
Abstract
Chronic kidney disease has a graded and independent inverse impact on cerebrovascular health. Both thrombotic and hemorrhagic complications are highly prevalent in chronic kidney disease patients. Growing evidence suggests that in chronic kidney disease patients, ischemic strokes are more common than hemorrhagic strokes. Chronic kidney disease is asymptomatic until an advanced stage, but mild to moderate chronic kidney disease incites various pathogenic mechanisms such as inflammation, oxidative stress, neurohormonal imbalance, formation of uremic toxins and vascular calcification which damage the endothelium and blood vessels. Cognitive dysfunction, dementia, transient infarcts, and white matter lesions are widespread in mild to moderate chronic kidney disease patients. Uremic toxins produced after chronic kidney disease can pass through the blood–brain barrier and mediate cognitive dysfunction and neurodegeneration. Furthermore, chronic kidney disease precipitates vascular risk factors that can lead to atherosclerosis, hypertension, atrial fibrillation, and diabetes. Chronic kidney disease also exacerbates stroke pathogenesis, worsens recovery outcomes, and limits the eligibility of stroke patients to receive available stroke therapeutics. This review highlights the mechanisms involved in the advancement of chronic kidney disease and its possible association with stroke.
Introduction
Progression of ischemic brain damage, effective revascularization, and recovery after a stroke depend on an array of existing comorbid conditions.1,2 Of these, chronic kidney disease (CKD) is an independent risk factor for both cardiovascular diseases and stroke.3–5 The Kidney Disease Outcomes Quality Initiative (KDOQI) and Kidney Disease Improving Global Outcomes (KDIGO) guidelines classified CKD into stages 1 to 5 based on the extent of kidney damage or a decreased glomerular filtration rate (GFR). 6 These guidelines indicate that irrespective of etiology, once the loss and/or reduction of the function of nephrons reaches a certain threshold, the remaining functional nephrons begin a process of irreversible nephrosclerosis that results in a progressive decline in the GFR. 7 As GFR alone does not manifest any clinical symptoms until stage 4 to 5, it is important to evaluate both GFR and albuminuria to improve prognostic accuracy of CKD progression.8,9 Several markers such as albuminuria, cardiac troponin T, cystatin C, β2 microglobulin, β-trace protein, C-reactive protein (CRP), and N-terminal pro-B-type natriuretic peptide currently serve to identify and predict vascular dysfunction in patients with CKD. 10 In CKD patients, stroke is the third most cause of death, and patients with end-stage renal disease (ESRD) show an up to 10-fold higher risk of hospitalization for ischemic and hemorrhagic strokes.11,12 The majority of CKD patients die due to CKD-induced cardiovascular diseases including stroke rather than end-stage CKD.13,14 The high death rate due to cardiovascular disease in patients with renal dysfunction suggests that vascular abnormalities begin in these patients at an early stage of kidney disease. A gradual decline in the GFR below 90 mL/min/1.73 m2 and the presence of proteinuria pose a substantial two-way risk to cerebrorenal microvasculature due to anatomical, vasoregulatory, and hemodynamic similarities.15,16 Several population-based studies indicate that a GFR of <60 mL/min/1.73 m2 is independently correlating with an increased risk of stroke and unfavorable long-term outcomes.17,18 Stroke risk is also known to increase progressively with the advancement of CKD that also exacerbates post-stroke brain damage, worsened functional outcomes, and higher mortality in CKD–stroke patients. 3 Meta-analyses studies have indicated that a GFR of ≤60 mL/min/1.73 m2 or over-imposed with albuminuria and proteinuria increases the stroke risk by 43% and 71–92%, respectively.19–21 Moreover, kidney dysfunction occurs in ∼40% of stroke survivors. 22 Recently, a meta-analysis of studies with over two million patients has indicated a strong correlation between declined renal function and increased stroke risk. 23 This study indicated that for every 10 mL/min/1.73 m2 decline in GFR, stroke risk increases by 7% and for every 25 mg/mmol increase in albuminuria, stroke risk increases by 10%. Albuminuria was also shown to have a strong positive correlation with post-stroke mortality. 23 Interestingly, a study that followed up patients for 15.5 years identified that albuminuria is a risk factor for stroke only in men, but not in women. 24 Many studies also indicated that CKD patients are more prone to ischemic strokes than hemorrhagic strokes. 25
In the context of kidney dysfunction after stroke, a meta-analysis of 13 studies with 5,147,754 patients identified a high-risk acute kidney injury (AKI) after ischemic stroke.
