
Abstract
We investigated the effects of sulforaphane (SFN), an isothiocyanate from cruciferous vegetables, in the regulation of cerebral blood flow using cranial windows in newborn pigs. SFN administered topically (10 µM–1 mM) or systemically (0.4 mg/kg ip) caused immediate and sustained dilation of pial arterioles concomitantly with elevated H2S in periarachnoid cortical cerebrospinal fluid. H2S is a potent vasodilator of cerebral arterioles. SFN is not a H2S donor but it acts via stimulating H2S generation in the brain catalyzed by cystathionine γ-lyase (CSE) and cystathionine β-synthase (CBS). CSE/CBS inhibitors propargylglycine, β-cyano-L-alanine, and aminooxyacetic acid blocked brain H2S generation and cerebral vasodilation caused by SFN. The SFN-elicited vasodilation requires activation of potassium channels in cerebral arterioles. The inhibitors of KATP and BK channels glibenclamide, paxilline, and iberiotoxin blocked the vasodilator effects of topical and systemic SFN, supporting the concept that H2S is the mediator of the vasodilator properties of SFN in cerebral circulation. Overall, we provide first evidence that SFN is a brain permeable compound that increases cerebral blood flow via a non-genomic mechanism that is mediated via activation of CSE/CBS-catalyzed H2S formation in neurovascular cells followed by H2S-induced activation of KATP and BK channels in arteriolar smooth muscle.
Keywords
Introduction
Sulforaphane (SFN), an organosulfur phytochemical isothiocyanate from cruciferous vegetables, has a wide spectrum of biological effects that involve transcriptional control of gene expression. Historically, SFN has attracted the attention of numerous researchers as a potential anti-cancer dietary agent. 1 Recently, anti-inflammatory and cardioprotective properties of the compound have been also described.2–8 The mechanisms of pleiotropic effects of SFN have been linked largely to upregulation of various antioxidant genes via activation of the transcription factor nuclear factor erythroid 2-related factor 2 (Nfr2).2–8 The cytoprotective properties of SFN via the Nrf2-mediated gene targeting mechanism require an extended time period to complete (at least several hours).
There is a paucity of information on the effects of SFN in the brain. Studies in mice suggest that the accumulation of orally administered SFN in the brain is limited as compared to its accumulation in the small intestine, kidney, lung, and prostate. 9 However, recent studies in rodents suggest that SFN supplements may protect the ischemic brain via a gene targeting mechanism.3–6,10,11
The mechanism that accounts for the biological activity of SFN remains elusive. The isothiocyanate moiety of SFN has been recognized as an H2S-releasing chemical group.12,13 However, no data are available to support the ability of SFN to spontaneously release H2S. Furthermore, there is no experimental evidence that H2S accounts for the biological effects of SFN. The gaseous messenger H2S has emerged as a vasoactive and cytoprotective gaseous mediator produced by many organs, including the brain, the heart, and kidneys.14–18 The pleiotropic effects of H2S in the systemic and cerebral circulation include vasodilator, anti-inflammatory, and antiapoptotic activities. We have pioneered the studies on the role of H2S as a physiologically important vasodilator in the cerebral circulation of newborn pigs produced in the brain via L-cysteine catabolism by cystathionine-γ-lyase (CSE) and cystathionine β-synthase (CBS).16,17,19–21 H2S-produced vasodilation involves activation of KATP and BK channels in cerebral arterioles17–20 and systemic circulation.14,18,22–24 H2S may directly activate plasma membrane KATP channels in vascular smooth muscle (VSM) cells from cerebral vessels19 and systemic arteries22,24 leading to elevation in the channel currents, membrane hyperpolarization, and vasodilation. Indirect activation of BK channels by H2S in cerebral arteriole smooth muscle cells involves Ca2+ spark activation, elevation in BK current frequency, membrane hyperpolarization, and vasodilatation. 20
To date, there are no reports on non-genomic effects of SFN in the brain. Our study was designed to investigate acute effects of SFN in intact cerebral circulation. We tested the hypothesis that SFN is a brain-permeable compound that increases cerebral blood flow via a mechanism that involves enzymatic H2S production by the brain. Using the closed cranial window technique in newborn pigs, we detected the effects of systemically and topically administered D, L-SFN on the diameter of pial arterioles and on H2S production by the brain. We report here, for the first time, that SFN administered systemically via intraperitoneal injection or topically placed onto the brain surface exhibits potent vasodilator properties that require an endogenous CSE/H2S-mediated mechanism that involves arteriolar VSM KATP and BK channels.
Materials and methods
Animals
Newborn piglets (one to five days old, 1.5–3.0 kg, either sex) were purchased from a commercial breeder. The Animal Care and Use Committee of the University of Tennessee Health Science Center reviewed and approved all procedures involving animals in compliance with National Institutes of Health Office of Laboratory Animal Welfare guidelines. All experiments in the study were conducted according to the ARRIVE guidelines for animal experiments.
Cranial windows for intravital microscopy of pial arterioles
The pigs were anesthetized with acepromazine/ketamine/xylazine (3.3/33/2 mg/kg im), intubated via tracheostomies, and placed on mechanical ventilation to maintain blood gases in a normal range (arterial PCO2: 30–40 mm Hg, arterial PO2: 70–90 mm Hg, and pH 7.3–7.4). Femoral arterial and venous catheters were inserted for monitoring of cardiovascular parameters and blood gases. Long-term anesthesia was achieved with α-chloralose (50 mg/kg iv initially plus 5 mg/kg maintenance as needed). The closed cranial window technique was used for observation of pial arterioles and collection of periarachnoid cerebrospinal fluid (pCSF) for detection of H2S production as described previously.21,25 The space under the window was filled with artificial cerebrospinal fluid (aCSF) (in mM): 3.0 KCl, 1.5 MgC2, 1.5 CaCl2, 132 NaCl, 6.6 urea, 3.7 dextrose, and 24.6 NaHCO3 equilibrated with 6% CO2-6% O2-88% N2 to pH 7.3–7.35 at 37℃. Pial arterioles are major cerebral resistance vessels that contribute to cerebral blood flow regulation. The diameter of pial arterioles was measured with a videomicrometer coupled to a television camera mounted on the microscope. Several medium-sized pial arterioles (40–80 μm diameter) were selected for observation in each piglet.
