
Abstract
Lipopolysaccharide (LPS) is a major component of the outer membrane of Gram-negative bacteria and a potent inflammatory stimulus for the innate immune response via toll-like receptor (TLR) 4 activation. Type 2 diabetes is associated with changes in gut microbiota and impaired intestinal barrier functions, leading to translocation of microbiota-derived LPS into the circulatory system, a condition referred to as metabolic endotoxemia. We investigated the effects of metabolic endotoxemia after experimental stroke with transient middle cerebral artery occlusion (MCAO) in a murine model of type 2 diabetes (db/db) and phenotypically normal littermates (db/+). Compared to db/+ mice, db/db mice exhibited an altered gut microbial composition, increased intestinal permeability, and higher plasma LPS levels. In addition, db/db mice presented increased infarct volumes and higher expression levels of LPS, TLR4, and inflammatory cytokines in the ischemic brain, as well as more severe neurological impairments and reduced survival rates after MCAO. Oral administration of a non-absorbable antibiotic modulated the gut microbiota and improved metabolic endotoxemia and stroke outcomes in db/db mice; these effects were associated with reduction of LPS levels and neuroinflammation in the ischemic brain. These data suggest that targeting metabolic endotoxemia may be a novel potential therapeutic strategy to improve stroke outcomes.
Introduction
Lipopolysaccharide (LPS), often referred to as endotoxin, is a major component of the outer membrane of Gram-negative bacteria. Lipopolysaccharide is a potent inflammatory stimulus for the innate immune response via toll-like receptor (TLR) 4 activation. 1 Inflammation plays important roles in the development of strokes and is also implicated in the pathophysiology of ischemic lesions, as well as in the overall outcome after stroke. 2 According to the Bruneck Study, an increased plasma LPS level constitutes a substantial risk factor for incident carotid atherosclerosis and cardiovascular disease. 3 Furthermore, a higher plasma LPS level is associated with worse short-term outcomes in patients with acute ischemic stroke. 4 Experimental studies have demonstrated that systemic LPS administration exacerbates brain damage after cerebral ischemia.5,6
The human intestine is home to a vast number of bacteria, and the intestinal barrier prevents under physiological conditions, the passage of harmful luminal contents such as LPS. 7 Recently, growing evidence has emerged that chronic exposure of the host to gut microbiota-derived LPS links to metabolic disorders, a condition referred to as metabolic endotoxemia. 8 Alteration of gut microbiota (gut dysbiosis) and impairment of intestinal barrier functions lead to translocation of LPS into the circulatory system and contribute to the pathogenesis of type 2 diabetes via the activation of proinflammatory cascades in adipose tissues. 8 Furthermore, increased intestinal permeability and increased circulating LPS levels have been reported to contribute to the pathogenesis of nonalcoholic steatohepatitis by inflammatory liver damage. 9 In patients with type 2 diabetes, increased serum levels of LPS and proinflammatory cytokines have been reported. 10 In addition, we have previously demonstrated increased inflammatory marker levels concurrent with gut dysbiosis and higher LPS-binding protein concentrations, which reflect circulating LPS levels. 11 These findings indicate the importance of gut microbiota-derived LPS as a trigger for inflammatory responses in the host in type 2 diabetes.
Stroke commonly occurs in patients with cardiovascular risk factors. Type 2 diabetes is observed in approximately 60% of patients with acute ischemic stroke. 12 Type 2 diabetes is not only associated with a significantly increased stroke risk but also with an early progression of ischemic lesions 13 and worse functional outcomes. 14 However, establishment of clinically effective neuroprotective therapies has been challenging.15,16
Hence, to study whether metabolic endotoxemia could be a therapeutic target to improve stroke outcome, we investigated the effects of metabolic endotoxemia on acute ischemic brain injury after experimental stroke in a murine model of type 2 diabetes. We hypothesized that metabolic endotoxemia will promote LPS-induced neuroinflammation and affect the outcome after stroke.
Materials and methods
Experimental animals
All animal experiments were approved by the Juntendo University Animal Ethics Committee (No. 1176) and were in accordance with Animal Research: Reporting in Vivo Experiments (ARRIVE) reporting guidelines for the care and use of laboratory animals. We strictly followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Leptin receptor-deficient (db/db) mice exhibit features of type 2 diabetes. 9 We purchased eight-week-old male db/db mice (n = 63) and lean, phenotypically normal littermates (db/+ mice) (n = 65) from Charles River Laboratories Japan, Inc. Mice were maintained on a 12-h light/dark cycle with ad libitum access to standard chow and tap water. In some animals, we used polymyxin B, a non-absorbable antibiotic, which selectively acts on Gram-negative bacteria. In the polymyxin B-treatment group, mice received 1 mg of polymyxin B (WAKO, Osaka, Japan) dissolved in 0.2 mL of distilled water once daily by oral gavage for one week. In the non-treatment group, mice received 0.2 mL of distilled water once daily by oral gavage for one week. The treatment schedule was identical for both groups. This treatment began when mice were 11 weeks old (Figure 1(a)).
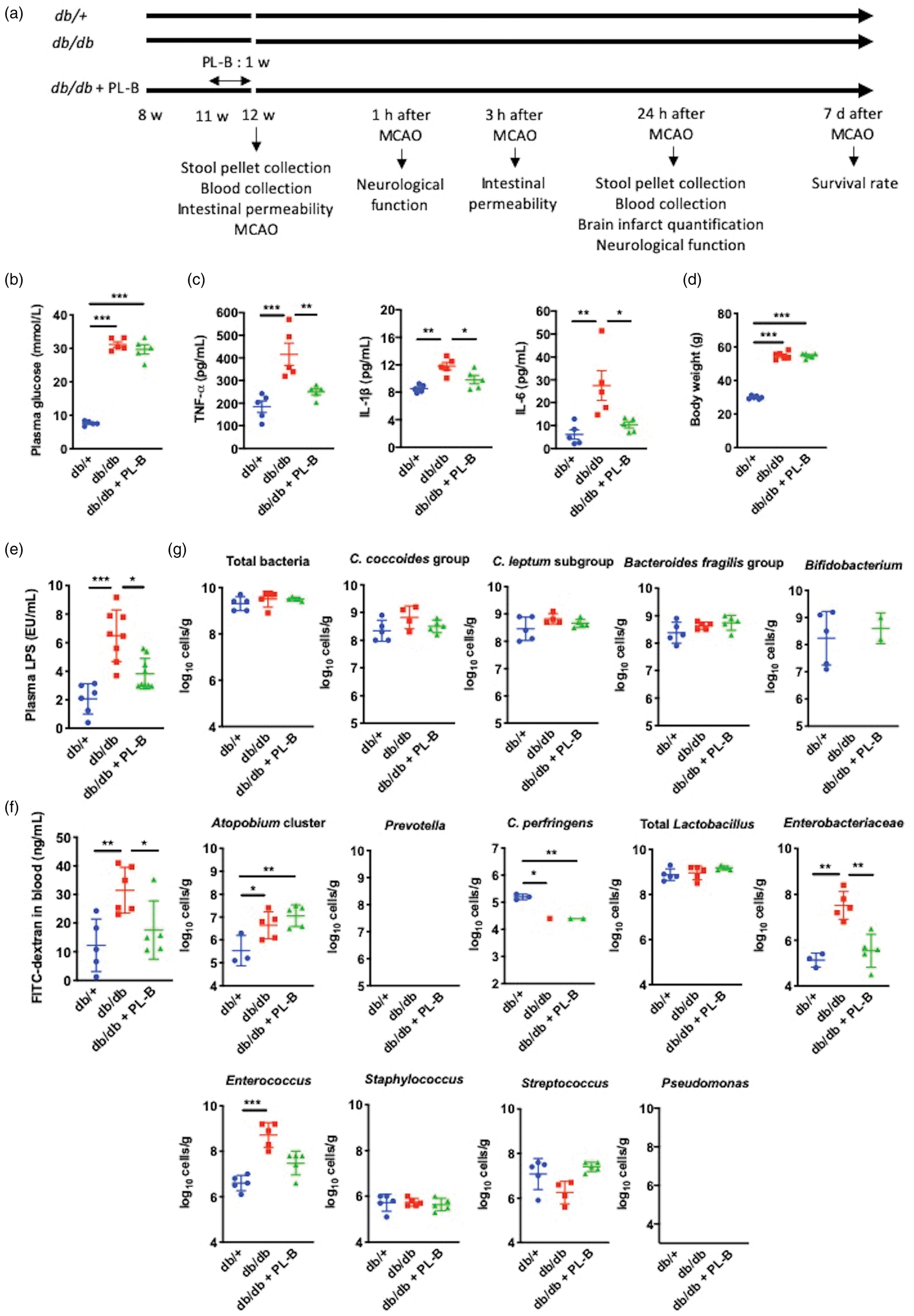
Biological parameters of db/+, db/db, and polymyxin B (PL-B)-treated db/db mice measured prior to stroke induction with middle cerebral artery occlusion (MCAO). (a) Experimental design. (b) Plasma glucose levels (n = 5 per group). (c) Plasma levels of tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6 (n = 5 per group). (d) Body weight (n = 6 per group). (e) Plasma lipopolysaccharide (LPS) levels (n = 6–9 per group). (f) Intestinal permeability assessed by quantitative analysis of fluorescein isothiocyanate (FITC)-dextran translocation (n = 5–6 per group). (g) Fecal bacterial counts analyzed by rRNA-targeted quantitative reverse transcription PCR (n = 5 per group). Bacterial counts below the threshold of detection were not plotted. Data are shown as mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001.