26
Furthermore, the same study identified that the prevalence of AKI is higher after intracerebral hemorrhage (ICH) than after ischemic stroke, but the development of AKI is an independent risk factor for mortality after ischemic stroke, but not after ICH. However, it is unclear if AKI is due to stroke onset or exacerbation of ongoing CKD. AKI and CKD are interconnected; CKD patients are at high risk of post-stroke AKI and also AKI exacerbates the ongoing CKD progression and promotes early renal failure.27,28 At the advanced stage of CKD, dialysis procedures in ESRD patients can induce ischemic stroke.29,30 CKD develops over time with five distinct stages (Figure 1). While most clinical studies highlighted the consequences of the advanced CKD (stage 4 and 5) in ischemic stroke patients, there is a lack of understanding in the implications of early (stage 1), mild (stage 2) and moderate (stage 3) CKD in ischemic pathogenesis. Several studies indicated that a slight decline in renal function manifests some degree of peripheral and central neurological complications due to a gradual increase in oxidative stress, inflammation, hemodynamic/vasoregulatory abnormalities, and uremic toxins in both humans and experimental models.31–35 Comorbid conditions like hypertension, diabetes, and atrial fibrillation in CKD patients also aggravates vascular complications (Figure 1). Even at an early stage of CKD, oxidative stress and low-grade inflammation make blood vessels and endothelium more vulnerable to even slight vascular shift, which in turn compromises the blood–brain barrier (BBB) integrity and facilitates infiltration of white blood cells (WBCs) and uptake of uremic toxins into central nervous system (CNS).
34
The proinflammatory molecules and reactive oxygen species (ROS) released by the invading WBCs and uremic toxins are known to manifest cognitive dysfunction in approximately 16–38% of advanced CKD patients.36,37 In both humans and rodents, CKD affects neurohormonal communication which results in a sympathetic overflow and activation of the tissue renin-angiotensin system (RAS).38,39 Several clinical and preclinical studies identified that in CKD, RAS-induced angiotensin-2 (ANG-II) mediates inflammation and induces vascular fibrosis.
40
In particular, experimental studies with rodents demonstrated that ANG-II acts as a promoter of proteinuria directly by efferent vasoconstriction and indirectly by TGF-β1-mediated impairment of afferent arteriole autoregulation that enhances capillary filtration pressure.41,42 ANG-II activates proinflammatory nuclear factor kappa-light-chain-enhancer of activated B (NF-κB) through AT1 and AT2 receptors in rat vascular smooth muscle cells
43
and by interacting with myeloid differentiation protein-2 and toll-like receptor-4 signaling in mice.
44
