
Abstract
Our previous studies indicate that function of ATP-dependent K+ channels (KATP) in cerebral arterioles is suppressed after ischemia. In the current study, we examined pial arteriolar responses to forskolin, dibutyryl-cAMP, NS-1619, and methionine (met)-enkephalin, activators of calcium-dependent K+ channels (KCa) before and 1 hour after 10 minutes of total, global ischemia in anesthetized piglets. Arteriolar diameters were measured using a closed cranial window and intravital microscopy. All pharmacologic agents were given topically. Baseline diameters were approximately 100 μm, and diameters had returned to normal by 1 hour after ischemia. Forskolin dilated arterioles by 9 ± 3%, 18 ± 4%, and 31 ± 12% at 5 × 10−8, 5 × 10−7, and 10−6 mol/L, respectively (P < 0.05, n = 10). In addition, dibutyryl-cAMP dilated arterioles by 8 ± 2% at 10−4 mol/L and 14 ± 2% at 3 × 10−4 mol/L (P < 0.05, n = 6). Also, NS-1619 increased diameter of arterioles by 9 ± 2% at 10−7 mol/L and 17 ± 9% at 10−5 mol/L (P < 0.05, n = 5). Finally, met-enkephalin dilated arterioles by 9 ± 2% at 10−8 mol/L and 16 ± 3% at 10−6 mol/L (P < 0.05, n = 5). At 1 hour after ischemia, arteriolar dilator effects to forskolin, dibutyryl-cAMP and NS-1619, and met-enkephalin were intact. Thus, in contrast to KATP, KCa in cerebral arterioles are resistant to ischemic stress.
Calcium-dependent K+ channels (KCa) have been identified in vascular smooth muscle membranes from a variety of arteries, including resistance vessels from brain (Faraci and Sobey, 1996; Nelson and Quayle, 1995). Activation of KCa causes relaxation of cerebrovascular smooth muscle cells to several brain metabolites, thereby suggesting an important role in the regulation of the brain circulation. In vitro experiments using patch-clamp approaches show that isoproterenol, forskolin, cAMP, and NS-1619 increase activity of these channels in cerebral arterioles (Song and Simard, 1995; Holland et al., 1996). In addition, application of selective inhibitors of KCa such as iberiotoxin or charybdotoxin to pial arterioles in vivo attenuates dilation of cerebral arterioles in response to adenosine, forskolin, methionine (met)-enkephalin, NS-1619, sodium nitroprusside, and cAMP (Taguchi et al., 1995; Armstead, 1997a, 1997b; Paterno et al., 1996).
Similar to KCa, ATP-dependent K+ channels (KATP) promote dilation in the cerebral circulation (Faraci and Sobey, 1996). We have shown that cerebral arterioles dilate to direct activation of KATP and that these responses are impaired after ischemia–reperfusion (Bari et al., 1996a,b; Louis et al., 1996). Thus, arteriolar dilator responses to application of aprikalim, a selective, pharmacologic activator of KATP, are drastically reduced after 10 minutes of total, global ischemia. Similar findings were obtained using iloprost and calcitonin gene-related peptide, which are receptor-linked activators of KATP. It is unknown whether KCa in cerebral arterioles are affected by ischemia.
This study examines the effects of ischemia on function of KCa in pial arterioles in newborn piglets. We tested the hypothesis that global cerebral ischemia would reduce arteriolar dilation to activators of KCa.
MATERIALS AND METHODS
Newborn (1- to 7-day-old) piglets of either sex with body weight of 0.9 to 1.4 kg were used in this study. Anesthesia was induced with sodium thiopental (30 mg/kg, intraperitoneally) followed by intravenous injection of α-chloralose (75 mg/kg, intraperitoneally). Supplemental doses of α-chloralose were given as needed to maintain a stable level of anesthesia. Animals were intubated with tracheotomy and artificially ventilated with room air. Body temperature was maintained at 37° to 38°C by a heating pad. Systemic arterial blood pressure was recorded using a cannula introduced to the right femoral artery connected to a pressure transducer. The right femoral vein was cannulated for drug administration. A closed cranial window was inserted as previously described (Bari et al., 1996c). Diameters of pial arterioles were measured using a microscope (Wild M36, Heerbrugg, Switzerland) equipped with a video camera (Panasonic, Secaucus, NJ, U.S.A.) and video microscaler (IV-550, For-A Co., Newton, MA, U.S.A.). After the surgical procedure, the cranial window was gently infused with artificial cerebrospinal fluid (aCSF) several times until a stable baseline was obtained.