Polymyxin B plasma concentration
Plasma samples of db/db mice were collected 4 h after oral administration of polymyxin B. Polymyxin B standard (Hycult Biotech, PA, USA) and samples were analyzed using an enzyme-linked immunosorbent assay (ELISA). Briefly, plasma samples were added to LPS-coated plates (Hycult Biotech) and incubated with 200 μL of blocking buffer for 1 h. Subsequently, anti-polymyxin B immunoglobulin M (IgM; Hycult Biotech) was added and incubated for 1 h. Anti-mouse IgM horseradish peroxidase (Hycult Biotech) was used as a secondary antibody. Finally, 3,3ʹ,5,5ʹ-tetramethylbenzidine (TMB) (Scy Tech, West Logan, UT, USA) and a stop solution were added to each well. Optical density values at 450 nm minus those obtained at 570 nm were analyzed.
Middle cerebral artery occlusion
Mice were anesthetized with 4.0% isoflurane (Abbott Japan Co., Ltd, Tokyo, Japan) and maintained with 1.0%–1.5% isoflurane in 70% N2O and 30% O2 using a small-animal anesthesia system. All surgical instruments were sterilized prior to surgery. Before any incision was made, the area was swabbed with ethanol. Transient cerebral focal ischemia was induced by MCAO for 60 min using an intraluminal filament technique as described previously (Figure 1(a)). 17 Briefly, the left common carotid artery and the left external carotid artery were exposed and ligated after a ventral midline neck incision. A silicon-coated nylon monofilament was inserted through the left common carotid artery into the left internal carotid artery to occlude the left MCA. After 60 min of occlusion, the monofilament was withdrawn for reperfusion. Regional cerebral blood flow (rCBF) was measured in the left temporal window on laser Doppler-flowmetry (FLOW-C1; Omegawave Inc., Tokyo, Japan) before, during, and 24 h after MCAO. We excluded mice (n = 5) in which the reduction in rCBF of the laser Doppler signal was below 60%, when compared with the preischemic state. During the procedure, a core body temperature of 37.0 ± 0.5°C was maintained using a heating pad.
Functional outcome testing and seven-day survival rate
Neurological severity was assessed 1 h and 24 h after MCAO on a scale of 0 (normal) to 18 (maximal deficit) using the Modified Neurological Severity Score, which is a composite of motor (muscle status, abnormal movement, and balance), sensory (visual, tactile, and proprioceptive), and reflex test scores. 18 In the severity scores of injury, 1 score point is awarded for the inability to perform the test or for the lack of a tested reflex; thus, the higher the score, the more severe the injury. The seven-day survival rate for each group was determined using the Kaplan–Meier analysis.
LPS administration
In a subgroup of polymyxin B-treated db/db mice, LPS (Escherichia coli 0111: B4, 100 µg/kg; Sigma-Aldrich Co., St. Louis, MO, USA) dissolved in sterile saline or vehicle (sterile saline) was injected intraperitoneally 30 min before the induction of MCAO as previously described.5,6
Biochemical assays
Blood samples were collected from 12-week-old db/+ and db/db mice treated with either distilled water or polymyxin B (Figure 1(a)). Plasma glucose levels before MCAO were measured using a blood glucose meter (Johnson & Johnson, New Brunswick, NJ, USA). Plasma cytokine levels before MCAO were measured using ELISA. These measured cytokines included tumor necrosis factor-α (TNF-α) (LifeSpan BioSciences, Seattle, USA; sensitivity 15.63 pg/mL), interleukin (IL)-1β (R&D Systems, Minneapolis, MN, USA; sensitivity 2.31 pg/mL), and IL-6 (R&D Systems; sensitivity 1.6 pg/mL). Plasma LPS levels were measured before and 24 h after MCAO. Plasma samples for LPS measurements were stored in LPS-free glass tubes and heated to 70°C for 5 min before LPS measurement. Plasma LPS concentrations were determined using a kit based on a Limulus amebocyte extract (LPS kit, Cusabio, USA).
Intestinal permeability
Intestinal permeability was measured as described previously.8,19 Briefly, mice that had fasted for 6 h received 4000 kDa fluorescein isothiocyanate (FITC)-dextran (440 mg/kg body weight, 100 mg/mL) dissolved in deionized water by oral gavage before or 3 h after MCAO. The mice were anesthetized 1 h after oral gavage, and a cardiac puncture was performed to collect blood. Plasma samples were diluted in equal volumes of phosphate-buffered saline (PBS; pH 7.4) and analyzed for FITC-dextran content with a fluorescence spectrophotometer (Mithras 2 LB 943 microplate reader, Berthold Technologies, Bad Wildbad, Germany) at an excitation wavelength of 485 nm and an emission wavelength of 528 nm.
16S and 23S rRNA-targeted quantitative reverse transcription-polymerase chain reaction
Fecal samples were collected from 12-week-old db/+ and db/db mice treated with either distilled water or polymyxin B, before and 24 h after MCAO (Figure 1(a)). The composition of fecal gut microbiota was determined via 16S and 23S rRNA-targeted qRT-PCR using the Yakult Intestinal Flora-SCAN analysis system (YIF-SCAN®, Yakult Honsha Co., Ltd, Tokyo, Japan) as previously described. 20 The sequences of primers used for these analyses are listed in Supplementary Table 1.
Tissue processing
Mice were anesthetized by an intraperitoneal injection of pentobarbital (50 mg/kg) 24 h after MCAO and transcardially perfused with 20 mL ice-cold PBS to remove blood from the brain capillaries as described previously. 21 The brains turned completely white. For histological analysis, brains were subsequently perfused with 4% paraformaldehyde in PBS, which was then removed, and brains were post-fixed in the same fixative overnight at 4°C. Subsequently, each brain was soaked overnight in 30% sucrose in PBS. For Western blot analysis and ELISA, brains were removed immediately after PBS perfusion and stored at −80°C.
Measurement of infarct volume
For infarct quantification, forebrains were coronally sectioned into 20-µm-thick sections on a cryostat (Model CM 1900, Leica, Germany) separated by 1-mm intervals. Sections were stained with Cresyl Violet and scanned using the AxioVision software (Carl Zeiss, Jena, Germany). Image analyses were performed using the ImageJ software (National Institutes of Health; https://imagej.nih.gov/ij/). Infarct volumes and hemispheric volumes were calculated using numerical integrations of the respective areas for all sections and the distance between them. Corrected infarct volumes were calculated to compensate for the effect of brain edema using the following equation: corrected infarct volume = infarct volume – (ipsilateral hemisphere volume – contralateral hemisphere volume). 22
Blood–brain barrier evaluation
To evaluate the permeability of the blood–brain barrier, brain sections (20-µm-thick) collected 24 h after MCAO were incubated in 3% H2O2 followed by blocking with 10% bovine serum albumin (Sigma-Aldrich) in PBS. Afterward, sections were incubated overnight at 4°C with donkey anti-mouse IgG (1:300 dilution; Vector Laboratories, Burlingame, CA, USA). Immunoreactivity was visualized using the avidin-biotin complex method (Vectastain ABC kit, 1:400 dilution; Vector Laboratories). IgG was quantified for blood–brain barrier disruption using Image J.