Collectively, these CKD-induced mechanisms compromise brain function and make the brain susceptible to even a slight ischemic insult through complex cerebrorenal mechanisms. In addition to the complexity of ischemic pathogenesis, CKD also alters the effect of treatments used in acute stroke and secondary stroke prevention.
45
However, substantial evidence to support the efficacy and safety of many of these medications in different stages of CKD is lacking, as these patients are excluded in many clinical trials due to safety issues.
46
Hence, understanding the mechanisms of stroke pathogenesis as a function of CKD progression is crucial to treat these patients, especially with medications that undergo renal clearance. This review highlights the mechanisms which need in-depth understanding across the stages of CKD and their possible role in ischemic pathogenesis.
CKD induces pre- and post-stroke complications. Abnormal changes in endogenous tPA/PAI-1, MMP-9, uremic toxins such as indoxyl sulfate, p-cresyl sulfate and guanidino substances and profibrotics like TGF-β1, Tenascin-C, PAI, ANG-II, and comorbid conditions like hypertension and diabetes lead to the progression of CKD from stage 1 to stage 5. Advancement of CKD also leads to progressive endothelial dysfunction, vascular stiffness, BBB disruption, and peripheral/central inflammation. These have severe consequences in the brain during both pre-stroke and post-stroke stages.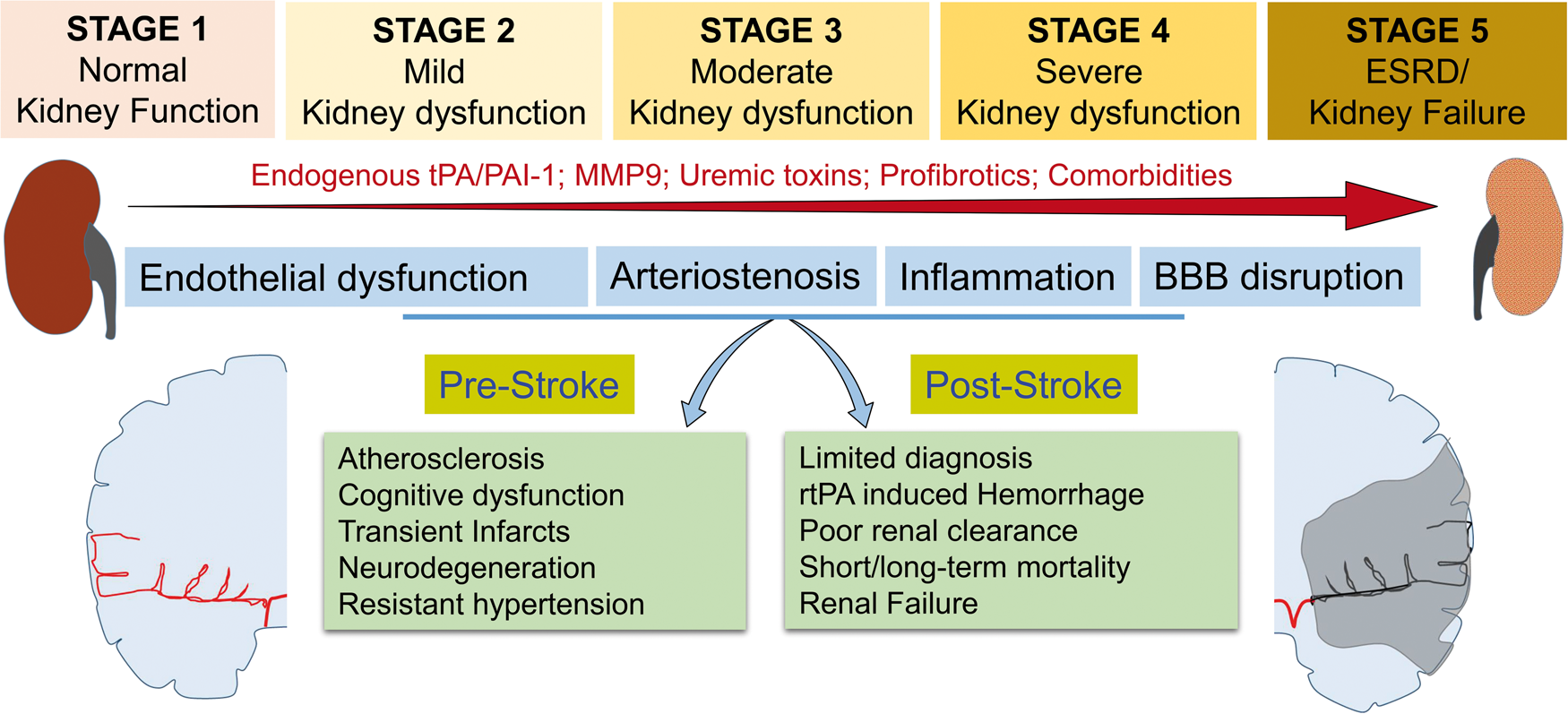
Epidemiology
CKD prevalence is highly diverse within and between various countries.47,48 In USA, CKD affects ∼15% of adults. 49 The incidence of CKD is much higher in people aged 65 years or older (38%) than in people aged 45–64 years (13%) and 18–44 years (7%). 49 A higher prevalence of CKD is observed in women (16.5%) compared to men (13%). 50 Atherosclerosis Risk in Communities (ARIC) study identified a significant risk of cardiovascular diseases including stroke in stage 2 CKD patients. 51 Heart Outcomes Prevention Evaluation Study (HOPE) showed that even a mild degree of renal dysfunction increases the risk of ischemic stroke or transient ischemic attack. 52 In pooled analyses of four prospective community-based cohorts, low GFR was shown to be significantly associated with increased risk of ischemic stroke, while low GFR + high albumin/creatinine ratio is associated with both ischemic and hemorrhagic stroke.53,54 Stroke remains the fifth leading cause of long-term disability in USA. 55 Combination of CKD and stroke has a substantial socioeconomic impact and results in a poor quality of life. Globally, the prevalence of stroke–CKD combination remains unknown, and very few studies evaluated the brain–kidney interactions in the stroke pathogenesis as influenced by CKD progression. In addition, the incidence of stroke and post-stroke disability in CKD patients with or without dialysis is also not systematically evaluated. Furthermore, the effect of age, sex, and comorbidities like hypertension and diabetes in promoting stroke in CKD patients is also not studied.