Cerebral ischemia–reperfusion injury
Cerebral ischemia–reperfusion injury was produced as described (Bari et al., 1996c). A hollow brass bolt was implanted in the right parietal cranium 20 mm rostral to the cranial window. Immediately after placement of the cranial window, a 3-mm hole was drilled in the skull using an electric drill with a toothless bit, and the dura was exposed. A hollow bolt was inserted and secured in place with cyanoacrylate ester (Super-Glue, S.P. Richards, Atlanta, GA) and dental acrylic. After implantation of the window and the bolt, aCSF was allowed to equilibrate with the peri arachnoid CSF under the window for 20 minutes. To induce ischemia, aCSF was infused to maintain intracranial pressure above mean arterial pressure so that blood flow through pial vessels was stopped. Venous blood was withdrawn as necessary to maintain mean arterial blood pressure near normal values. At the end of the 10-minute period of ischemia, the infusion tube was clamped, and the intracranial pressure was allowed to return to preischemia values.
Experimental protocols
The animals were carefully monitored during the α-chloralose anesthesia. At the beginning of each experiment, the cranial window was flushed with aCSF several times, and a stable baseline arteriolar diameter was achieved. The animals then were divided into five groups.
In four groups of piglets, we examined responses of cerebral arterioles to topical application of activators of KCa before and 1 hour after cerebral ischemia. These KCa activators were as follows: forskolin (5 × 10−8, 5 × 10−7, and 5 × 10−6 mol/L), dibutyryl-cAMP (10−4 and 3 × 10−4 mol/L), met-enkephalin (10−8 and 10−6 mol/L), and NS-1619 (10−7 and 10−5 mol/L). We also examined arteriolar responses to forskolin at 2 hours after ischemia. We chose to focus on 1 to 2 hours after ischemia because the greatest reductions in dilator responses to KATP activation and other agents were during this period (Busija et al., 1996; Bari et al., 1996c; Louis et al., 1996). We used NS-1619 as a direct, selective activator of KCa (Edwards et al., 1994; Oleson et al., 1994). Armstead (1997b) has shown that pial arteriolar dilation to NS-1619 in piglets is almost totally dependent on activation of KCa.
In a fifth group of piglets, we examined arteriolar responses to forskolin in the absence and presence of charybdotoxin (10−7 mol/L).
Statistical analysis
All values are presented as mean ± standard deviation. Data were analyzed using repeated measures analysis of variance in all groups except for the NS-1619 group. In that group, not all animals were represented before and after ischemia, so a non-repeated measures analysis of variance was used. Pair-wise comparisons were made using the Student-Newman-Kuels test. A significance level of P < 0.05 was used.
RESULTS
Forskolin caused dose-dependent arteriolar dilation, which was unaffected by ischemia (Fig. 1). Baseline arteriolar diameter was 103 ± 6 μm, and diameter increased to 112 ± 7 μm at 5 × 10−8 mol/L, 122 ± 8 μm at 5 × 10−7 mol/L, and 135 ± 15 μm at 5 × 10−6 mol/L (n = 10; P < 0.05, compared with baseline). At 1 hour after recovery from ischemia, baseline arteriolar diameter was 100 ± 7 μm, and forskolin increased diameter to 109 ± 10 μm, 117 ± 10 μm, and 131 ± 12 μm at 5 × 10−8, 5 × 10−7, and 5 × 10−6 mol/L (n = 10; P < 0.05, compared with baseline; not significant when compared with preischemia responses). At 2 hours after ischemia, arteriolar diameter was 105 ± 7 μm, and forskolin increased diameter to 114 ± 3 μm (8 ± 2%), 120 ± 8 μm (14 ± 3%), and 131 ± 14 μm (24 ± 9%) at 5 × 10−8, 5 × 10−7, and 5 × 10−6 mol/L respectively (n = 10; P < 0.05, compared with baseline; not significant when compared with preischemia or 1 hour postischemia responses).