Experimental protocols
Effects of topical administration of SFN on cerebral circulation
D, L-SFN was dissolved in DMSO (100 mM stock) and diluted with aCSF for applications. To detect dose-dependent effects of topical SFN on pial arteriolar diameter, brain H2S production, and systemic cardiovascular parameters, SFN in ascending concentrations from 10−6 M to 10−3 M was applied directly to the brain surface through the cranial window ports in consecutive 10-min intervals. The diameter of pial arterioles was recorded, and the samples of pCSF (0.5 ml) were collected through the port of the cranial window in 10-min intervals for H2S measurements.
To investigate whether CSE/CBS activities contribute to the effects of topical SFN, we used the CSE-selective inhibitors propargylglycine (PPG, 5 mM) and β-cyano-L-alanine (BCA, 10−4 M), and the CBS/CSE inhibitor aminooxyacetic acid (AOA, 10−3 M) placed on the brain surface under the cranial window. 21 To determine whether arteriolar potassium channels are involved in the mechanism of SFN-induced cerebral vasodilation, we used the KATP blocker glibenclamide (10−7 M), and the BK channel blockers paxilline (4 × 10−5 M) and iberiotoxin (10−7 M) administered topically on the brain surface.17,19–21 To investigate the effects of the inhibitors on the responses to topical SFN, we used a repeated measurements protocol in the following groups (n = 4 animals in each group): (1) control group (10−6–10−3 M SFN in aCSF); (2) PPG-treated group (SFN in aCSF with 5 mM PPG); (3) BCA-treated group (SFN in aCSF with 0.1 mM BCA); (4) AOA-treated group (SFN in aCSF with 1 mM AOA); (5) glibenclamide-treated group (SFN in aCSF with 10−7 M glibenclamide); (6) paxilline-treated group (SFN in aCSF with 4 × 10−5 M paxilline), and (7) iberiotoxin-treated group (SFN in aCSF with 10−7 M iberiotoxin). After recording the control dose-dependent responses to SFN (10−6–10−3 M) dissolved in aCSF, the cranial window was flushed with aCSF for 30 min to allow pial arterioles to return to the baseline. The inhibitors were then topically applied to the brain for 30 min, and the dose-dependent responses to SFN in the presence of each inhibitor were repeatedly recorded. Each animal received one control and one experimental treatment with SFN and the inhibitor. The H2S-independent vasodilator sodium nitroprusside (SNP, 10−6 M) was used at the end of the experiment to assess overall cerebral VSM function. The samples of pCSF (0.5 ml) were collected through the port of the cranial window in 10-min intervals for H2S measurements. Systemic cardiovascular parameters (mean arterial blood pressure, heart rate, blood gases, and body temperature) were continuously recorded.
Effects of systemic administration of SFN on cerebral circulation
We detected time-dependent effects of systemic SFN on pial arteriolar diameter, brain H2S production, and systemic cardiovascular parameters. SFN (0.4 mg/kg) was dissolved in DMSO/5 ml saline (1:100) and injected aseptically intraperitoneally via a syringe equipped with a 0.2 µm Millipore filter. To investigate whether CSE activity and potassium channels contribute to the effects of systemic SFN, we used the following groups: (1) control group (SFN 0.5 mg/kg ip alone; n = 7); (2) topical 0.1 mM BCA-treated group (n = 4); (3) topical 1 mM AOA-treated group (n = 4); (4) topical 10−7 M glibenclamide-treated group (n = 4); and (5) topical 10−5 M paxilline-treated group (n = 4). Pial arteriolar diameter and systemic parameters were recorded before and after SFN administration in control and inhibitor-treated animals, and the samples of pCSF were collected in 10-min intervals for H2S measurements for a period of up to 2 h. The H2S-independent vasodilator SNP (10−6 M) was used at the end of the experiment to assess cerebral VSM function in all groups.
Detection of enzymatic H2S production by the brain cortex tissue
The samples of the brain cortex homogenate in Krebs buffer (1:10) were incubated in sealed vials in the absence or presence of the inhibitors of enzymatic H2S production in the brain BCA (0.1 mM) or AOA (1 mM) under baseline conditions (Krebs alone) and with 20 µM SFN for 1 h at 37℃. The samples were placed in an ice bath to stop the reaction, precleared by centrifugation, and analyzed for H2S. The H2S values were normalized to the total amount of protein in each sample of the brain homogenate.
Measurements of H2S
We used a H2S-selective electrode (Lazar Research Laboratories) on a Jenko Model 6230 microcomputer-based pH/mV/Temp meter (Jenko Electronics, LTD) to measure H2S levels in pCSF samples as described elsewhere. 21 Sodium sulfide (Na2S) (0.5–10 µM) was used for a calibration curve, as suggested by the manufacturer. Using the standard calibration curve, H2S sample readings in mV were normalized to baseline and expressed in µM. The H2S detection limit was 0.4 ± 0.2 µM H2S.
Materials
Hydrogen sulfide gas (99.5%) was from NexAir (Memphis, TN). Paxilline was from R@D Systems (Minneapolis, MN), iberiotoxin was from Tocris Bioscience (Minneapolis, MN). D, L-Sulforaphane and all other reagents were purchased from MilliporeSigma (Burlington, MA).
Statistical analysis
Data are presented as mean ± SD of absolute values or percentage of control. Data comparison was performed using Student's t-test and one-way analysis of variance (ANOVA). P values of < 0.05 were considered statistically significant.
Results
Systemic cardiovascular parameters
Effects of topical and systemically administered sulforaphane on systemic cardiovascular parameters in newborn pigs.