Oxidative stress and inflammation in CKD
Chronic inflammation and oxidative stress are known to be associated with CKD progression. Markers of oxidative stress such as mitochondrial superoxide, oxidized low-density lipoprotein (LDL), homocysteine, superoxide dismutase, and glutathione were shown to be involved in CKD progression.56–59 Uremic toxins such as indoxyl sulfate, p-cresyl sulfate, advanced glycation end products (AGEs), and activated NADPH oxidase (NOX) promote oxidative stress and endothelial dysfunction in both CKD patients and preclinical rodent models of CKD.60–62 In addition, ANG-II plays a central role in the pathophysiology of arterial hypertension through the activation of NOX. 63 Oxidative stress leads to arteriosclerosis and arterial stiffness in CKD. 64 In particular, uremic toxins have been shown to induce arterial stiffness through increased oxidative stress.65–67 Elevated levels of ROS are known to lead to cerebral sympathoexcitation in CKD. 68 Further, indoxyl sulfate and p-cresyl phosphate also activate leukocyte adhesion and cerebral endothelial dysfunction. 35 Treatment with the antioxidant tempol was shown to improve cognitive dysfunction in uremic mice. 69 In a rat model of hypertensive stroke, renal denervation attenuated ischemic brain injury by inhibiting oxidative stress, indicating its role in kidney disease-associated exacerbation of stroke pathogenesis. 70
Chronic low-grade inflammation can be seen in patients at all stages of CKD. 71 Elevated levels of proinflammatory molecules IL-6, TNF-α, osteoprotegerin, osteocalcin, osteopontin, and fibroblast growth factor 23 were reported in the blood even in patients in early stage 2 CKD. 72 Elevated plasma levels of fibrinogen, IL-6, and TNF-α and decreased serum albumin in CKD patients were shown to be independently associated with CKD progression. 73 Following hemodialysis, CKD patients showed higher mortality if the levels of circulating proinflammatory mediators (IL-1, IL-6, and TNF-α) are higher compared to those with higher anti-inflammatory mediators (IL-2, IL-4, IL-5, IL-12, CH50, and T-cell number). 74 ANG-II-induced hypertension is characterized by inflammation that in turn contributes to renal fibrosis. 75 Inflammation is also shown to be a strong predictor of poor clinical outcome and mortality in advanced CKD patients. 76 Recent studies showed that gut-derived uremic toxins also play a major role in systemic inflammation in CKD patients and experimental models.77,78 Uremic toxins indoxyl sulfate and guanidino compounds enter CNS via organic anionic/cationic transporters and promote neuroinflammation.79,80 Uremic toxins also promote macrophage polarization toward the deleterious proinflammatory M1 phenotype and activate the resident microglia to promote post-stroke brain damage. 35 All these studies suggest that uremic toxins formed in CKD precipitate secondary brain damage by amplifying post-stroke oxidative stress and inflammation. Treatment with β-hydroxy β-methylglutaryl-CoA reductase inhibitors (statins), angiotensin-converting enzyme inhibitors and AT1-receptor blockers reduce vascular oxidative stress and proteinuric nephropathy, thereby preventing cardiovascular disease progression.81–84
Early vascular changes in CKD
CKD is commonly considered as a “silent killer,” as it does not manifest any clinically detectable symptoms until an advanced stage of the disease. 85 In many patients, CKD is generally identified while screening for other disease-related diagnostic procedures. The decline in kidney function incites changes in the central as well as peripheral neuronal activity and manifests several neurological complications by synergistic mechanisms that include vascular calcification and uremic toxins in both humans and rodents.86,87 The metabolic changes start at an early stage of CKD by abnormal calcium-phosphate metabolism and inflammation that collectively damage the endothelium.88,89 Levels of fibroblast growth factor 23, a hormone that regulates the phosphorus metabolism has a positive graded correlation with stages of CKD and have a strong association with a higher risk of cardioembolic stroke.90,91 Recent human studies indicate that even a mild impairment of kidney function is associated with higher plasma levels of gut-derived uremic toxins. 92 High levels of microbial metabolite Trimethylamine-N-oxide (TMAO), uremic toxins like indoxyl sulfate, p-cresyl sulfate and guanidino substances that induce oxidative stress are found in the brain tissue from CKD patients particularly in the regions that play a determinant role in cognition, such as the thalamus, mammillary bodies, and cerebral cortex.93–95 High TMAO levels associated with the stage of CKD and disease progression. 96 Uremic toxins induce endothelial dysfunction leading to BBB disruption and leukocyte infiltration into the damaged rat brain. 34 Furthermore, experimental studies with rodents demonstrated that the uremic toxins amplify the release of proinflammatory cytokines and ROS by infiltrated macrophages, influencing the microglia and astrocytes in the brain.93,97 These deleterious mechanisms synergistically exacerbate post-stroke pathogenesis and account for poor stroke recovery in CKD patients. The deleterious role of uremic toxins in the acceleration of ischemic pathogenesis is further corroborated by a retrospective analysis of the three to five years of treatment with AST-120, an adsorbent claimed to remove gut uremic toxins and decrease the prevalence of stroke events in pre-dialysis CKD patients. 98 Further, chronic mild to moderate renal insufficiency is recognized as a clinical situation posing significant risks to endothelial integrity. 