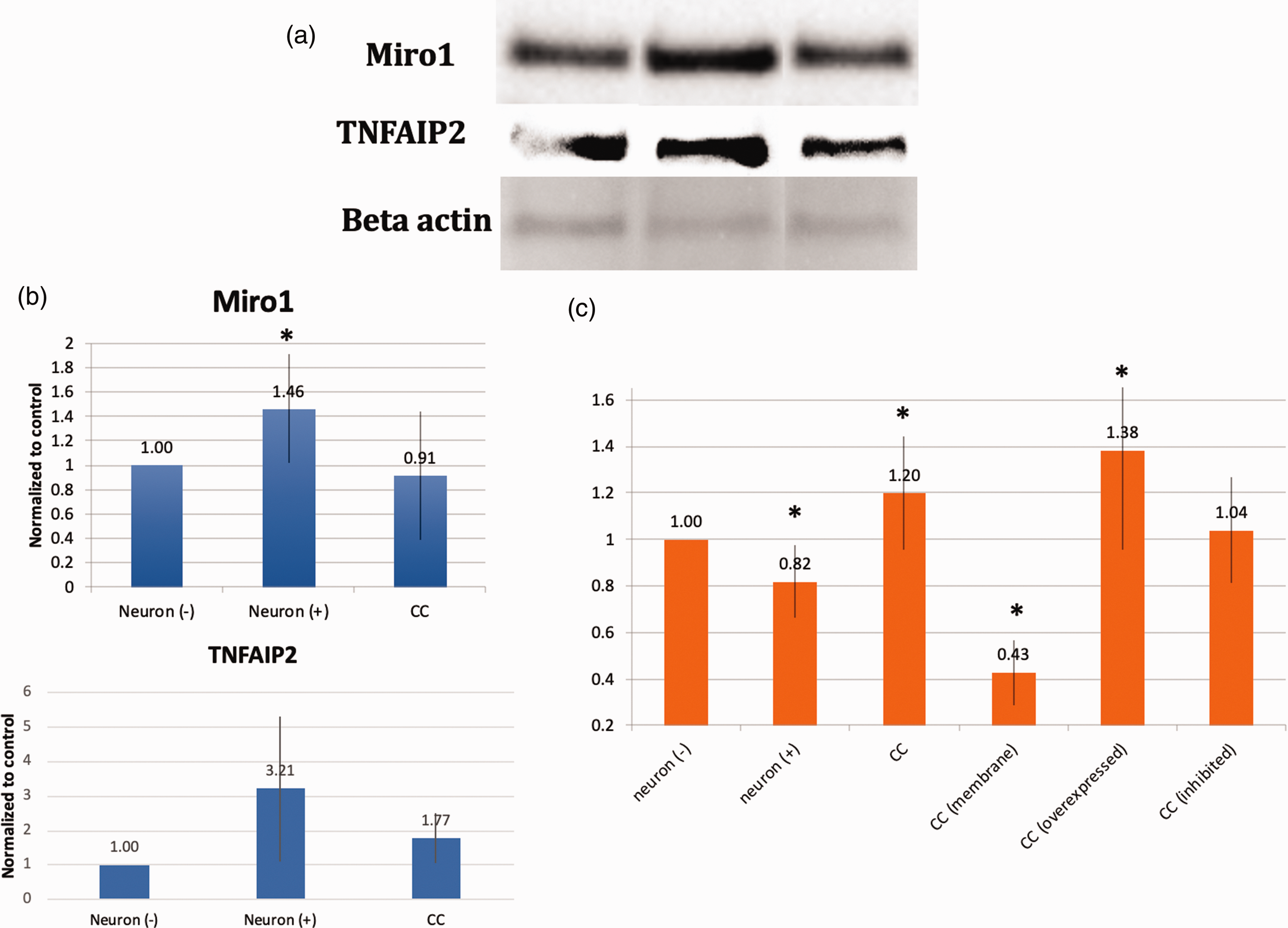
(a) Western Blot of Miro1 and TNFAIP2 expression before (Neuron (−)) or after (Neuron (+)) oxidant injury with H2O2 as well as after co-culture with MSC (CC). (b) Expression levels normalized to actin in Lanes 1–3 bar graphs, 25 ug protein per lane. Lane 1, untreated neurons (Neuron (−)). Lane 2, neurons after H2O2 treatment at 150 µM for 2 h (Neuron (+)). Lane 3, oxidant-damaged neurons co-cultured overnight with MSC (CC). Both Miro1 and TNFAIP2 expression were increased in primary cortical neurons after hydrogen peroxide exposure. This expression returned toward normal when oxidant-damaged neurons were co-cultured with MSC. (c) MTT assay. Some data repeated from Figure 2 for comparison purposes. Oxidant injury (neuron (+)) resulted in more neuronal cell death that recovered with co-culture with MSC (CC), but not if MSC were separated from neurons with a semi-porous membrane (CC (membrane)). This benefit is also decreased if Miro 1 was inhibited in MSC (CC (inhibited)) and enhanced if Miro 1 was overexpressed (CC (overexpressed)), implicating Miro 1 in the metabolic benefit of MSC co-culture with neurons. *Significant difference from control.