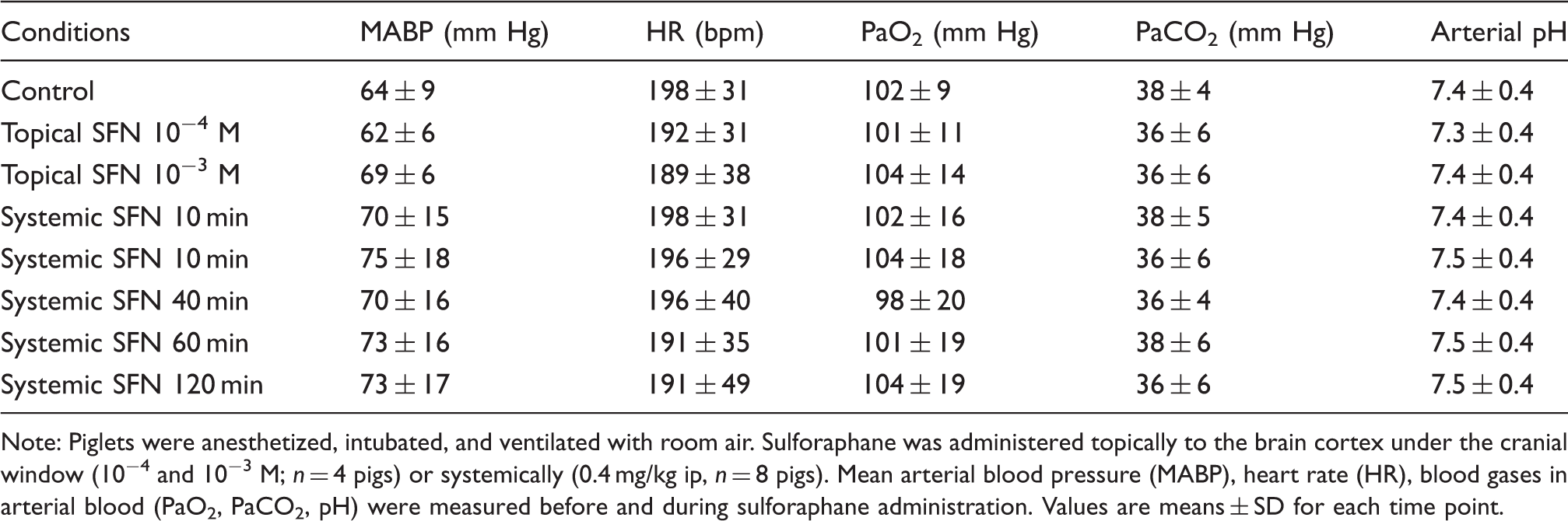
Note: Piglets were anesthetized, intubated, and ventilated with room air. Sulforaphane was administered topically to the brain cortex under the cranial window (10−4 and 10−3 M; n = 4 pigs) or systemically (0.4 mg/kg ip, n = 8 pigs). Mean arterial blood pressure (MABP), heart rate (HR), blood gases in arterial blood (PaO2, PaCO2, pH) were measured before and during sulforaphane administration. Values are means ± SD for each time point.
Vasodilator effects of topically administered SFN
SNF (10−6–10−3 M) placed on the brain surface under the cranial window produced dose-dependent dilation of pial arterioles (EC50 ∼ 5 × 10−5 M, Figure 1(a)). The maximal vasodilator effect (30–40% above the baseline) was produced by 10−3 M SFN. The vasodilator effect of topical SFN was observed within 5–10 min and sustained for at least 60 min after the application. The vasodilator response of pial arterioles to topical SFN was readily reversible after flushing the brain surface with aCSF for 10–20 min (Figure 1(a)).
Vasodilator effects of topically (a) and systemically (b) administered sulforaphane on pial arterioles. (a) Sulforaphane at consecutively increasing concentrations (10−6–10−3 M) was applied on the brain surface under the cranial window (n = 14 pigs). Each concentration of the drug was placed for 10 min, and the diameter of pial arterioles was measured. At the end of the experiment, the brain surface was flushed with aCSF for 20 min to demonstrate reversibility of the vasodilator effect of sulforaphane. (b) Sulforaphane (0.4 mg/kg) was administered via intraperitoneal injection (n = 9 pigs). The diameter of pial arterioles was measured every 10 min during a 2-h period after the administration. Values are means ± SD. *P < 0.05 compared with the baseline diameter.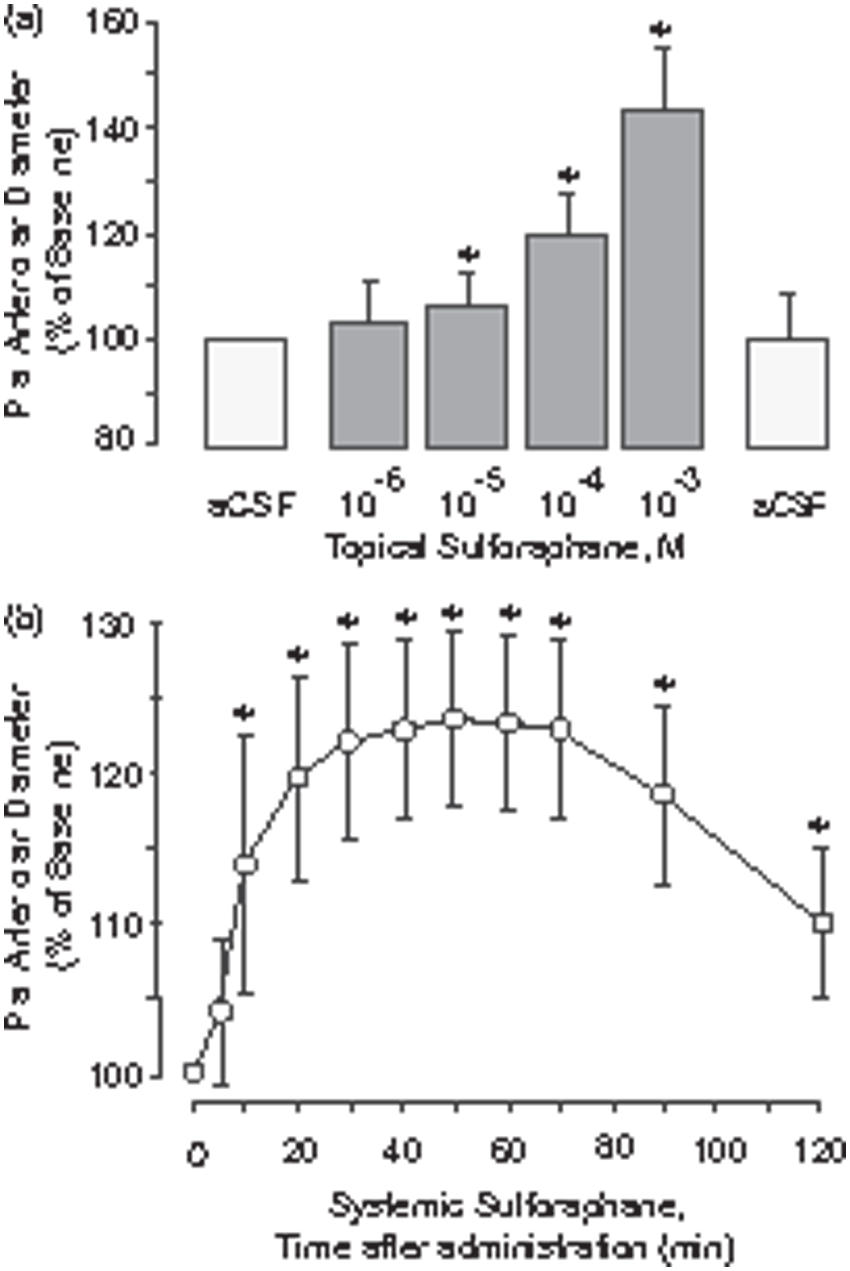
Vasodilator effects of systemically administered SFN
Systemic administration of SFN (0.4 mg/kg ip) caused vasodilation of pial arterioles that occurred 5–10 min after the injection and was sustained for over 2 h (Figure 1(b)). The maximal vasodilator response to SFN (∼25% above the baseline) was observed 20–80 min after the administration.