Immunohistochemical analysis
Immunohistochemical analysis was performed on brains harvested 24 h after MCAO. Sections of forebrains (20-µm-thick) were incubated with anti-ionized calcium-binding adapter molecule antibody (Iba1, 1:500 dilution; WAKO), treated with biotinylated secondary antibody (1:300 dilution; Vector Laboratories), and subsequently processed with an avidin-biotinylated peroxidase complex (Vectastain ABC kit; 1:400 dilution; Vector Laboratories). To assess microglia/macrophage activation, the total number of Iba1-stained cells as well as the Iba1-stained area were calculated at the ischemic boundary area (0.25 mm2) 24 h after MCAO using ImageJ as previously described. 23
Double immunofluorescence staining was performed by simultaneous incubation of the sections with the following primary antibodies overnight at 4°C: anti-Escherichia coli LPS (1:100 dilution; Abcam) and anti-E. coli K99 (1:100 dilution; Lifespan, Providence, RI, USA), anti-TLR4 (1:100 dilution; Santa Cruz Biotechnology), anti-Iba1 (1:100 dilution; Abcam), anti-NeuN (neuron) (1:100 dilution; Abcam), anti-CD31 (endothelial cells) (1:25 dilution; Abcam), anti-glial fibrillary acidic protein (GFAP) (1:500 dilution; Abcam), and anti-transmembrane protein 119 (TMEM119) (1:150 dilution; Abcam). Thereafter, sections were incubated with a FITC-conjugated secondary antibody (Jackson ImmunoResearch, Baltimore, USA) for the identification of E. coli LPS (1:250 dilution), E. coli K99 (1:250 dilution), or TLR4 (1:500 dilution), and a Cy™3-conjugated secondary antibody (Jackson ImmunoResearch) for the identification of Iba1 (1:1000 dilution), NeuN (1:500 dilution), CD31 (1:500 dilution), GFAP (1:500 dilution) or TMEM119 (1:1000 dilution). Sections were covered with Vectashield mounting medium (Vector Laboratories), and immunofluorescent images were obtained using a laser-scanning microscope (model LSM780, Carl Zeiss). In immunohistochemical analyses, positively stained cells in the ischemic boundary area (0.25 mm2) were counted in three sections from each mouse using the ZEN 2011 software (Carl Zeiss). TLR4 expression was quantified by measuring the percentage of TLR4 immunostaining area in Iba1-, NeuN-, and CD31-positive cells, and the percentage of TMEM119-positive microglia in Iba1-positive cells was quantified using ImageJ. All immunohistochemical data were analyzed in the ipsilateral hemisphere in three coronal sections at +0.4 mm, +0.8 mm, and +1.2 mm from the bregma. Immunopositive cells were counted manually by a researcher who was blinded to the experimental conditions. The average cell number of all the sections was used as the cell number per mouse.
Western blot analysis
Mouse brain samples were obtained 24 h after reperfusion. Samples were homogenized with lysis buffer (CelLytic MT Cell Lysis Reagent; Sigma-Aldrich Co., St. Louis, MO, USA) containing a protease inhibitor cocktail (Complete Mini, EDTA-free; Roche). Homogenized samples were centrifuged at 12000 × g for 20 min, and pellets were discarded. Aliquots of supernatants containing 25 μg of protein were subjected to 4%–15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride membranes (Bio-Rad Laboratories, Hercules, CA, USA). The membranes were blocked in Brockace (Dainichi-Seiyaku, Gifu, Japan) for 60 min at room temperature. Subsequently, membranes were incubated overnight at 4°C with antibodies against TLR4 (1:500 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA), LPS (1:500 dilution; Abcam, Cambridge, MA, USA), and β-actin (1:5000 dilution, Abcam) followed by incubation with secondary antibodies and visualization via enhanced chemiluminescence (GE Healthcare UK, Little Chalfont, Buckinghamshire, UK).
Brain inflammatory cytokine measurement
Mouse brain samples were obtained 24 h after reperfusion. Samples were homogenized with PBS and centrifuged at 5000 × g for 5 min. The supernatant was collected and assayed immediately. Levels of inflammatory cytokines including TNF-α (LifeSpan BioSciences; sensitivity 15.63 pg/mL), IL-1β (R&D Systems; sensitivity 2.31 pg/mL), and IL-6 (R&D Systems; sensitivity 1.6 pg/mL) were measured using ELISA according to the manufacture’s protocol.
Statistical analysis
Power estimates were calculated with the values α = 0.05 and β = 0.8 to obtain group sizes appropriate for detecting effect sizes in the range of 30%–50% for in vivo models. Goodness-of-fit to a normal distribution was assessed by the Shapiro–Wilk test. Data are presented as mean ± standard deviation (SD). One-way analysis of variance with Tukey’s HSD post hoc tests was used to determine the significant differences between groups. Wilcoxon rank-sum test was used to determine the significant differences in neurological severity scores. Statistical analyses were performed using JMP 12.0.1 software (SAS Inc., Cary, NC, USA). A value of P < 0.05 was considered to indicate statistical significance. All experiments and measurements, including behavior outcome assessment, infarct volume measurement, and histological analysis, were performed in a blinded and randomized manner.
Results
Metabolic endotoxemia is associated with a worse stroke outcome
Male 12-week-old db/db mice showed higher levels of blood glucose (Figure 1(b)); inflammatory cytokines such as TNF-α, IL-1β, and IL-6 (Figure 1(c)); and body weight (Figure 1(d)) than db/+ mice before stroke induction. We orally administered polymyxin B and measured its plasma concentrations in db/db mice and confirmed that polymyxin B was not absorbed from the intestine (data not shown). Db/db mice treated with polymyxin-B had glucose levels (Figure 1(b)) and body weight (Figure 1(d)) similar to those in untreated db/db mice and higher than those in db/+ mice. However, the levels of TNF-α, IL-1β, and IL-6 in polymyxin B-treated db/db mice were similar to those in db/+ mice and lower than those in untreated db/db mice (Figure 1(c)). Furthermore, db/db mice showed higher plasma LPS levels than db/+ mice or db/db mice treated with polymyxin B (Figure 1(e)), accompanied by an increased intestinal permeability (Figure 1(f)) reflecting metabolic endotoxemia. To evaluate changes in gut microbiota composition, we analyzed bacterial 16S and 23S rRNA in feces of db/db and db/+ mice (Figure 1(g), Supplementary Figure 1(a), and Supplementary Table 2). The fecal counts for Atopobium cluster, Enterobacteriaceae, and Enterococcus were higher, whereas the count for Clostridium perfringens was lower in db/db mice than in db/+ mice. Bifidobacterium was undetectable in db/db mice. Polymyxin B administration attenuated the fecal count of Enterobacteriaceae in db/db mice. Although, the number of some bacteria differ among db/+, db/db, and polymyxin-B treated db/db mice, the total bacterial count was not different among these mice. This is due to the fact that the bacterial number of predominant bacteria including the Clostridium coccoides group, Clostridium leptum subgroup, Bacteroides fragilis group, and Lactobacillus that corresponds to approximately 80% to 90% of all bacteria, was not different among the groups (Supplementary Figure 2).
Subsequently, we induced transient MCAO in db/+, db/db, and polymyxin B-treated db/db mice. Physiological parameters such as weight loss (Supplementary Figure 3(a)) and rCBF (Supplementary Figure 3(b)) did not differ among the groups. Plasma LPS levels (Supplementary Table 3) and intestinal permeability (Supplementary Table 4) after MCAO were higher than those before MCAO in all groups. In addition, db/db mice showed higher plasma LPS levels (Figure 2(a)) and intestinal permeability (Figure 2(b)) after MCAO among the groups. Differences were observed in bacterial number and detection rates in some bacteria between those before and after MCAO (Supplementary Figure 4). Particularly, the bacterial number of Lactobacillus was higher after MCAO than before MCAO in all groups. Furthermore, the bacterial number and detection rates of Bifidobacterium in db/+ and polymyxin B-treated db/db mice, Enterococcus in db/db mice, and Atopobium cluster, Clostridium perfringens, and Enterobacteriaceae in all groups were lower after MCAO compared to those before MCAO. However, the gut microbiota compositions after MCAO were not different among db/+, db/db, and polymyxin B-treated db/db mice (Figure 2(c), Supplementary Table 5 and Supplementary Figure 1(b)).