99 Endothelial cells play a vital role in maintaining vascular homeostasis by regulating vasodilatation, fibrinolysis, and thrombosis mechanisms through the synthesis and release of substances such as nitric oxide, prostacyclin, thrombomodulin, and tissue plasminogen activator (tPA).100,101 Thus, endothelium dysfunction mediated by intercellular and vascular adhesion molecules, endothelial selectin and von Willebrand Factor (vWF) is a critical factor in establishing a strong correlation with renal insufficiency and vascular health.102,103 Moreover, a strong association between adipokines and endothelial dysfunction was found in many CKD preclinical and clinical studies.104,105 Proinflammatory molecules, interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) induced by endothelial dysfunction damage the vasculature indirectly by stimulating the synthesis of CRP. 106 CRP is an indicator of inflammation, and its levels show a positive correlation with the progression of ischemic stroke. CRP levels within 12 h of ischemic stroke are also an independent prognostic factor of poor outcome at three months. 107 Higher CRP concentrations are associated with a higher risk of ischemic stroke, but not hemorrhagic stroke, particularly in hypertensive males. 108 Thrombomodulin, a novel biomarker of endothelial dysfunction, correlates with CKD stage, kidney function parameters (urea, creatinine, and cystatin C) as well as with oxidative stress, hypertension, and left ventricular hypertrophy.109,110 Increased levels of circulatory endothelial cells is a clinical sign of the development of vascular disease in patients with CKD and indicate the long-term outcome of vascular dysfunction in these patients. 111 Association of endothelial dysfunction with increased serum creatinine in patients with a preserved steady GFR after percutaneous coronary procedure suggests that endothelial damage starts even with a mild decline in kidney function. 112 Moreover, low GFR is associated with higher chances of other potential diseases such as atherosclerosis, atrial fibrillation, coronary heart disease, peripheral artery disease, heart failure, bone disease, anemia which are all again potential risk factors for stroke.113,114 Furthermore, cardiovascular complications and venous thrombosis that mainly dominate at the early stages of CKD suggest a prothrombotic state, while procoagulant state persists with episodes of bleeding due to the strong association of uremic toxins with platelet dysfunction at advanced stages of CKD.115,116 These observations suggest that mild to moderate CKD patients are more prone to ischemic strokes, while advanced CKD patients are more prone to hemorrhagic strokes. Taken together, vascular changes in progressive CKD patients act as a double-edged sword which results in either ischemic or hemorrhagic stroke.
Prothrombotic mechanisms in CKD
The gradual destruction and irreversible loss of kidney function in CKD change the dynamics of body fluids, which result in an ionic imbalance and alterations in the blood composition. 117 Even a mild CKD impairs the endothelial barrier and promotes vascular stiffness due to changes in the proinflammatory and prothrombotic microenvironment. Furthermore, damaged endothelium releases abnormal thrombotic as well as antithrombotic mediators such as tissue factor, circulating coagulation factor fVIII, thrombomodulin, endothelial protein C receptor, protease-activated receptor-type receptors, and vWF that changes the blood composition and rheological properties. 118 The hemodynamic changes in the body worsen with the severity of renal dysfunction and promote thrombosis, brain hemorrhages, or mixed cerebrovascular events. ESRD was associated with bleeding, whereas early stages of CKD with moderate preserved renal function are associated with thrombosis. 115 The risk of thromboembolism in CKD patients starts as early as at GFR <75 mL/min/1.73 m2 with perceived normal kidney function. 119 In addition, venous thrombosis is more common in severe CKD, but also highly prevalent across CKD stages 1–3 in the presence of albuminuria. 120 CKD increases the circulatory levels of proinflammatory mediators such as CRP, IL-6, plasma tissue factor-induced NF-κB, and protease-activated receptor-1121 which leads to the induction of several coagulation factors like fibrinogen, factor VIII, and vWF. 122 The ensuing increased coagulation is further complicated by CKD-induced atherosclerotic plaque rupture, which recruits more platelets to vulnerable areas leading to abnormal aggregation of platelets. 123 These aggregated platelets attract other blood cells to form a highly stable thrombus through intracellular calcium oscillation. 124 Several studies in patients and patient blood samples suggest that thrombus induced by profibrotic molecules acts in synergy with vWF and is much stronger and not easily dissociated by endogenous tPA.124,125 In addition, activation of the RAS in CKD increases levels of various profibrotic markers and proinflammatory cytokines in addition to alterations in mean arterial blood pressure. 41 Furthermore, plasminogen activator inhibitor-1 (PAI-1) inhibits extracellular matrix turnover, stimulates macrophages, promotes myofibroblast infiltration, and vascular fibrosis with the progression of CKD. 126 Moreover, PAI-1 also inhibits the activation of the fibrinolytic system by inhibiting the tPA and urokinase. 127 Interestingly, it is not understood how CKD patients can concurrently experience both thrombosis and hemorrhage in the brain. It is also not clear how the same renal disease that initiated predominantly prothrombotic mechanisms mediates hemorrhagic transformations after thrombolytic therapy. However, these mechanisms collectively suggest that exogenous tPA used after acute ischemic stroke might cause a mechanistic shift from CKD–modified ischemic events to post-stroke bleeding complications.