Topically and systemically administered SFN increases brain H2S production via CSE/CBS activation
Topical administration of SFN at the vasodilator concentrations (10−5–10−3 M) caused a 3- to 4-fold increase in the H2S level in the pCSF (Figure 2(a)) concomitantly with cerebral vasodilation (Figure 2(b)) observed within a 10-min period. Topical SFN-elicited H2S elevation and the vasodilation response of pial arterioles were completely blocked by the selective CSE inhibitors PPG and BCA, administered topically (PPG, 5 mM; BCA, 0.1 mM) or systemically (BCA, 1 mg/kg ip) (Figure 2(a) and (b)). The CBS inhibitor AOA (5 mg/kg ip) also abrogated H2S elevation and cerebral vasodilation responses to SFN (n = 3; data not shown).
Activation of enzymatic production of H2S by the brain is involved in the vasodilation of pial arterioles in response to topical sulforaphane. Sulforaphane at consecutively increasing concentrations (10−5–10−3 M) was applied under the cranial window either alone (Control, n = 8 pigs) or in combination with the CSE inhibitors propargylglycine (topical PPG, 5 mM, n = 4 pigs) or β-cyano-L-alanine (topical BCA, 0.1 mM, n = 4 pigs). In separate experiments, systemic BCA (1 mg/kg ip, n = 4 pigs) was administered 30 min before testing the responses to topical sulforaphane. The samples of periarachnoid corticospinal fluid (pCSF) were collected for H2S detection (a), and pial arteriolar diameter was measured (b) in 10-min intervals following the placement of each concentration of sulforaphane. Values are means ± SD. *P < 0.05 compared with the baseline values.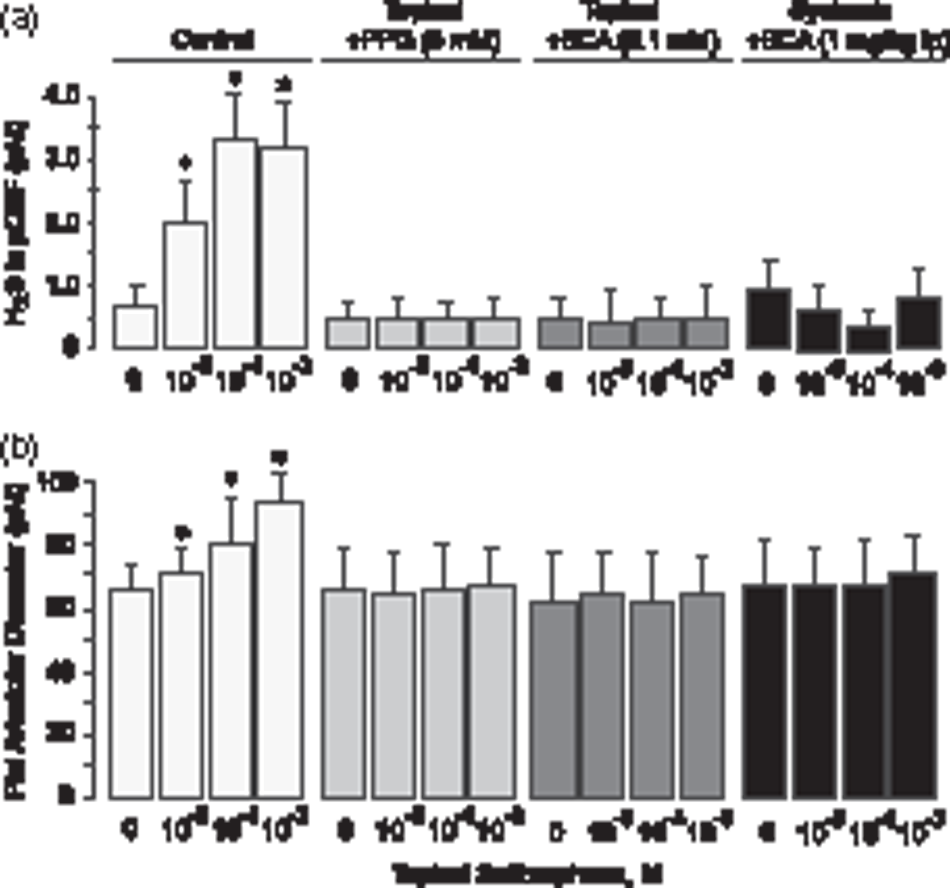
Systemic administration of SFN (0.4 mg/kg ip) produced a rapid increase in H2S level in the pCSF (Figure 3(a)) and caused vasodilation of pial arterioles (Figure 3(b)). The 3- to 5-fold elevation of H2S above the baseline in pCSF and the concomitant cerebral vasodilator response were sustained during the 20–90 min period after the administration (Figure 3(a) and (b)). The topically applied selective CSE inhibitor BCA (0.1 mM) and the CBS inhibitor AOA (1 mM) greatly attenuated the H2S elevation and blocked the vasodilation caused by systemic SFN (Figure 3(a) and (b)). These data suggest that vasodilator responses of pial arterioles to topical and systemic SFN require stimulation of endogenous production of H2S in the brain via CSE/CBS activation.