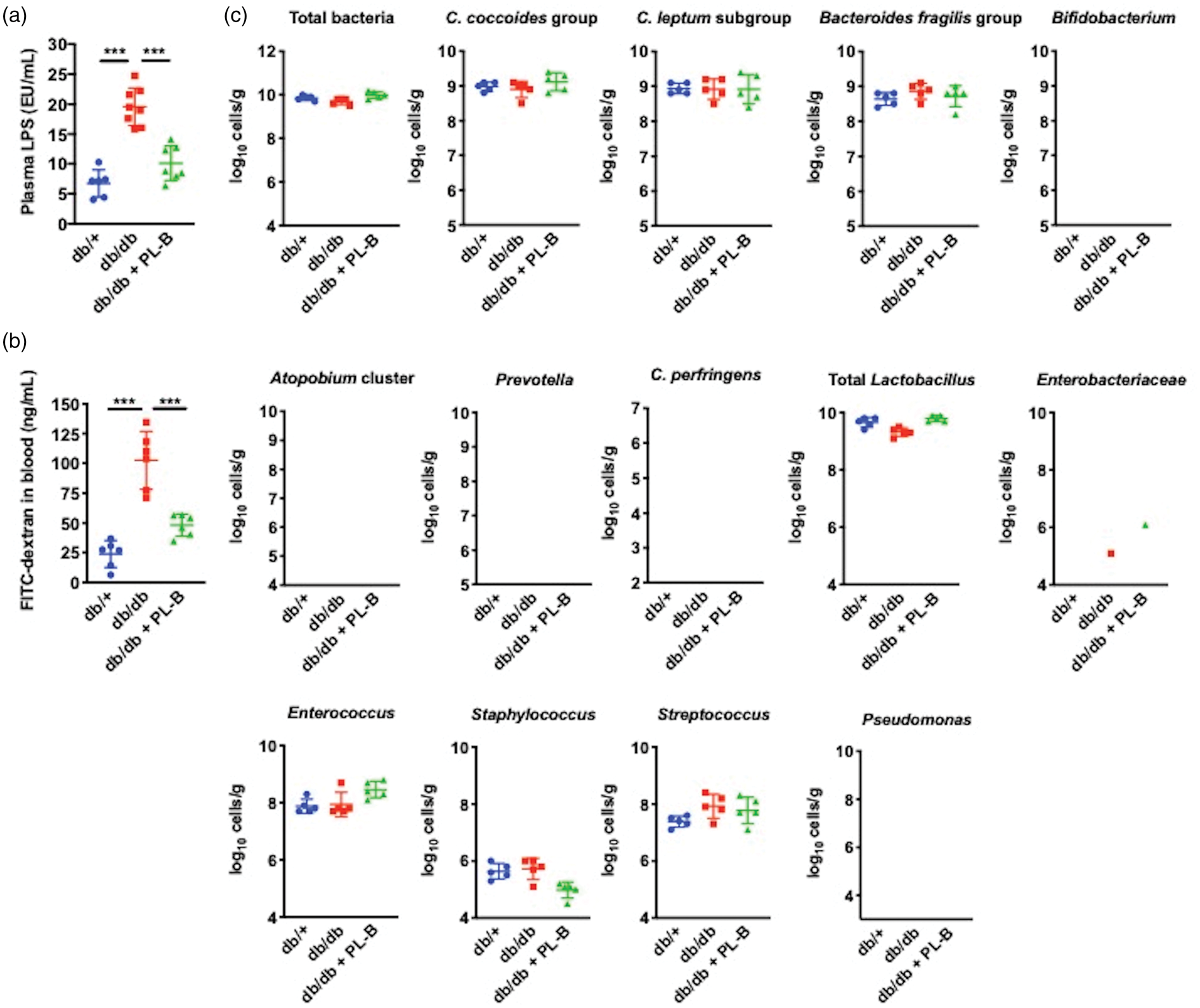
Biological parameters of db/+, db/db, and polymyxin B (PL-B)-treated db/db mice measured after stroke induction via middle cerebral artery occlusion (MCAO). (a) Plasma LPS levels after MCAO (n = 6–9 per group) (b) Intestinal permeability after MCAO (n = 5 per group). (c) Fecal bacterial counts after MCAO analyzed by rRNA-targeted quantitative reverse transcription PCR (n = 5 per group). Bacterial counts below the threshold of detection were not plotted. Data are shown as mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001.
Infarct volumes (Figure 3(a)) and blood–brain barrier damage (Figure 3(b)) were greater in db/db mice than in db/+ mice 24 h after MCAO. In addition, Iba1-positive microglia/macrophages in peri-infarct areas in db/+ mice were highly ramified, whereas those in db/db mice exhibited a less ramified and even amoeboid cell morphology (Figure 3(c)). The number of Iba1-positive cells and Iba1-immunoreactive areas were higher in db/db than in db/+ mice in peri-infarct areas 24 h after MCAO (Figure 3(c)). To analyze Iba1-positive cells in more detail, we performed immunohistochemical analysis using TMEM119, which is a specific marker of the tissue resident microglia (Figure 3(d)). Total number of both TMEM119-positive and TMEM119-negative cells was higher in db/db than db/+ mice in the peri-infarct area 24 h after MCAO (Figure 3(e)). The percentage of TMEM119-positive cells in the Iba1-positive cells was approximately 60% in both groups (Figure 3(e)). Additionally, db/db mice showed higher neurological severity scores 24 h after MCAO (Figure 3(f)) and a poorer seven-day survival rate (Figure 3(g)) than db/+ mice. However, polymyxin B treatment partially reduced the infarct volumes (still increased in comparison to db/+ mice) (Figure 3(a)) and blood–brain barrier damage (Figure 3(b)), as well as changed the morphology of Iba1-positive cells to a ramified phenotype (Figure 3(c), decreased Iba1-positive cell number and areas (Figure 3(c)), and improved neurological function 24 h after MCAO (Figure 3(f)) and seven-day survival (Figure 3(g)) in db/db mice.