Discussion
Mitochondrial transfer occurred from MSC to neurons after oxidative stress in vitro (Figure 1). These observations are consistent with those from other groups such as Ahmad et al., 11 who demonstrated that MSC transfer of mitochondria to lung epithelial cells protected against oxidative stress-induced lung injury. It has also recently been demonstrated to occur between neurons and other cells by other groups.38,44 The mitochondrial transfer we observed occurs robustly and within a relatively short (several hours) period of time. Mitochondrial transfer was not observed when MSC and neurons were separated by a porous trans-well membrane, indicating that this transfer is dependent on cell-to-cell contact (Figure 2(b)).
In order to assess any metabolic benefits that occurred as a result of mitochondrial transfer in vitro, we assayed survival and metabolic function. Oxidant-damaged neurons had lower rates of survival and ATP production than control neurons, and overall worse metabolic function. When co-cultured with MSC, we observed an improvement in survival, ATP production and overall cellular metabolism that was dependent on cell-to-cell contact, as metabolic benefits were not observed when these co-cultured cells were separated by a porous trans-well insert (data not shown). This suggests that benefits conferred from MSC to neurons are not only due to the release of various paracrine factors from MSC, but also from mitochondrial transfer. These observations link MSC-mediated neuroprotection at least partially to improved metabolic function in vitro.
Both Miro1 and TNFAIP2 expression were increased in primary neurons after hydrogen peroxide exposure (Figure 3). The expression of these genes has previously been demonstrated to be important in intercellular mitochondrial transfer, and our observations link the expression of these genes to ischemic damage11,39 Furthermore, microarray analysis showed a number of inflammation-related genes and mitochondria motility genes and their upstream effectors all increase expression in neurons after hydrogen peroxide treatment (Figure 3). Studies have shown that Miro1 expression regulates the trafficking of mitochondria, and its overexpression enhanced mitochondrial transfer, leading to improved rescue of alveolar epithelial cells after injury. 11 TNFAIP2 is required for TNT formation and is believed to be a major mechanism for mitochondrial transfer to occur.11,39 The increase in TNFAIP2 expression links the phenomenon of mitochondrial transfer to the inflammatory response, a response that is activated in vivo following cerebral ischemia and leads to recruitment of cells, including MSC, to the brain.2,7,14,39 It has previously been demonstrated that mitochondrial transfer from stem cells to neurons improves behavioral outcomes after stroke. 44 Upregulation of Miro1 in MSC improves cell viability of ischemic neurons after co-culture. Inhibition of Miro1 in MSC, on the other hand, limits any benefit. This demonstrates a potential mechanism of action of mitochondrial transfer in neurons related to the mitochondrial motility pathway (see Figure 5) and suggests a potential treatment to improve neuronal survival after brain ischemia.