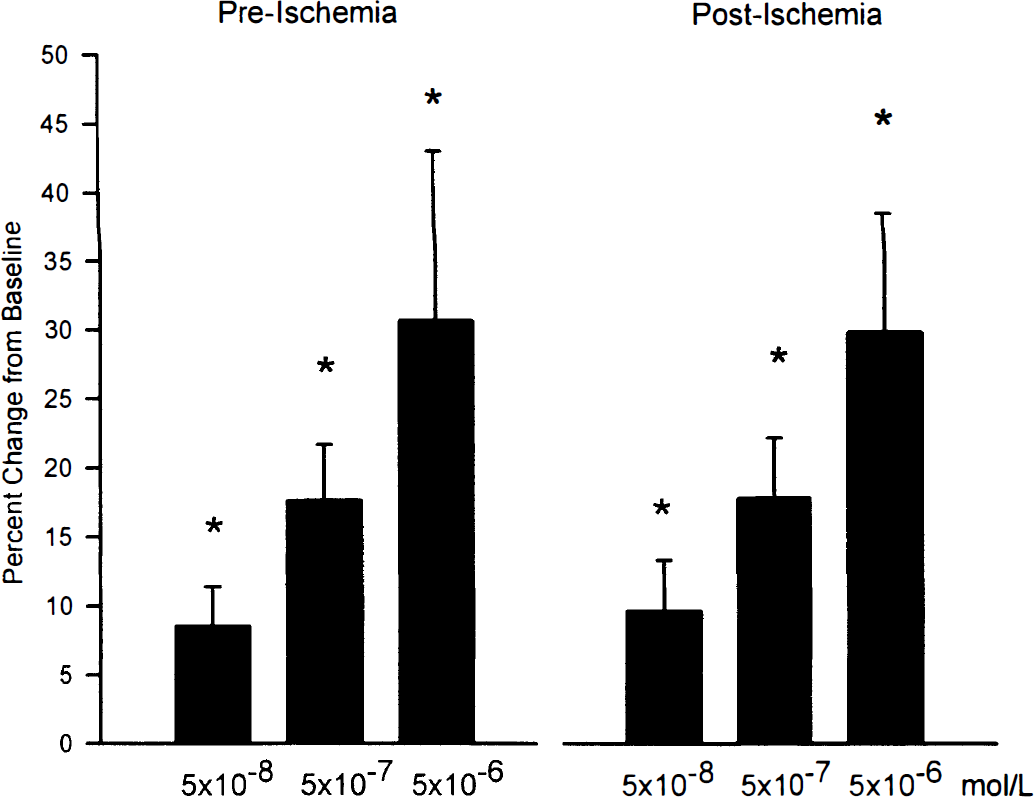
Effects of ischemia on cerebral arteriolar dilation to forskolin. Arteriolar dilator responses were intact after ischemia. Values are mean ± SD for 10 animals. *P < 0.05, compared with respective baseline.
Dibutyryl cAMP caused dose-dependent arteriolar dilation, which was unaffected by ischemia (Fig. 2). Baseline arteriolar diameter was 102 ± 2 μm, and diameter increased to 111 ± 2 μm at 10−4 mol/L and 117 ± 2 μm at 3 × 10−4 mol/L (n = 6; P < 0.05, compared with baseline). At 1 hour after recovery from ischemia, baseline arteriolar diameter was 101 ± 3 μm, and dibutyryl cAMP increased diameter to 109 ± 4 μm and 116 ± 2 μm at 10−4 and 3 × 10−4 mol/L (n = 6; P < 0.05, compared with baseline; not significant when compared with preischemia responses).