Thrombolysis after acute ischemic stroke in CKD
One of the clinically challenging decisions after acute ischemic stroke in CKD patients is to determine the patient's eligibility for thrombolysis with recombinant tPA as well as the risk associated with mechanical thrombectomy. The presence of subclinical manifestations such as transient infarcts, lacunar infarcts, and microbleeds make the thrombolysis more complex in CKD patients. There are a few population-based studies that evaluated the significance of CKD involvement in thrombolysis and post-stroke recovery, but many of them are with small sample size, lack of matched controls, not focused on stages of CKD.11,128–132 Even in those cases, the prediction of outcomes is complex as ESRD patients suffer from several other co-comorbidities such as neurogenic hypertension, diabetes, anemia, and atrial fibrillation. However, these studies collectively identified that CKD interferes with a diagnosis associated contrast agents used for multi-model tomography imaging, 133 alters the efficacy of thrombolysis, 134 increases the bleeding risk,135,136 leads to the high frequency of dehydration, 137 and poses a safety issue for post-stroke management medications which need renal clearance.131,132,138 Although several clinical findings imply that the presence of CKD alone is not a barrier to the administration of recombinant tPA, thrombolysis in CKD is associated with a high rate of in-hospital mortality and other unfavorable outcomes.135,139–141 Drug information for recombinant tPA formulations includes a warning sign that its administration must be with caution in case of the severe renal disease. It is evident that recombinant tPA-induced thrombolysis in CKD patients presents a substantial risk of hemorrhagic transformations as well as the poor clinical outcome. CKD patients exhibit high levels of endogenous circulatory tPA, thrombotic mediators, endothelial damage, platelet dysfunction, and infiltration of inflammatory cells into the brain. All these cumulatively could aggravate the bleeding complications after recombinant tPA administration in stroke–CKD patients.
Previous studies indicated that both urokinase-type plasminogen activator and its soluble receptor (uPA/suPAR) and tPA/PAI-1 concentrations were higher in chronic renal failure.
142
Impaired renal function leads to decreased clearance of uPA/suPAR complexes which determinate fibrinolytic activity by accelerated conversion of plasminogen into plasmin.127,143 A recent study identified a graded positive association between uremic toxin anthranilic acid and uPA/suPAR in the mild-to-moderate CKD subgroup and an inverse relationship with tPA/PAI-1 in the severe-to-end-stage CKD subgroup.
144
Furthermore, overexpression of PAI-1 in renal tissue induces RAS hyperactivity-related alterations to coagulation/fibrinolytic systems and promotes CKD progression.
145
Moreover, mechanistic functions of exogenous as well as endogenous tPA during acute ischemic stroke in CKD other than thrombolysis need to be identified based on tPA functional domains.
146
The finger domain of tPA is essential to promote fibrinolytic activity at low plasminogen activator concentrations.
147
In addition to fibrinolysis, functional domains of tPA interact with LDL receptor-related proteins to support the BBB crossing, astrocytic clearance, or microglial activation.
148
Endogenous tPA promotes BBB permeability by inducing matrix metalloproteinase-9 (MMP-9), N-methyl-D-aspartate receptor, platelet-derived growth factor receptors, and LDL receptor-related proteins in endothelial cells and astrocytes leading to disruption of tight junctions and BBB leakage that results in intracranial hemorrhage (Figure 2). In addition to high levels of endogenous tPA in CKD, exogenous tPA administration in the presence of acute stroke-induced excitotoxicity and energy failure results in abnormal levels of cellular and circulatory tPA, which collectively aggravates hemorrhagic transformations (Figure 2). In support of the deleterious effects of tPA after AIS, clinical studies identified an independent mechanism of exogenous tPA-induced seizures and worsening of outcome after thrombolytic therapy.149,150 In addition, brain imaging studies revealed that exogenous tPA promotes BBB leakage in patients with stroke.