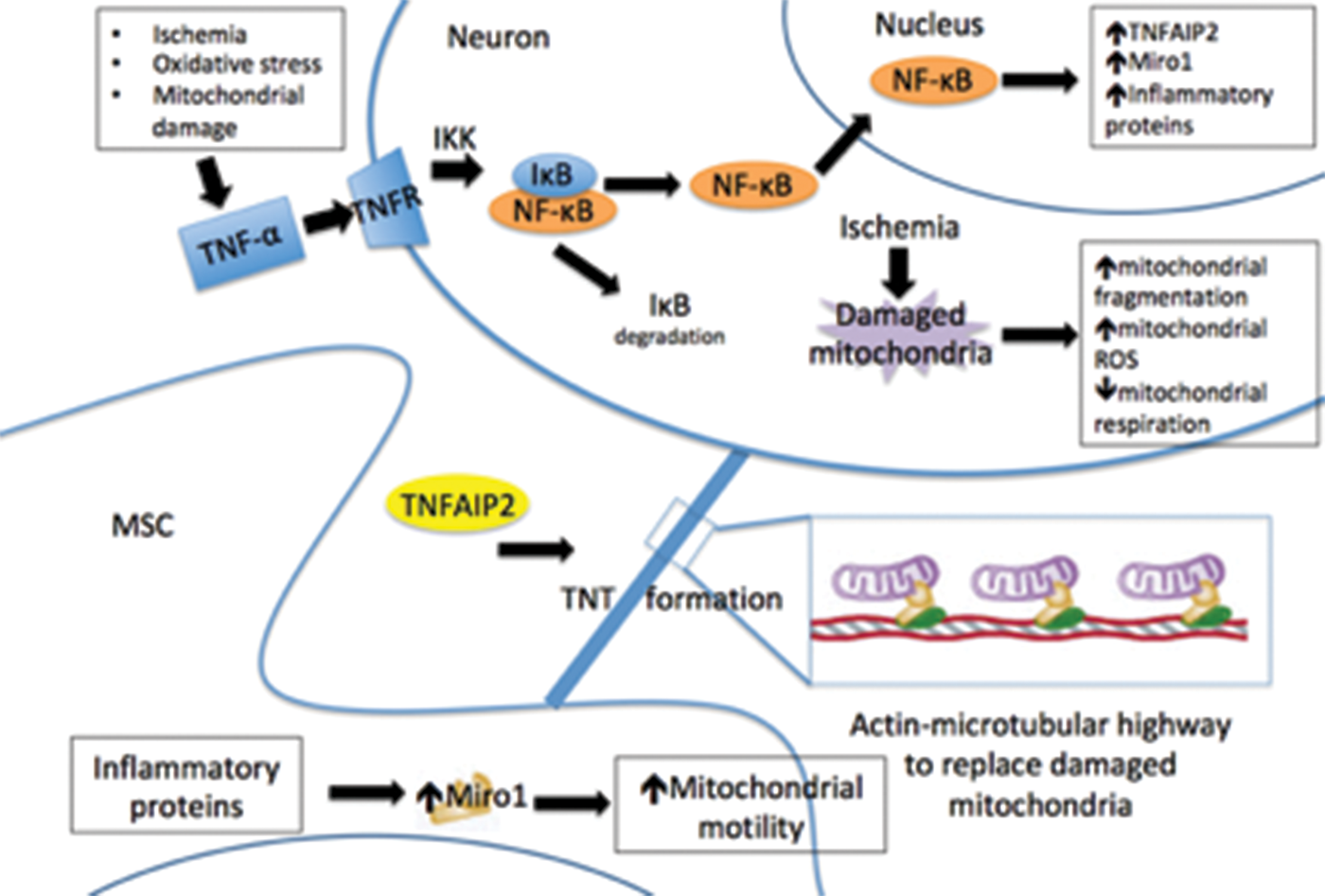
Proposed mechanism of intercellular transport of mitochondria from mesenchymal stem cells (MSC) to neurons via tunneling nanotubes (TNT). Ischemia damages neuronal mitochondria, but also induces an inflammatory response that increases TNFAIP2 and Miro 1 production. This, in turn, increases mitochondrial motility and the transfer of healthy mitochondria from MSC to neurons, potentially sparing neurons from apoptosis.
Limitations
In vitro experimentation often is a useful way to study molecular mechanisms of action. However, findings may not translate to in vivo mechanisms of disease and repair. While H2O2 damages cells primarily through oxidation of its targets, this is only a portion of the mechanism of neuronal damage after brain ischemia, although it does mimic reperfusion injury. Ultimately, in vivo studies will be important in the future to validate in vitro findings.
Conclusions
MSC transfer healthy mitochondria to damaged neurons and improve neuronal metabolism after oxidant injury in vitro. Miro1 plays a role in this process, with its expression in MSC correlating to cell viability after co-culture. This represents an exciting potential therapy to mitigate neuronal cell death following ischemia. Further work needs to be done to better understand whether these findings can be duplicated in vivo, and if so, what underlying molecular mechanisms control mitochondrial transfer. With a deeper understanding of the mechanisms controlling intercellular mitochondrial transfer, it may be feasible to artificially enhance the number of mitochondria that are transferred to injured cells and improve functional outcomes after cerebral ischemia.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X20928147 - Supplemental material for Mitochondrial transfer from mesenchymal stem cells improves neuronal metabolism after oxidant injury in vitro: The role of Miro1
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X20928147 for Mitochondrial transfer from mesenchymal stem cells improves neuronal metabolism after oxidant injury in vitro: The role of Miro1 by Nancy Tseng, Scott C Lambie, Christopher Q Huynh, Bridget Sanford, Manisha Patel, Paco S Herson and D Ryan Ormond in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by a research grant from the American Heart Association.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
Conceived and designed the experiments DRO.
Performed the experiments SCL, NT, CQH, BS.
Analyzed the data DRO, SCL, NT, PH, MP, BS.
Wrote the manuscript DRO, SCL, NT, PH, MP.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.