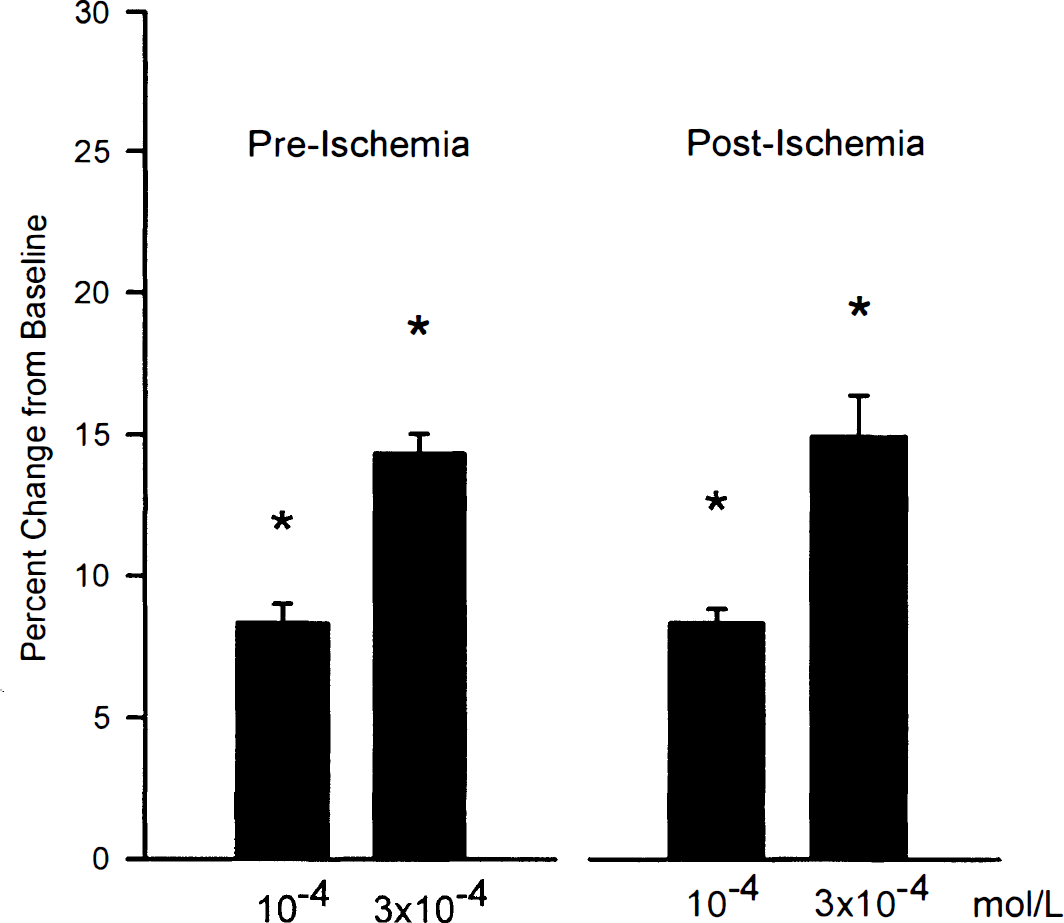
Effects of ischemia on cerebral arteriolar dilation to dibutyryl cAMP. Arteriolar dilator responses were intact after ischemia. Values are mean ± SD for six animals. *P < 0.05, compared with respective baseline.
NS-1619 caused dose-dependent arteriolar dilation, which was unaffected by ischemia (Fig. 3). Baseline arteriolar diameter was 107 ± 12 μm, and diameter increased with NS-1619 to 117 ± 15 μm at 10−7 mol/L and 125 ± 16 μm at 10−5 mol/L (n = 5; P < 0.05, compared with baseline). At 1 hour after recovery from ischemia, baseline arteriolar diameter was 108 ± 14 μm, and NS-1619 increased diameter to 115 ± 14 μm and 127 ± 13 μm at 10−7 and 10−5 mol/L (n = 5; P < 0.05, compared with baseline; not significant when compared with preischemia responses).