151
On the other hand, a recent study on the impact of CKD in patients undergoing surgical carotid artery revascularization reported substantial differences in the procedures used for revascularization (carotid artery stenting/carotid artery endarterectomy) as a function of other disorders such as diabetes mellitus, hypertension, hyperlipidemia, and coronary artery disease.
152
This study also showed that among CKD cohorts, in-hospital major adverse cardiovascular and cerebrovascular events were higher for patients who underwent carotid artery stenting. Since the recombinant tPA-induced thrombolysis is the best available option currently, an understanding of the bleeding complications and hemorrhagic transformation due to endogenous as well as exogenous tPA is necessary. A recent study identified a significant correlation between uremic toxin anthranilic acid and fibrinolytic molecules such as uPA, tPA, suPAR, PAI-1, and PAP at different stages of CKD.
144
In particular, this study identified the baseline differences in tPA among patients with mild to moderate and severe CKD. Non-fibrinolytic mechanisms of tPA suggest that CKD patients are highly vulnerable to tPA-induced complications, even at an early stage of CKD.
153
Although there are no evidence-based studies to confirm the association between hemorrhagic transformations after thrombolytic therapy in acute ischemic stroke in early CKD patients, studies demonstrated that the hemostatic abnormalities start as early as the first stage of CKD.
120
Possible mechanism of hemorrhagic transformations after thrombolysis. Cumulative endogenous tPA in circulation due to abnormalities in tPA/PAI ratio and exogenous administration affects the influx of excess circulatory tPA into the ischemic tissue. Excess accumulation of extracellular matrix components, uremic toxins, energy failure, oxidative stress, and excitotoxicity influences the effects of cellular as well as circulatory tPA. In stroke–CKD cases, bleeding complications are mediated by MMP-9, synergistically with cytokine activity of tPA, uremic toxins, and altered ECM leading to hemorrhagic transformation.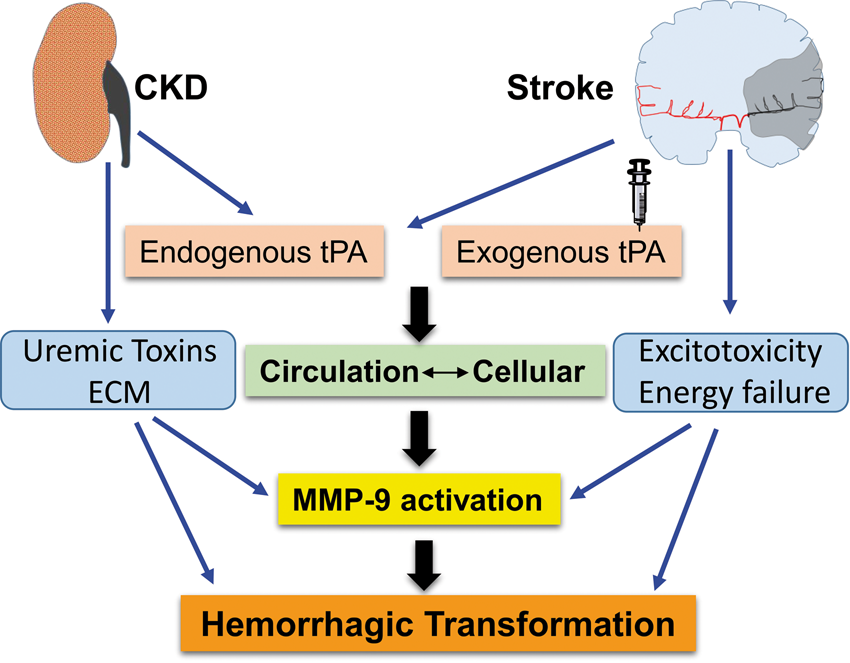
Current challenges in treating acute ischemic stroke in CKD
Age-specific challenges
CKD in the elderly is fatal than progression to ESRD when compared with younger patients.154–157 Studies from healthy kidney donors revealed an age-dependent exacerbation of nephrosclerosis from 2.7% in 30 years old, 58% in 60–69 years old, and 73% in 70 years old humans.158,159 Currently available CKD markers do not correlate well with CKD progression in aged individuals due to age-associated changes in GFR, albuminuria, body mass index, serum cholesterol, glucose, and uric acid levels. 158 In older CKD patients and those with existing comorbid conditions like diabetes and hypertension, vascular complications are predominantly higher and affect sodium-glucose reabsorption.162–165
Sex-specific challenges
CKD progression is faster in men than women, but women with CKD are at a higher risk of disabling and fatal strokes than men.160,161 Women have higher renovascular resistance, lower GFR, lower renal plasma flow, and distinct RAS activity compared with men.166–168 Lower bone mineral density associated with higher coronary artery calcification, and vascular calcification were seen in females, but not males, with ESRD. 169 Currently, there are no sensitive diagnostic tests to accurately identify sex-specific CKD progression and sex-specific stroke therapies at different stages of CKD.