Activation of enzymatic production of H2S by the brain is involved in the vasodilation of pial arterioles caused by systemic sulforaphane. Sulforaphane (0.4 mg/kg) was administered via intraperitoneal injection alone (Control, n = 4 pigs) or in combination with the CSE-specific inhibitor β-cyano-L-alanine (BCA, 0.1 mM, n = 4 pigs) or the non-selective CSE/CBS inhibitor aminooxyacetic acid (AOA, 1 mM, n = 4 pigs). The samples of periarachnoid corticospinal fluid were collected for H2S detection (a), and pial arteriolar diameter was measured (b) in 20-min intervals following the injection of sulforaphane. Values are means ± SD. *P < 0.05 compared with the baseline values.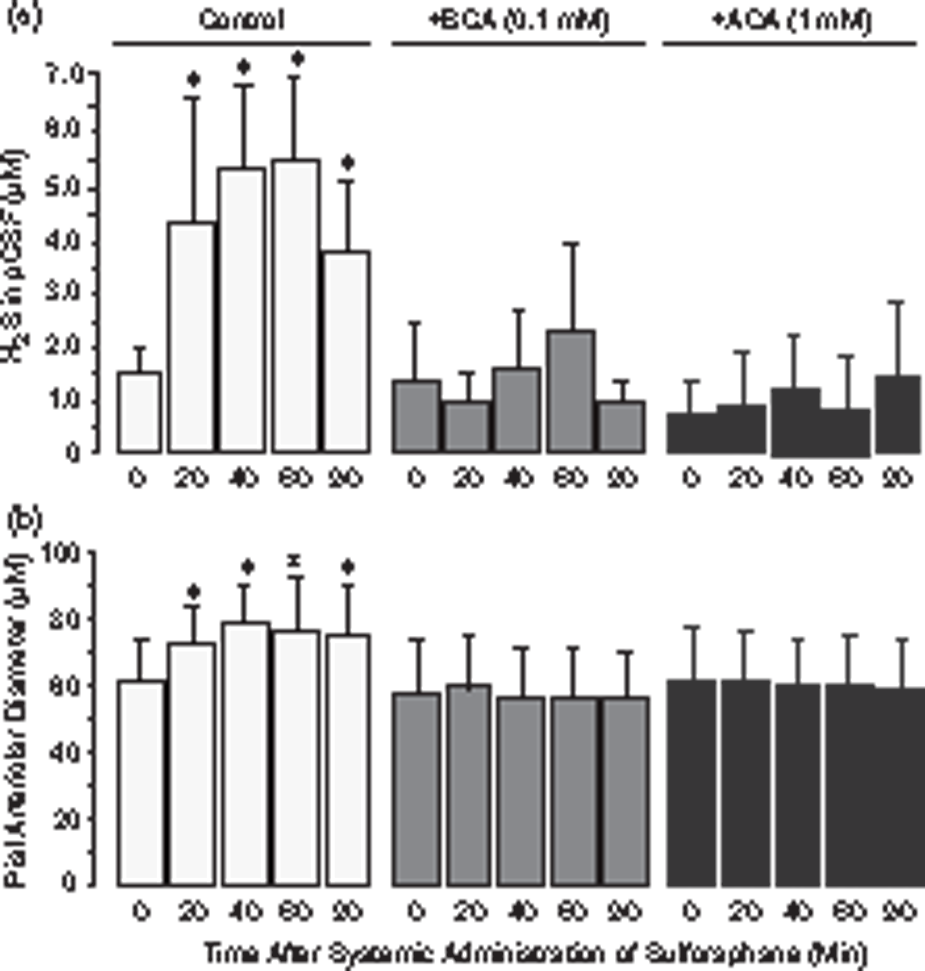
We investigated whether SFN is a donor molecule capable of spontaneously releasing H2S. SFN dissolved in aqueous solutions at high concentration (10 mM) released 2.5 ± 0.5 µM and 6.0 ± 1.0 µM H2S following 1- and 5-h incubation at 37℃, respectively (< 0.05% of the SFN moiety). At the maximal vasodilator concentration (1 mM), SFN released only 0.5 ± 0.2 µM H2S when incubated for 5 h at 37℃. These data demonstrate that although SFN is capable of spontaneous generation of H2S, it is a very weak and slow H2S donor. Considering that H2S causes dilation of cerebral arterioles at concentrations above 10 µM,16,17,19 we may conclude that the vasodilator potency of SFN cannot be attributed to the spontaneous H2S release mechanism.
SFN stimulates enzymatic CSE/CBS-catalyzed formation of H2S in the brain tissue
We tested the possibility that SFN may elicit H2S production in the neurovascular unit that expresses H2S-generating enzymes CSE and CBS.14–17,21 We detected H2S production by freshly prepared total homogenate of the brain cortex (Figure 4). Basal H2S production by the brain tissue was 6.5 ± 1.0 µM/mg protein/h at 37℃. In the presence of SFN at the vasodilator concentration (20 µM), a substantial increase in H2S production in the brain was observed (9.0 ± 0.9 µM/mg protein, P < 0.005) that accounted for ∼10% of the SFN conversion. Basal and SFN-elicited H2S formation was reduced by the CSE/CBS inhibitors BCA (0.1 mM) and AOA (1 mM). These data suggest that SFN rapidly enhances enzymatic production of H2S by the brain via a non-genomic mechanism.