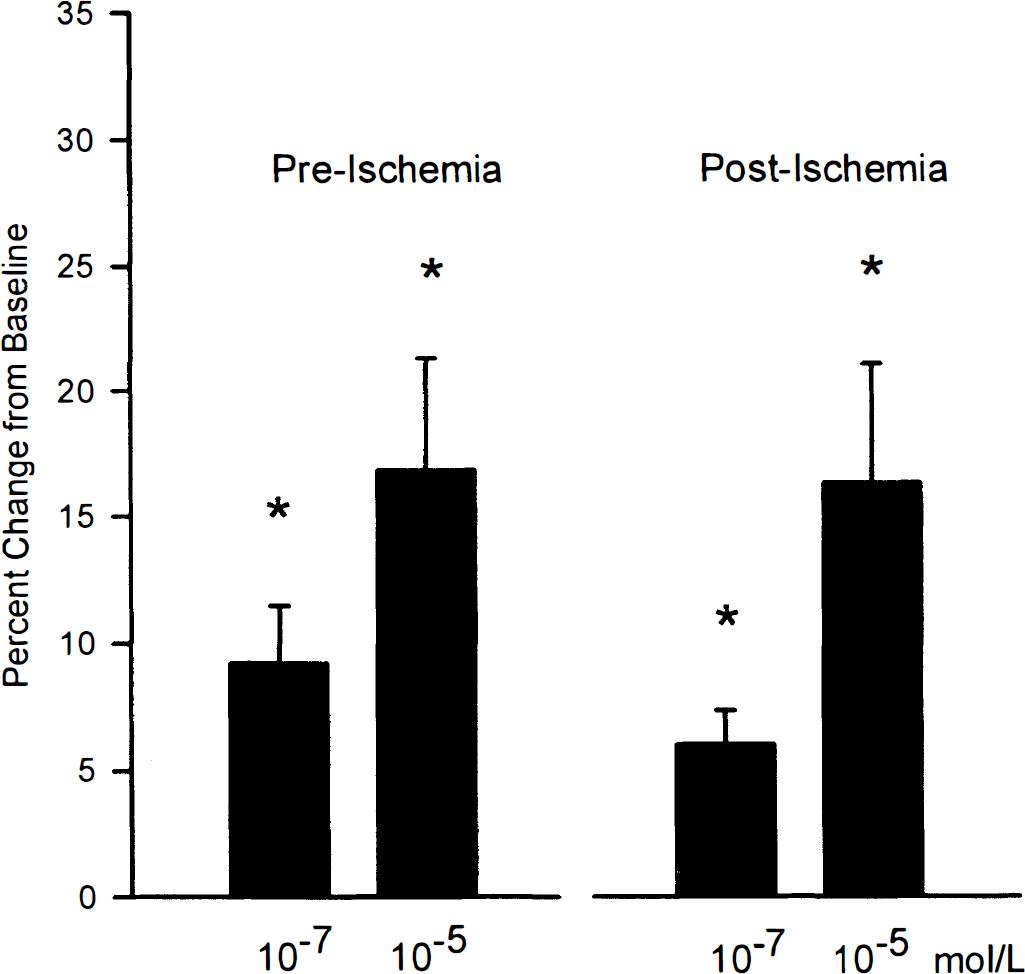
Effects of ischemia on cerebral arteriolar responses to NS-1619. Arteriolar dilation was unaffected by ischemia. Values are mean ± SD for five animals. *P < 0.05, compared with respective baseline.
Methionine-enkephalin also resulted in dilation of pial arterioles, which was unaffected by ischemia (Fig. 4). Baseline arteriolar diameter was 106 ± 4 μm, and diameter increased to 115 ± 7 μm at 10−8 mol/L and 123 ± 7 μm at 10−6 mol/L (n = 5; P < 0.05, compared with baseline). At 1 hour after recovery from ischemia, baseline arteriolar diameter was 108 ± 6 μm, and met-enkephalin increased diameter to 116 ± 7 μm and 127 ± 9 μm at 10−8 and 10−6 mol/L (n = 5; P < 0.05, compared with baseline; not significant when compared with preischemia responses).