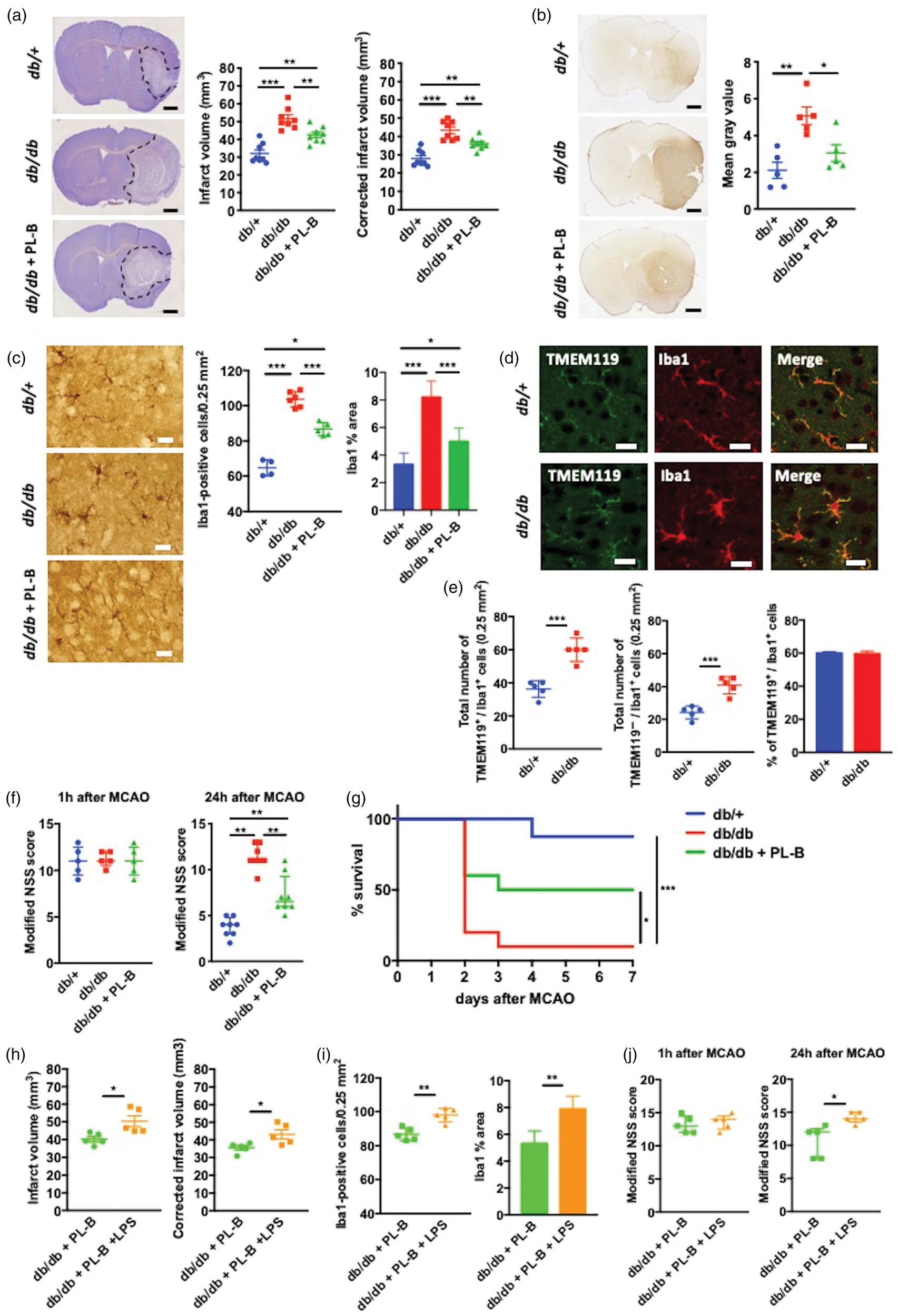
Stroke outcomes in db/+, db/db, and polymyxin B (PL-B)-treated db/db mice. (a) Representative images of Cresyl Violet-stained brain sections (left) and quantification of infarct volumes (right) 24 h after middle cerebral artery occlusion (MCAO). Total hemispheric infarct volumes were corrected for edema (n = 8 per group). Scale bars = 1 mm. (b) Representative images (left) and quantification (right) of blood–brain barrier damage assessed by IgG immunoreactivity 24 h after MCAO (n = 5 per group). Scale bars = 1 mm. (c) Representative images of Iba1-positive cells, cell count of Iba1-positive cells, and percentage of Iba1-positive area in the peri-infarct area 24 h after MCAO (n = 4–6 per group). Scale bars = 20 μm. Data are shown as mean ± SD. (d) Double-immunofluorescence of Iba1 and anti-transmembrane protein 119 (TMEM119) in the peri-infarct area 24 h after MCAO in db/+ and db/db mice. Scale bars = 20 μm. (e) Total number of TMEM119-positive cells and TMEM119-negative cells in Iba1-positive cells, and percentage of TMEM119-positive cells in Iba1-positive cells in the peri-infarct area 24 h after MCAO in db/+ and db/db mice (n = 5 per group). (f) Modified neurological severity scores (NSS) 1 h and 24 h after MCAO (n = 8 per group). Data are shown as median and interquartile range. (g) Seven-day survival after MCAO (n = 8–10 per group). Kaplan–Meier curve followed by the log rank test. (h) Infarct volumes 24 h after MCAO in polymyxin B-treated db/db mice with and without intraperitoneal lipopolysaccharide (LPS) administration. Total hemispheric infarct volumes were corrected for edema (n = 5 per group). (i) Cell count of Iba1-positive cells and percentage of Iba1-positive area in the peri-infarct area 24 h after MCAO in polymyxin B-treated db/db mice with and without intraperitoneal LPS administration. (n = 5 per group). Data are shown as mean ± SD. (j) Modified NSS 1 h and 24 h after MCAO in polymyxin B-treated db/db mice with and without intraperitoneal LPS administration (n = 5 per group). Data are shown as median and interquartile range. *P < 0.05, **P < 0.01, and ***P < 0.001.
Because polymyxin B treatment attenuated the circulating LPS levels and ameliorated the stroke outcome in db/db mice, we examined the direct impact of LPS on stroke outcome in db/db mice. In polymyxin B-treated db/db mice, intraperitoneal LPS administration before MCAO increased the infarct volume (Figure 3(h)) and number of Iba1-positive cells and Iba1-immunoreactive areas in the peri-infarct area (Figure 3(i)), as well as the neurological severity score (Figure3(j)), 24 h after MCAO.
In the normoglycemic control db/+ mice, infarct volume (Figure 3(a)) and stroke outcomes such as neurological severity score (Figure 3(f)) and survival rate after MCAO (Figure 3(g)) were comparable to those in C57BL/6 mice, which has been reported in our previous study. 17 Oral gavage of polymyxin B did not show any effects on plasma LPS levels (Supplementary Figure 5(a)), intestinal permeability (Supplementary Figure 5(b), infarct volumes 24 h after MCAO (Supplementary Figure 5(c)) and neurological functions 1 h and 24 h after MCAO (Supplementary Figure 5(d)) in db/+ mice. Additionally, the bacterial counts did not change between db/+ and polymyxin B-treated db/+ mice 24 h after MCAO (Supplementary Figure 5(e)).
LPS and neuroinflammation in the ischemic brain
Since we found that metabolic endotoxemia was associated with worse stroke outcome, we analyzed the presence of LPS in the brain 24 h after ischemia. Western blot analysis revealed that LPS levels were higher in the ischemic hemispheres of db/db mice compared to those of db/+ mice; LPS levels were attenuated by polymyxin B treatment in db/db mice (Figure 4(a)). In all groups, LPS was not detected in the contralateral, non-ischemic hemispheres (Supplementary Figure 6(a)). Next, we examined the expression levels of TLR4, an LPS receptor, and inflammatory cytokines in the ischemic brain. Expression levels of TLR4 (Figure 4(b)), TNF-α, IL-1β, and IL-6 (Figure 4(c)) were higher in db/db mice than in db/+ mice, but lower in polymyxin B-treated db/db mice than in untreated db/db mice. The expression levels of TLR4 and these inflammatory cytokines in the contralateral hemispheres did not change among these groups (Supplementary Figure 6(b) and (c)).
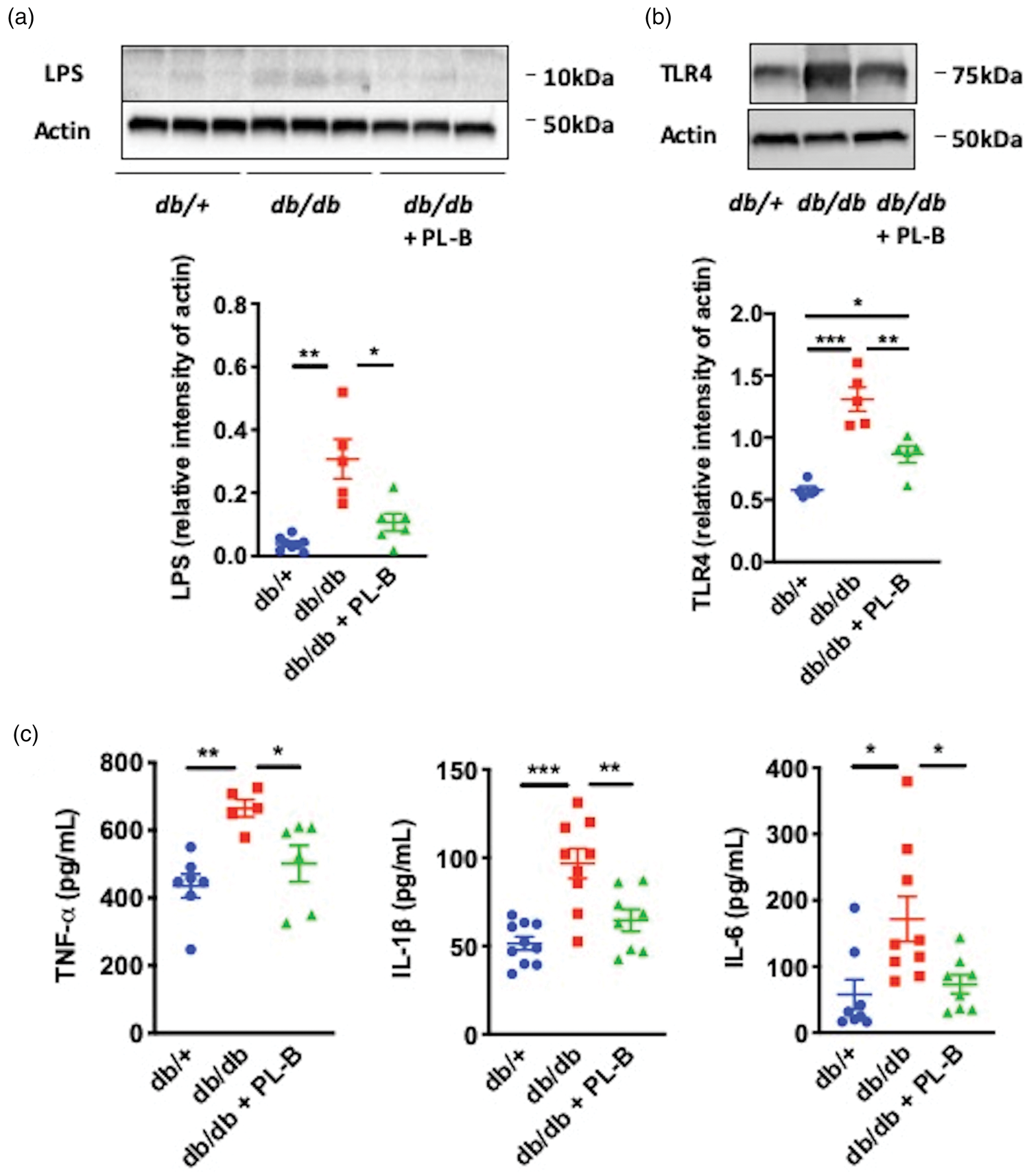
Lipopolysaccharide (LPS), toll-like receptor (TLR) 4, and inflammatory cytokines in the ischemic hemispheres of db/+, db/db, and polymyxin B (PL-B)-treated db/db mice. Western blot analyses of (a) LPS and (b) TLR4 expression. (c) Enzyme-linked immunoassay results for tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6. All data were analyzed in mice sacrificed 24 h after middle cerebral artery occlusion (n = 5–10 per group). Data are shown as mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001.