Sulforaphane enhances enzymatic H2S production by the brain tissue. H2S concentration was measured in samples of the brain cortex homogenate after incubation for 1 h at 37℃. The CSE-specific inhibitor β-cyano-L-alanine (BCA, 0.1 mM) and the non-selective CSE/CBS inhibitor aminooxyacetic acid (AOA, 1 mM) were used to detect enzymatic H2S production by the brain cortex tissue in the absence and in the presence of 20 µM sulforaphane. Values are means ± SD. *P < 0.05 compared with the control baseline values. †P < 0.05 compared with the corresponding control values.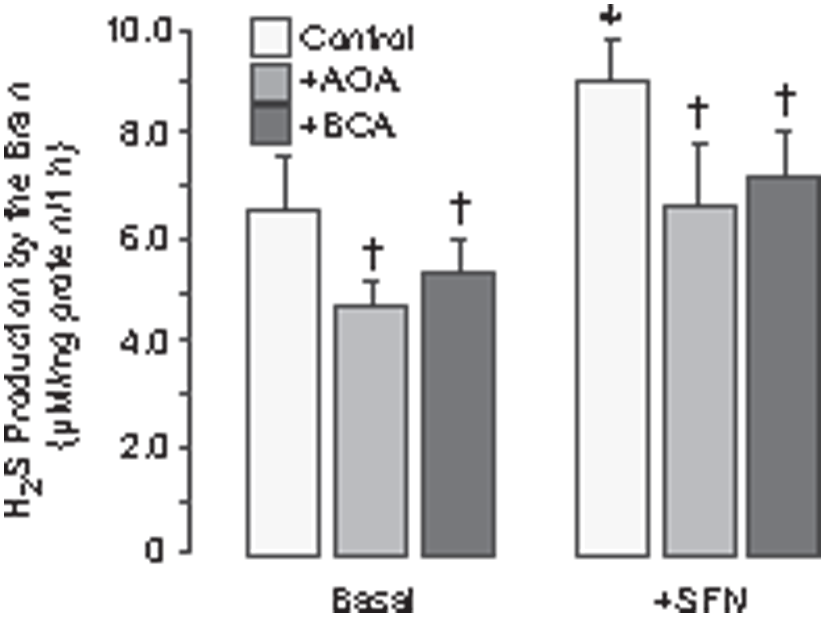
Activation of KATP and BK channels is required for cerebral vasodilation caused by topical and systemic SFN
We wished to determine the causal connections between the ability of SFN to increase brain H2S production and to produce cerebral vasodilation. It has been established that the vasodilator potency of H2S in cerebral circulation requires activation of KATP and BK channels in arteriolar VSM.16–20 Using selective pharmacological inhibitors of the potassium channels, we tested the hypothesis that brain-produced H2S mediates the cerebral vasodilator response to SFN. First, we tested the effects of KATP and BK inhibitors on brain H2S production and cerebral vasodilator response to topically applied SFN (Figure 5(a) and (b)). The KATP inhibitor glibenclamide (10−6 M) and the BK channel inhibitors paxilline (4 × 10−5 M) and iberiotoxin (10−7 M) topically applied to the brain cortex did not change the baseline pial arteriolar diameter but completely blocked the cerebrovascular dilator responses to SFN (Figure 5(b)). Importantly, the KATP and BK channel inhibitors did not reduce SFN-elicited H2S elevation (Figure 5(a)), indicating that the ability of SFN to increase the enzymatic production of H2S by the brain was not affected. Second, we tested the effects of the KATP and BK inhibitors on cerebral vasodilation caused by systemically administered SFN. In control pigs, SFN (0.4 mg/kg ip) caused dilation of pial arterioles observed during the 10–60-min period after the administration (Figure 6). In the presence of topical glibenclamide (10−6 M) and paxilline (4 × 10−5 M), the vasodilator responses of pial arterioles to systemic SFN were completely abrogated (Figure 6). Alternatively, the potassium channel inhibitors did not reduce the dilator responses of pial arterioles to the cGMP-dependent vasodilator SNP (Figures 5(b) and 6) indicating that VSM remained fully functional.
The inhibitors of KATP and BK channels block cerebral vasodilation in response to topically administered sulforaphane. Sulforaphane at consecutively increasing concentrations (10−5–10−3 M) was applied under the cranial window either alone (Control, n = 8 pigs) or in combination with the KATP channel inhibitor glibenclamide (10−7 M, n = 4 pigs), and the BK channel inhibitors paxilline (4 × 10−5 M, n = 4 pigs) and iberiotoxin (10−7 M, n = 4 pigs). At the end of each experiment, sodium nitroprusside (SNP, 10−6 M) was applied topically to test the general vasodilatory function of the arteriolar smooth muscle. The samples of periarachnoid corticospinal fluid (pCSF) were collected for H2S detection (a), and pial arteriolar diameter was measured (b) in 10-min intervals following the placement of each concentration of sulforaphane. Values are means ± SD. *P < 0.05 compared with the corresponding baseline values. The inhibitors of the KATP and BK channels block cerebral vasodilation caused by systemically administered sulforaphane. Sulforaphane (0.4 mg/kg ip) was administered to control pigs (n = 8) and to pigs treated with the KATP channel inhibitor glibenclamide (10−7 M, n = 4) or the BK channel inhibitor paxilline (4 × 10−5 M, n = 4) topically placed on the brain cortex for the duration of the experiment. Pial arteriolar diameter was measured in 20-min intervals during the 60-min observation period after the sulforaphane administration. At the end of each experiment, sodium nitroprusside (SNP, 10−6 M) was applied topically to test the general vasodilatory function of the arteriolar smooth muscle. Values are means ± SD. *P < 0.05 compared with the corresponding baseline diameters.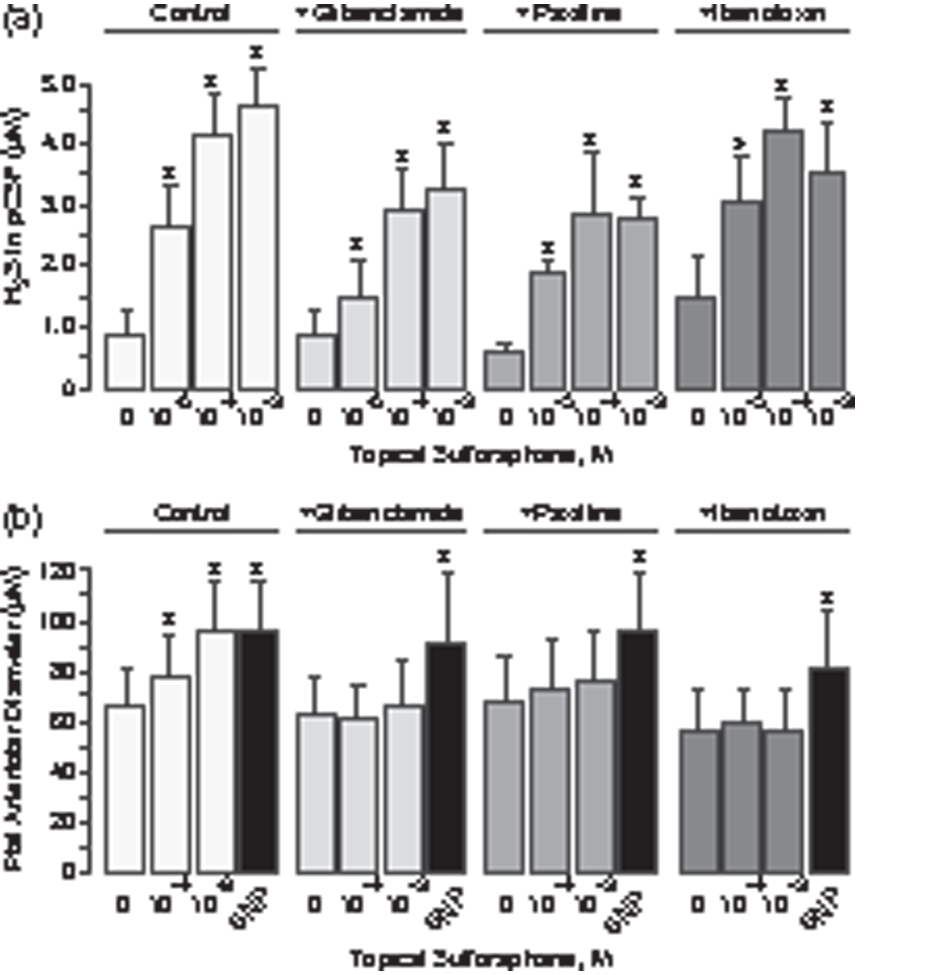
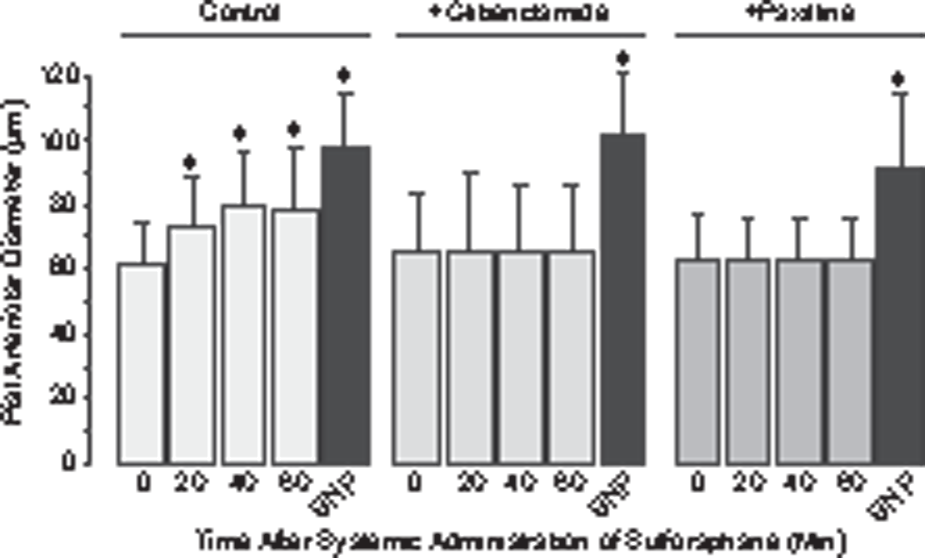
Overall, these data suggest that brain-produced H2S mediates the cerebral vasodilator response to SFN via activation of KATP and BK channels in arteriolar VSM.
Discussion