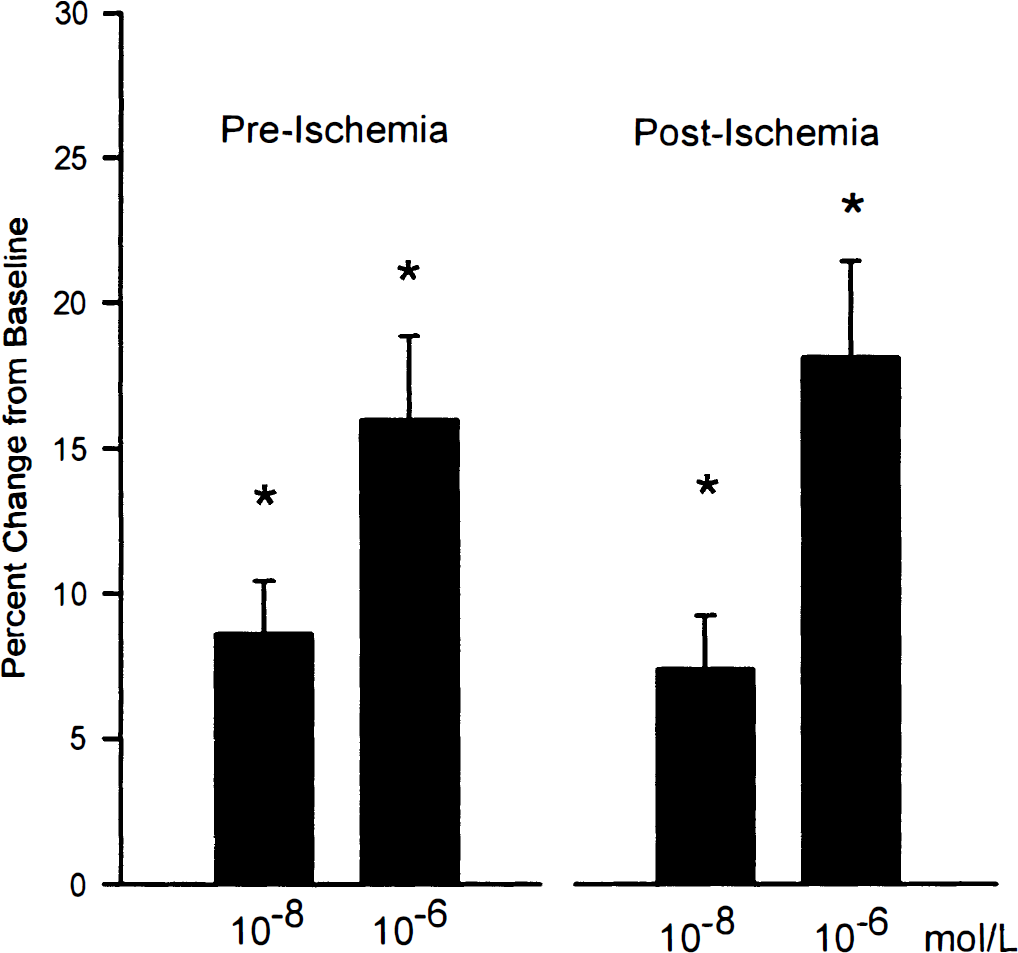
Effects of ischemia on cerebral arteriolar responses to met-enkephalin. Arteriolar dilation was unaffected by ischemia. Values are mean ± SD for five animals. *P < 0.05, compared with respective baseline.
Coapplication of charybdotoxin reduced arteriolar dilation to both doses of forskolin (Fig. 5). Before ischemia, baseline was 102 ± 3 μm, and diameter increased to 109 ± 4 μm at 5 × 10−8 mol/L and 131 ± 6 μm at 5 × 10−6 mol/L (n = 7; P < 0.05, compared with baseline). With coapplication of charybodtoxin, baseline diameter was 100 ± 5 μm, and diameter was only 103 ± 5 μm at 5 × 10−8 mol/L and 122 ± 7 μm at 5 × 10−6 mol/L (P < 0.05, compared with corresponding control).