Localization of LPS, E. coli K99 pili protein, and TLR4 in the ischemic brain
We examined the localization of LPS and E. coli (which belongs to the Enterobacteriaceae family) K99 pili protein in the brain 24 h after ischemia. The immunohistochemical analysis demonstrated that LPS (Figure 5(a)) and E. coli K99 pili protein (Figure 5(b)) were localized in the peri-infarct area in Iba1-positive microglia/macrophages, neurons, and endothelial cells, but not in astrocytes. High magnification images showed that LPS (Figure 5(a)) and E. coli K99 pili protein (Figure 5(b)) were adherent on the surface of Iba1-positive microglia/macrophages, neurons, and endothelial cells. Neither LPS nor E. coli K99 pili protein was detected in the contralateral hemispheres (data not shown). We also observed in the peri-infarct area, TLR4 immunoreactivity in Iba1-positive microglia/macrophages, neurons, and endothelial cells, but not in astrocytes (Figure 5(c)). The percentages of TLR4-immunoreactive areas in these cells were higher in db/db mice compared to db/+ mice and polymyxin B-treated db/db mice (Figure 5(d)).
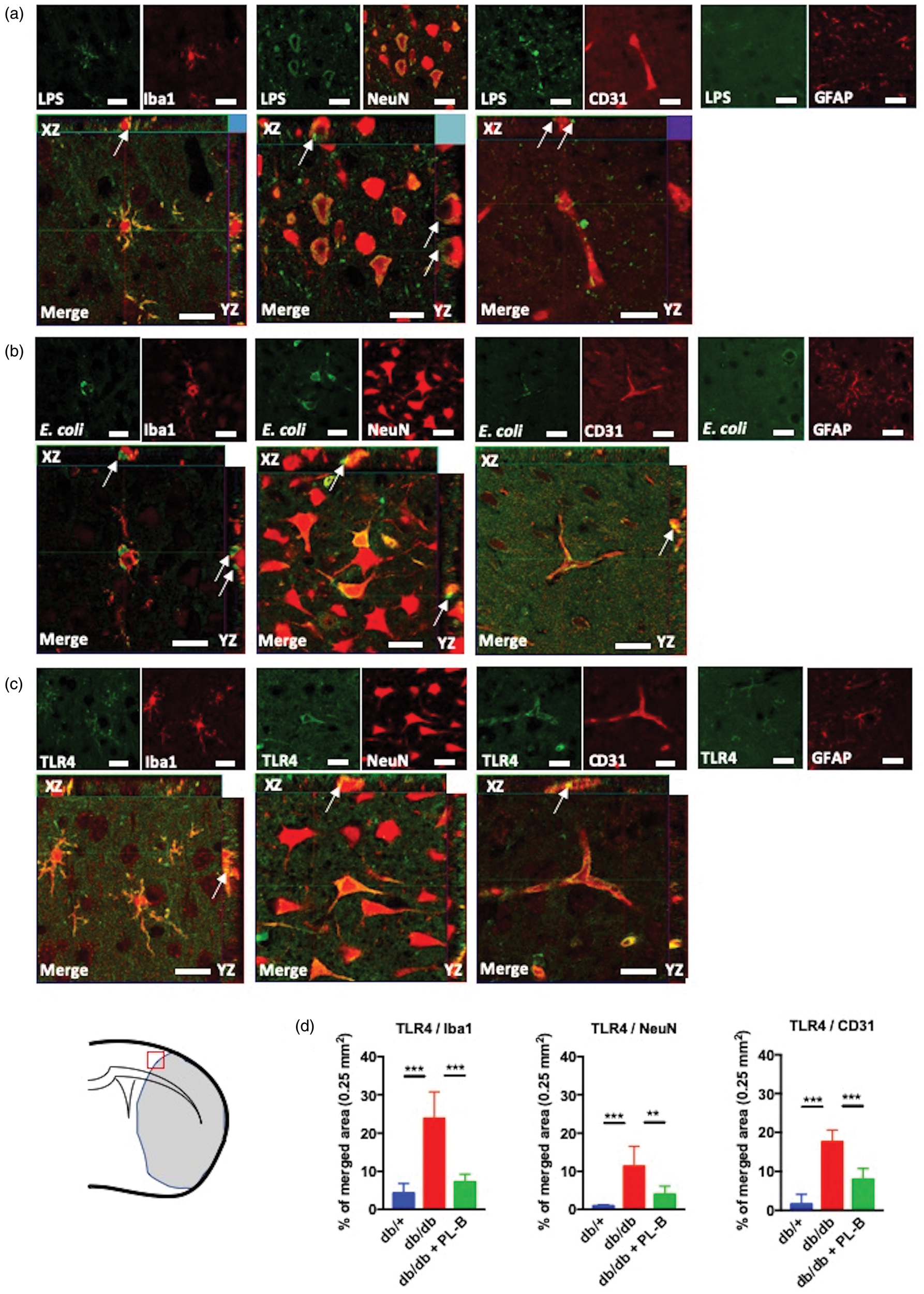
Cellular localization of lipopolysaccharide (LPS), E. coli K99 and toll-like receptor 4 (TLR4) in the peri-infarct areas of ischemic brain. Double-immunofluorescence of (a) LPS, (b) E. coli K99, and (c) TLR4 in microglia/macrophages (Iba1), neurons (NeuN), endothelial cells (CD31), and astrocytes (GFAP) in the peri-infarct area 24 h after transient middle cerebral artery occlusion in db/db mice. High magnification images show that (a) LPS and (b) E. coli K99 were adherent on the surface of microglia/macrophages, neurons and endothelial cells (arrows). (c) TLR4 was colocalized with Iba1, NeuN and CD31 (arrows). Scale bars = 20 μm. A red square in the brain illustration shows the region of analysis. (d) The percentages of TLR4-immunoreactive areas in Iba1, NeuN, and CD31-positive cells in db/+, db/db, and polymyxin B (PL-B)-treated db/db mice (n = 6 per group). Data are shown as mean ± SD. **P < 0.01 and ***P < 0.001.