We detected acute non-genomic effects of phytochemical isothiocyanate sulforaphane in the cerebral circulation of healthy newborn pigs. Our major novel finding is that sulforaphane is a brain-permeable compound that has acute vasodilator effects in the cerebral circulation following systemic administration. We provide evidence that the mechanism of the cerebral vasodilator potency of topically and systemically administered SFN includes its ability to stimulate endogenous enzymatic production of the vasodilator mediator H2S via acute activation of CSE/CBS in the cerebrovascular unit. In turn, SFN-elicited enzymatic H2S formation in the brain activates BK and KATP channels, leading to vasodilation of cerebral arterioles.
Sulforaphane (1-isothiocyanate-(4R)-(methylsulfinyl)butane), a dietary phytochemical from cruciferous vegetables attracted tremendous interest from researchers and clinicians after the discovery of its anti-cancerous and antioxidant properties. For the last 20 years, the main focus has been directed toward the gene-targeting potency of synthetic isothiocyanates.1–11 The ability of SFN to stimulate the Nrf2-dependent gene transcription mechanism leading to upregulation of the antioxidant defense machinery has been demonstrated in the cardiovascular system and in the brain.2–8,10,11,26 SFN administered in high doses (5–50 mg/kg ip) to rodents during ischemia, hypoxic insults, and inflammation enhanced the transcription of a battery of cytoprotective genes in the brain encoding the antioxidant enzymes superoxide dismutase (SOD1), heme oxygenase-1, and NAD(P)H quinone oxidoreductase (NQO1) and frequently improved the outcome of the disease.3,5,9,10,26 The clinical significance of these findings is currently being tested by several trials designed to assess the anticancer and cardioprotective effects of SFN administered to patients at daily doses of 0.5–50 mg.
Astoundingly, the acute non-genomic effects of SFN remain unexplored. We, for the first time, provide evidence that SFN has potent immediate cerebral vasodilator effects when applied to the brain cortex or administered by systemic intraperitoneal injection. We used the cranial window technique in intact healthy newborn pigs of both sexes to investigate the effects of SFN on the diameter of pial arterioles, the major resistance vessels that regulate cerebral blood flow. The dose-dependent vasodilator effects of low concentrations of topical SFN (10−5–10−3 M) were observed immediately (within 5–10 min) and were sustained for at least for 2 h after the placement. The effects of SFN were readily reversible after a brief flush of the brain surface with physiological solutions. A single systemic injection of a low dose of SFN (0.4 mg/kg ip) also caused an immediate cerebral vasodilation that reached a maximal plateau (20–25% above the baseline) within 20–90 min and was sustained even after 2 h after the injection. Remarkably, systemic SFN had no effect on arterial blood pressure or other systemic cardiovascular parameters, and it did not affect body temperature.
The ability of intraperitoneally administered SFN to immediately produce cerebral vasodilation suggests that SFN can readily penetrate a non-compromised BBB in healthy newborn pigs. The question of brain permeability of SFN remains disputable, however. The analysis of tissue distribution of SFN given orally to normal mice showed that the brain accumulated only a very low amount of the supplement, as compared with major accumulation in the small intestine, kidney, and lung. 9 In normal mice, only a minor temporary rise in SFN in the brain (30 ng/g tissue) was detected by high-performance liquid chromatography within 15–30 min after the injection of a very high dose of SFN (50 mg/kg ip). 26 However, the ability of SFN to exhibit neuroprotective effects in the ischemic rodent brain suggests that it can penetrate a compromised BBB.3,5,10,26
What is the mechanism of the cerebral vasodilation caused by SFN? The functional isothiocyanate group is a potential source of H2S. H2S has emerged as a vasoactive physiologically important mediator produced in the brain.16,17,19–21 We detected a significant increase in the level of H2S in the pCSF during treatment with topical and systemic SFN. Remarkably, the dose- and time-dependent elevations in brain H2S occurred concomitantly with the vasodilator effects of SFN. These findings indicate that SFN increases cerebral blood flow via a H2S-mediated mechanism.