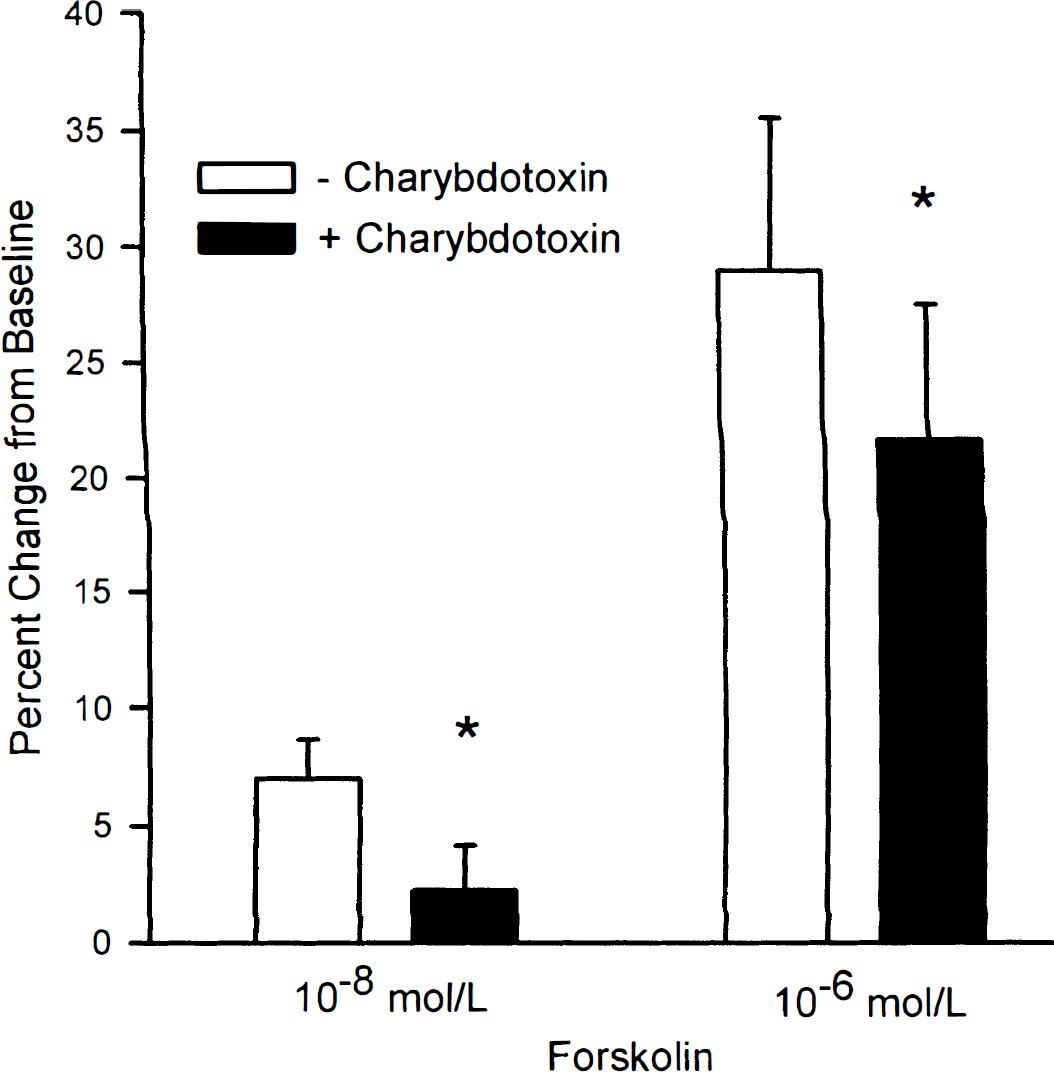
Effects of coapplication of charybdotoxin on arteriolar dilation to forskolin. Arteriolar dilation was reduced by charybdotoxin at both doses of forskolin. Values are mean ± SD for seven animals.