Discussion
The present study demonstrates that metabolic endotoxemia is negatively associated with stroke outcome. Increased intestinal permeability and circulating LPS levels were associated with increased levels of LPS, TLR4, and inflammatory cytokines in the ischemic brain in diabetic mice (Figure 6). Moreover, gut microbiota modulation with a non-absorbable antibiotic polymyxin B attenuated LPS levels in the circulation, thereby in the ischemic brain and improved the stroke outcome. Notably, LPS administration attenuated the positive effects of gut microbiota modulation with the antibiotic on stroke outcomes. Additionally, polymyxin B treatment had no effect on stroke outcomes in control mice, in which circulating LPS levels were much lower than in diabetic mice. These findings suggest an association between metabolic endotoxemia and the pathophysiology of acute ischemic brain injury via the Gram-negative bacterial toxin LPS in diabetic mice.
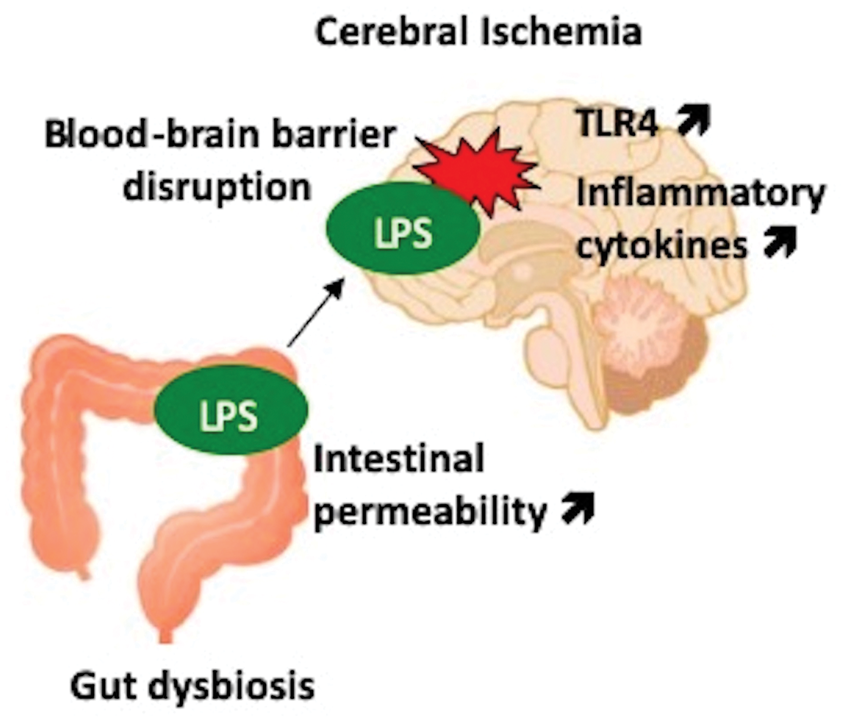
Proposed mechanism of ischemic brain injury induced by metabolic endotoxemia. Gut dysbiosis in type 2 diabetes causes increased intestinal permeability and the translocation of microbiota-derived lipopolysaccharide (LPS) into the circulatory system. Disruption of the blood–brain barrier after cerebral ischemia allows an influx of LPS into brain parenchyma, where the induction of toll-like receptor 4 (TLR4), and inflammatory cytokines exacerbate ischemic brain injury.
In the present study, db/db mice showed enrichment of fecal Enterobacteriaceae. Enterobacteriaceae are a large family of Gram-negative bacteria that includes opportunistic pathogens such as E. coli, Klebsiella, and Salmonella. Consistent with the results of our study, a previous study in db/db mice also showed an increase in Proteobacteria (this phylum contains Enterobacteriaceae). 24 An increase in the number of Proteobacteria is observed during gut dysbiosis in several diseases including inflammatory bowel disease, colorectal cancer, metabolic syndrome, and type 2 diabetes. 25 In addition, a previous clinical study showed that acute stroke patients had a more abundant presence of Enterobacter (which belongs to the Enterobacteriaceae family). 26 However, dysbiosis with expansion of the Burkholderiaceae family, which belongs to the Proteobacteria, is associated with neuroprotective effects on ischemic cerebral injury in mice. 27
Polymyxin B is one of the primary classes of antibiotics with activity against most Gram-negative bacteria. 28 In our bacterial analysis, Gram-negative bacteria included the Bacteroides fragilis group, Prevotella, Enterobacteriaceae, and Pseudomonas. However, Prevotella and Pseudomonas were undetectable in all groups. Polymyxin B attenuated Enterobacteriaceae. However, polymyxin B had no effects on the bacterial counts of Bacteroides fragilis, known to be resistant to this antibiotic. 29 We showed that increased fecal levels of Enterobacteriaceae were associated with increases in circulating LPS levels concurrent with increased intestinal permeability before stroke induction. Emerging evidence has revealed that gut microbiota has potent effects on intestinal permeability. The proliferation of Enterobacteriaceae leads to an increase in luminal LPS content in the gut, which results in changes in tight junction proteins and an increase in intestinal permeability through TLR4-induced inflammation. 30 The substantial changes in expression/distribution of tight junction proteins with enhanced intestinal permeability have been shown to lead to an abnormal leakage of bacterial LPS into the circulatory system in db/db mice. 9 We also found that higher plasma LPS levels were associated with increasing inflammatory cytokine levels, while gut microbiota modulation reduced circulating LPS levels and inflammatory signaling. These findings are consistent with a previous report investigating endotoxemia and systemic inflammation using db/db mice. 9 Furthermore, our study found that Bifidobacterium was undetectable in feces of db/db mice before stroke induction. Decreased Bifidobacterium levels have been implicated in intestinal barrier dysfunction and LPS translocation.31,32 Therefore, the reduction in Bifidobacterium may also have increased the levels of circulating LPS in db/db mice in our study. Additionally, our study showed that the number of Clostridium perfringens in db/db mice was lower than that in db/+ mice. This finding is in line with a previous study, which showed a lower amount of Clostridium perfringens in obese subjects compared to normal-weight people. 33 Thus, a decrease in the number of Clostridium perfringens may be associated with an obese phenotype. However, these Gram-positive bacteria were not affected by polymyxin B treatment in db/db mice, suggesting Clostridium perfringens may not have contributed to endotoxemia before stroke induction in these mice.
Lately, special focus has been placed on the gut–brain axis in ischemic stroke pathophysiology. Gut microbiota have been implicated in ischemic brain injury after stroke via the regulation of intestinal T cells.27,34 Additionally, stroke causes specific changes in gut microbiota, which in turn contributes to stroke outcomes.34–36 Stroke also promotes the translocation and dissemination of bacteria from host gut microbiota as a mechanism leading to post-stroke infection. 19 Recently, Gram-negative bacteria-derived LPS has been implicated in the neuropathology of the human brain. For example, LPS and the K99 pili protein of Gram-negative E. coli bacteria were detected at higher levels in brains of patients with Alzheimer’s disease compared to controls and were colocalized with amyloid β in amyloid plaques and around vessels. 37 However, the origin of these bacterial molecules in the brain is currently not well understood.
In the current study, we determined the presence of LPS and E. coli K99 pili protein in the ischemic brain 24 h after cerebral ischemia. Lipopolysaccharide levels in the ischemic brain were attenuated by oral gavage of the non-absorbable antibiotic, polymyxin B in db/db mice. These findings were associated with suppression of fecal Enterobacteriaceae counts, intestinal permeability, and circulating LPS levels before stroke induction. We also found that intestinal permeability was further enhanced after MCAO in all group mice. These findings were in line with a previous study investigating the intestinal permeability in mice subjected to MCAO. 19 Importantly, our study showed that db/db mice had the most prominent increase of plasma LPS levels and intestinal permeability after MCAO among groups. Our study also showed that stroke induced certain changes in gut microbiota, particularly an increase in Lactobacillus, which is in line with a previous study. 36 Additionally, some non-predominant bacteria were reduced or undetectable after MCAO. Reduction in species diversity is a key feature of dysbiosis after stroke. 34 However, the gut microbiota compositions after MCAO were not different among db/+, db/db, and polymyxin B-treated db/db mice. Therefore, our findings suggest that stroke has a greater impact on gut microbiota composition than the phenotypes such as type 2 diabetes.