Comorbid condition-specific challenges
Comorbidities such as diabetes (one in three) and hypertension (one in five) frequently manifest together with a substantial overlap of CKD etiology and mechanisms.170,171 Sympathetic activation plays a crucial role in the pathogenesis of hypertension associated with CKD. 172 Insulin resistance and RAS-induced sympathetic activation in CKD are the key mechanisms which compromises both peripheral and brain sodium-glucose handling. The decline in GFR affects renal sodium-glucose reabsorption due to failed sodium-glucose cotransporters. Further, one of the unique variations in CKD progression in comorbid subjects is the abnormal sodium-glucose reabsorption and water retention, which results in resistant hypertension and impaired glucose metabolism.173–175 Recent studies with sodium-glucose cotransporter-2 inhibitor empagliflozin identified a common therapeutic effect to treat diabetes, cardiac disease, and stroke.176,177 The multiple therapeutic benefits of empagliflozin suggests the importance of identifying cerebrorenal common mechanisms to fight the brain damage in CKD/stroke patients. Treating CKD patients with stroke in the presence of diabetes/hypertension is challenging, as these subsets of patients are more prone to drug interactions due to existing multisystem disease pathology.
In addition, older adults are generally considered more prone to stroke. However, the incidence of ischemic stroke in younger patients is increasing dramatically.178–180 Importantly, comorbid conditions like CKD pose a potential threat to cerebrovascular health, as its advancement triggers many common vascular complications and promotes the failure of other organs. The complexity of vascular injury in CKD at all stages may act as a disease multiplier and limits the post-stroke recovery. Currently, there are no evidence-based studies to assess the severity of strokes in CKD patients at different stages. Furthermore, the effectiveness and safety of thrombolytic therapy for acute ischemic stroke are unknown for patients with mild to moderate CKD. On the other hand, post-stroke management with anticoagulants, antiplatelet agents and lipid-lowering agents to prevent recurrent strokes presents the risk of undesirable side effects in CKD patients with reduced renal clearance. Future studies are needed to understand the molecular mechanisms of a stroke at each stage of CKD, and their impact on hemodynamics, endothelium, and brain functions. The major challenge of treating stroke in CKD patients is the overlapping comorbidities including atherosclerosis, diabetes mellitus, and hypertension which precipitate stroke onset.181,182 Although blood profile is be routinely checked before thrombolysis in acute stroke patients, sensitive biomarkers to identify the progressive CKD are still absent. Furthermore, it is also important to identify biomarkers that change due to stroke during kidney disease. 183 Further studies are needed to determine the early CKD mechanisms which directly influence brain function and to establish an independent correlation of how those mechanisms differ in the presence of vascular abnormalities. However, the practicality of establishing such correlations in patients is limited, as early CKD clinical manifestations are undetectable, and importantly, the progression of CKD is heterogenic. Hence, studies to identify clinically detectable symptoms as biomarkers of progressive CKD at early stages and the clinical manifestations of how CKD leads to stroke pathogenesis are needed. In particular, identification of potential biomarkers with diagnostic value and establishment of a threshold level for effective thrombolysis to limit bleeding complications at each stage of CKD might help to minimize post-stroke morbidity and mortality in these patients efficiently. Furthermore, stroke also influences many peripheral organs, including kidneys. Hence, studying kidney function in stroke patients is also of paramount importance.
Conclusions and limitations
The relationship of brain and kidney is two-way, an in-depth understanding of peripheral/central vascular changes and the various actions of tPA at each stage of CKD are needed to manage the outcomes of stroke in these patients. Furthermore, understanding the effect of stroke on kidney dysfunction is also needed. This poses a difficulty as many CKD patients and stroke patients also show other organ dysfunction and comorbidities. Controlled preclinical studies with models of CKD over-imposed with stroke might offer an opportunity to understand this devastating condition and test novel therapies.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is partially supported by grants from NIH (RO1 NS099531, RO1 NS101960 and RO1 NS109459) and Veterans Association (I01 BX002985 and I01 BX004344) and the Department of Neurological Surgery, University of Wisconsin.
Acknowledgements
The authors thank Dr. Suresh L. Mehta for critical suggestions.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.