DISCUSSION
There are two major findings of this study. First, we confirm that activators of KCa dilate pial arterioles in piglets. And second, we present new information that function of KCa in cerebral resistance vessels is not significantly affected by ischemic stress. Thus, KCa are important mediators of cerebrovascular responses to several stimuli, and arteriolar dilator responses to forskolin, met-enkephalin, dibutyryl cAMP, and NS-1619 are intact after ischemia.
The relative importance of KCa in mediating arteriolar dilation varies among forskolin, met-enkephalin, dibutyryl cAMP, and NS1619. Armstead (1997a; 1997b) has shown KCa accounted for ∼25% of the dilation to met-enkephalin, ∼80% of the dilation to a cAMP analogue, and virtually all of the dilation to NS-1619. Further, Taguchi and others (1995) have shown that iberiotoxin and charybdotoxin attenuate cerebral arteriolar dilation to forskolin by approximately 50% in rabbits. Our results with charybdotoxin are qualitatively similar to those of Taguchi and associates (1995) and indicate in piglets that the activation of KCa contributes to forskolin-induced arteriolar dilation, especially at lower doses. Thus, cAMP analogues and NS-1619 appear to rely more on KCa in mediating dilation than met-enkephalin and forskolin, where other mechanisms such as activation of KATP are involved (Kitazono et al., 1993; Shankar and Armstead, 1995).
Our previous studies show that anoxic stress is able to impair function of two types of ion channels relevant to control of the cerebral circulation. First, arteriolar dilator responses to pharmacologic (aprikalim) and physiologic (iloprost, calcitonin gene-related peptide) activators of KATP are reduced for 1 to 2 hours by ischemia (Bari et al., 1996c; Louis et al., 1996). Second, arteriolar dilator responses to activation of neuronal N-methyl-
Since KATP as well as KCa contribute to arteriolar dilation to met-enkephalin in piglets (Armstead, 1997a; Shankar and Armstead, 1995), an unexpected finding was that the arteriolar response to this opioid was intact after ischemia. We have shown previously that arteriolar dilation to aprikalim, iloprost, and calcitonin gene-related peptide is almost totally dependent on activation of KATP, and that ischemic stress can reduce dilation to these agonists by approximately 90%. In contrast, arteriolar dilations to arterial hypoxia and topical adenosine, which are only partially dependent on KATP activation, are totally intact after ischemia (unpublished observations). At least two possibilities may explain these results. First, alternative dilator mechanisms may have compensated for decreased function of KATP. If KCa are relatively resistant to ischemia, then their contribution to met-enkephalin–induced dilation may have increased and thus compensated for loss of KATP function. And second, remaining KATP function may be sufficient to allow normal arteriolar responsiveness. Thus, low levels of KATP function after ischemia may be sufficient when operating in conjunction with other mechanisms to allow normal responsiveness to occur. Permissive effects have been suggested for nitric oxide (Armstead, 1995) and prostaglandins (Leffler et al., 1992).
The resistance of KCa to ischemia compared with KATP has several important implications. First, at least in the neonate, retained KCa function after hypoxic/anoxic stress would promote cerebrovascular dilation directly during the immediate resuscitation period. Second, normal KCa function would restrain effects of constrictor agents on the cerebral circulation during and after hypoxic/anoxic stress. For example, Paternò et al. (1996) has shown that KCa activation restrains effects of constrictor agents on cerebral arterioles. And third, the resilience of KCa to oxidative stress reinforces the view that oxygen radicals may exert arterial dilation through activation of these channels. For example, oxygen radicals appear to dilate cerebral arterioles through reversible activation of KCa (Faraci et al., 1997).
In summary, activators of KCa dilate cerebral arterioles of piglets, and KCa-dependent responses in cerebral arterioles are resistant to ischemic stress.