Toll-like receptor 4 plays an important role in post-stroke inflammation and contributes to the progression of brain damage, whereas genetic deletion of TLR4 significantly reduces the infarct volume. 38 Damage-associated molecular patterns released from injured neurons such as high mobility group box 139 and peroxiredoxin family proteins 40 activate TLR4 in the brain after cerebral ischemia. Among the members of the TLR family, TLR4 is mainly involved in LPS-mediated inflammatory responses. 1 In the present study, TLR4 and inflammatory cytokine levels in the ischemic brain were significantly higher in db/db compared to db/+ mice. These findings were associated with LPS levels in ischemic brains; the reduction in LPS levels in polymyxin B-treated db/db mice decreased TLR4 and inflammatory cytokine levels. We also found that db/db mice showed higher levels of plasma inflammatory cytokines compared with db/+ and polymyxin B-treated db/db mice. Therefore, although ischemic injury increases TLR4 activation,39,40 it is also possible that influx of LPS, as well as extravasation of plasma inflammatory cytokines into the ischemic brain, also contributes to neuroinflammation.
In the peri-infarct area, we found that LPS and E. coli K99 pili protein were adherent on the surface of Iba1-positive microglia/macrophages, endothelial cells, and neurons, but not in astrocytes. Furthermore, Iba1-positive microglia/macrophages, endothelial cells, and neurons, except astrocytes expressed TLR4. Immunoreactive areas of TLR4 in these cells were higher in db/db mice compared to db/+ and polymyxin B-treated db/db mice, suggesting TLR4 activation was pronounced in db/db mice. Among the major non-neuronal cell types in the brain, microglia expressed high levels of TLR4, whereas astrocytes expressed none. 41 Furthermore, microglia are the only glial cells that bind LPS. 41 These findings suggest that microglia/macrophages contribute to LPS-induced neuroinflammation. The question of TLR4 expression in astrocytes has not been undeniably clarified, since in different studies TLR4 was either absent41,42 or present. 43 These inconsistencies in the reported expression patterns may be related to differences in the experimental paradigms and immunoprobes used. Endothelial cells also express TLR444 and produce cytokines in response to LPS stimulation. 45 Additionally, LPS has been reported to induce inflammatory cytokine production in neurons via TLR4 activation. 46
Our study showed that Iba1-positive cells in db/+ mice were characterized by a highly ramified shape, whereas those in db/db mice showed a less ramified or amoeboid cell morphology, reflecting their activation status. 47 The origin of cells identified by Iba1 could either be activated brain-resident microglia or macrophages derived from circulating monocytes. 47 The TMEM119 is a recently identified highly specific cell-surface marker of microglia that is not expressed by macrophages or other immune or neural cell types. 48 Our study showed that approximately 60% of Iba1-positive cells expressed TMEM119 in both db/+ and db/db mice. This is in line with a pathological study of human brains showing that 43% of Iba1-positive cells at the edge of acute ischemic lesions expressed TMEM119. 49 Altogether, these findings suggest that Iba1-positive cells consist in part of resident microglia, and in part of infiltrating macrophages in per-infarct areas of acute cerebral ischemia.
Although systemic LPS administration has been shown to disrupt the blood-brain barrier, 50 we did not see significant differences in IgG staining in the contralateral, non-ischemic hemispheres among db/+, db/db, and polymyxin B-treated db/db mice. Indeed, LPS was not detected and levels of TLR4 and inflammatory cytokines were not different in the non-ischemic hemispheres in all these mice. This might be explained by differences in experimental models (systemic LPS administration vs. metabolic endotoxemia), which may result in different effects on the blood–brain barrier.
Interestingly, LPS preconditioning provides neuroprotective effects via several mechanisms including proinflammatory cytokine production. Induction of TNF-α by LPS administration before stroke causes ischemic tolerance by suppressing the TNF-α response to cerebral ischemic injury. 51 Furthermore, LPS pre-treatment reduces N-methyl-D-aspartate-mediated cerebral injury by inducing nitric oxide and cGMP. 52 Thus, LPS can be neuroprotective when it is administered under optimal conditions (dose and timing). However, our study indicated that chronic exposure to LPS may be deleterious by increasing neuroinflammation after stroke.
Among the cardiovascular risk factors, type 2 diabetes is considerably associated with poor functional outcomes after stroke. 14 Yet, controlling post-stroke hyperglycemia has no effect on the outcome in experimental animal models 15 or stroke patients. 16 In the present study, we demonstrated that the reduction in circulating LPS levels was associated with an attenuation of inflammatory cytokine production, as well as with improvements in neurological functions and survival after stroke, without influencing plasma glucose levels in a mouse model of type 2 diabetes. Although our study demonstrated that polymyxin B was effective to reduce the bacterial counts of Enterobacteriaceae and thereby decrease circulating LPS levels, antibiotic treatment may not be suitable for the clinical setting due to potential undesirable side effects associated with antibiotic treatment and the risk of emergence of antibiotic-resistant bacteria. Experimental studies have shown that supplementation of Bifidobacterium 31 or oligofructose 32 decreases plasma LPS levels. Thus, the administration of pro- or prebiotics may be a potential treatment option to improve stroke outcomes by preventing LPS translocation in patients with type 2 diabetes.
Although interspecies differences should always be considered, the murine gut has successfully been used as a model to investigate the role of gut microbiota in complex human disorders, including post-stroke infections, obesity, and diabetes.8,19
The present study had some limitations. First, although we used sterile techniques, LPS could have contaminated tissue samples. However, LPS levels were consistently higher in db/db compared to db/+ and polymyxin B-treated db/db mice, despite using the same techniques in all groups. In addition, LPS was not detected in non-ischemic hemispheres. Second, the effects of other bacteria, not included in our microbiota analysis, need to be further investigated. However, the rRNA-targeted qRT-PCR used in this study enables high-resolution quantification of targeted bacterial populations including potentially pathogenic bacteria which might not be quantified efficiently, by either routine DNA-based PCR or next-generation sequencing methods due to their low sensitivity.53,54 Third, we focused on LPS in the present study, but further studies are needed to examine the potential importance of other bacteria-derived molecules such as peptidoglycan in stroke pathophysiology. Additionally, as gut microbiota contributes to ischemic cerebral injury by regulating peripheral immune cells,27,34 analyzing the impact of gut dysbiosis and polymyxin B treatment on peripheral immune cells is crucial. Fourth, since parameters were measured post-stroke and correlated with the size of the lesion, they did not address the direct role of endotoxemia. Experiments with TLR4 knock-out mice would provide more details about the interactions between endotoxemia and neuroinflammation. Additionally, our study showed that db/db mice had greater ischemic damage compared to db/+ mice 24 h after MCAO. To elucidate how the infarct develops over time, further investigations are needed at earlier time points. Finally, our results are limited to young male mice and may not be applicable to old or female mice. In addition, the 12-week-old db/db mice used in the current study had extremely high glucose levels, which could be responsible for the poor survival rate after stroke, and this limited the ability to conduct long-term studies on the effect of polymyxin B or the effects of polymyxin B administration after stroke. Particularly, it would be of interest to examine whether LPS that remains in the brain, contributes to poststroke cognitive decline in a similar manner to Alzheimer’s disease. 37 In this regard, a milder diabetic model such as high-fat diet fed-mice would be useful in future research.
In conclusion, our study suggests that metabolic endotoxemia is associated with neuroinflammation after transient focal cerebral ischemia in type 2 diabetes. Targeting metabolic endotoxemia might be a novel potential therapeutic strategy to improve stroke outcomes.
Supplemental Material
JCB899577 Supplemental Material - Supplemental material for Metabolic endotoxemia promotes neuroinflammation after focal cerebral ischemia
Supplemental material, JCB899577 Supplemental Material for Metabolic endotoxemia promotes neuroinflammation after focal cerebral ischemia by Naohide Kurita, Kazuo Yamashiro, Takuma Kuroki, Ryota Tanaka, Takao Urabe, Yuji Ueno, Nobukazu Miyamoto, Masashi Takanashi, Hideki Shimura, Toshiki Inaba, Yuichiro Yamashiro, Koji Nomoto, Satoshi Matsumoto, Takuya Takahashi, Hirokazu Tsuji, Takashi Asahara and Nobutaka Hattori in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was funded by the JSPS KAKENHI grant no. JP15K07439.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
NK, KY, TK, RT, TU, YY, NH: Conception and experimental design; NK, KY, TK, YU, NM, MT, HS, TI, KN, SM, TT, HT, TA: Experimental work; NK, KY: Interpretation of data and writing of manuscript. All authors discussed the results and provided input regarding the manuscript. All the authors have approved the final version of the manuscript.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.