We provide evidence that SFN itself is not a H2S donor molecule. The measurements of H2S formation from SFN at a wide concentration range (0.1–100 mM) demonstrate that the ability of SFN to generate H2S is extremely limited (<0.05% of the SFN moiety in 1–5 h at 37℃) and cannot account for the vasodilator activity of the compound. Although some reports suggest that isothiocyanate derivatives are capable of releasing H2S under certain “triggering” conditions,12,13 no convincing experimental data support this notion. Martelli et al. reported that H2S formation from several tested isothiocyanate derivatives is negligible, but it is stimulated in the presence of L-cysteine.12,13 However, the ability of L-cysteine alone to release H2S has not been taken into account.
Which mechanism contributes to SFN-elicited H2S formation by the brain in vivo? We provide evidence that SFN activates a naturally occurring enzymatic mechanism that catalyzes the production of H2S in the brain. H2S is a product of L-cysteine catabolism by cystathionine γ-lyase (CSE) and cystathionine β-synthase (CBS) that are highly expressed in the brain. CSE is the major source of H2S production in the cerebral vasculature and is essential in the regulation of cerebral vascular tone, whereas CBS is localized largely to astrocytes and neurons.16,17,21 To test the contribution of endogenously produced H2S to the SFN-elicited vasodilator responses, we used CSE-selective inhibitors PPG and BCA, and the CBS inhibitor AOA that also may inhibit CSE. 27 All the tested CSE/CBS inhibitors administered either topically or systemically, blocked the cerebral vasodilatory responses to topically and systemic SFN. Furthermore, the inhibitors completely blocked or greatly reduced H2S elevations in the brain caused by topical or systemic SFN. These findings suggest that SFN produces cerebral vasodilation via stimulating the enzymatic production of H2S in the brain. In the in vitro experiments, we confirmed the ability of SFN to rapidly activate enzymatic H2S production in the brain tissue. It appears that CSE expressed in the neurovascular unit can be activated by SFN via a non-genomic mechanism that may involve posttranslational modifications of the enzyme. We cannot exclude the possibility that CBS, which is highly expressed in cortical astrocytes and neurons,17,21 also may respond to SFN by activating H2S production, as suggested by the ability of the CBS inhibitor AOA to block the vasodilation and H2S elevation caused by systemic SFN. However, the insufficient selectivity and low affinity of the currently available inhibitors14,27 does not allow to more precisely detect the involvement of CBS in the vasodilator responses to SFN. Overall, the rapid activation of enzymatic production of H2S in the brain via CSE/CBS-catalyzed reaction is the major mechanism that leads to SFN-elicited vasodilation of cerebral arterioles.
We present experimental evidence that the cerebral vasodilation responses to topical and systemic SFN require the involvement of KATP and BK channels in cerebral arterioles that are activated by endogenously produced H2S. It has been established that H2S causes cerebral vasodilation via activation of the potassium channels in cerebral arteriole VSM, including the ATP-sensitive potassium channel (KATP) and the Ca2+- and voltage-gated potassium channel (BK).18–20,22–24 To provide the in vivo evidence that endogenously produced H2S is the mediator of the vasodilator potency of SFN, we used the KATP channel inhibitor glibenclamide and the BK channel inhibitors paxilline and iberiotoxin, as in previous reports.19–21,28 Glibenclamide, paxilline, and iberiotoxin applied under the cranial window completely blocked the cerebral vasodilator responses to topical SFN. In contrast, the elevations in H2S production were not reduced, suggesting that the inhibitors did not interfere with the ability of SFN to stimulate enzymatic H2S production by the brain. The vasodilator responses of cerebral arterioles to systemic SFN were also completely eliminated in the presence of topical glibenclamide and paxilline. Confirming the specificity of the targeting of H2S-mediated vasodilatory effects, the inhibitors did not reduce the overall VSM function tested by the cGMP-mediated dilator SNP.
Which neurovascular components are activated by SNF-elicited brain-derived H2S to produce cerebral vasodilation? Physiological responses of cerebral arterioles to vasoactive stimulation may include intercellular communication in the neurovascular unit, including VSM and endothelial cells, astrocytes, pericytes, and neurons.29–31 Arteriolar VSM cells have been identified as the major cells that respond to H2S by activation of KATP and BK channels, leading to elevation in the channel currents, membrane hyperpolarization, and vasodilation.14,17–20,22–24 Currently, only limited information is available on the contribution of non-VSM BK channels to the vasodilator responses to H2S. Reports on mesenteric arteries indicate that endothelial cells express functional BK channels and may contribute to H2S-induced vasodilation.23,32 However, endothelium-denuded preparations of cerebral arterioles maintained the ability to respond to H2S by BK channel activation and vasodilation, 12 suggesting vascular bed-specific differences in endothelial BK channel functions. Distinct cell-specific structural and pharmacological differences between the types of BK channels may allow us to pinpoint selected neurovascular cells targeted by H2S elevations in response to SFN. VSM and endothelial cells express type I BK channels containing a β1 subunit and are inhibited by both paxilline and iberiotoxin.20,23,32 Astrocytes and neurons express type II BK channels containing a tissue-specific β4 accessory subunit that confers BK channel resistance to venom blockers including iberiotoxin.30,33,34 In our experiments, the responses to SFN were completely blocked by both paxilline and iberiotoxin, suggesting that arteriolar VSM BK activation accounts for the vasodilator response of cerebral arterioles to SFN. The contribution of endothelial BK channels is unlikely, because endothelium-denuded cerebral arterioles maintain the ability to respond to H2S by BK channels activation and vasodilation. 12 The contribution of astrocyte BK channels to the vasodilator responses to SFN is unlikely because we did not observe the phenomenon of iberiotoxin resistance combined with paxilline sensitivity in blocking SFN-induced H2S-mediated vasodilation. However, astrocytes as the main sites of enzymatic H2S synthesis in the brain are essential components of the vasodilator potency of SFN.
Overall, we provide evidence that the cerebral vasodilation responses to SFN involve acute non-genomic stimulation of endogenous H2S production in the neurovascular unit followed by activation of H2S-responsive KATP and BK channels in cerebral VSM and vasodilation of cerebral resistance arterioles. We anticipate that SFN supplements may be used as a promising therapeutic approach to increase blood flow to the brain.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the NIH R01NS101717 (HP) and NIH R01NS105655 (HP).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors' contributions
HP and JL contributed to the experimental concept and design; DH, JL, and AF performed experiments; HP, DH, and JL took part in the data analysis; HP, DH, and JL were involved in the interpretation of result; HP and JL prepared figures; HP, DH, JL, and AF edited and approved the final version of the manuscript.