
Abstract
Glioblastoma is a highly aggressive and treatment resistant primary brain tumor. Features of glioblastoma include peritumoral cerebral edema, the major contributor to neurological impairment. Although the current clinical approach to edema management is administration of the synthetic corticoid dexamethasone, increasing evidence indicates numerous adverse effects of dexamethasone on glioblastoma burden at the molecular, cellular and clinical level. The contradictions of dexamethasone for glioblastoma and brain metastasis therapy are discussed in this article. Finally, alternative strategies for cerebrovascular edema therapy with vascular stabilizing, anti-permeability agents that are either approved or in clinical trials for diabetic retinopathy and macula edema, are addressed.
Keywords
Introduction
Patients with central nervous system (CNS) malignancies such as glioblastoma (GBM) and brain metastasis usually present with peritumoral vasogenic edema, the pivotal contributor to morbidity and mortality in neuro-oncology. Peritumoral edema is caused by breakdown of the blood-brain barrier (BBB) with subsequent leakage of plasma components into the cerebral extracellular space and consequent elevation of intracranial pressure (ICP).1–4 The increase in ICP leads to functional impairment of the BBB associated with neurological deficits and poor penetration of chemotherapeutics such as temozolomide (TMZ), which at least in part contribute to poor clinical outcome in GBM. 5 To date, the neurosurgical and neuro-oncological drug of choice in peritumoral edema management is the synthetic corticoid dexamethasone (DEX), an agent with a pleiotropic action profile and 30-fold greater glucocorticoid effect compared to the endogenous hormone cortisol. Although clinically effective in vasogenic edema management, DEX administration is associated with numerous systemic side effects including insomnia, psychiatric alteration, tremor, hyperglycemia, muscle atrophy, cushingoid appearance, hypertension, gastrointestinal perforation and notably, immunosuppression. 6 Despite decades of treatment, the mechanism by which DEX reduces peritumoral edema remains poorly understood. The current hypotheses include the DEX-mediated improvement of BBB function via upregulation of tight-junction proteins and inhibition of inflammatory pathways, resulting in reduced vessel permeability.7–10 This brief opinion article highlights the state of knowledge regarding the impact of DEX on GBM burden and discusses alternative therapeutic approaches targeting the vascular endothelial growth factor (VEGF) and Tie2 pathways, shown to reduce edema by improving BBB function.3,11,12 We emphasize the utilization (“repurposing”) of drugs that are approved or in clinical trials for edema management in retinal vascular diseases that show defects in the blood-retina barrier.13,14
BBB dysfunction in GBM
One of the major hallmarks of GBM is the dysfunction of the BBB, a critical barrier that maintains CNS homeostasis. The BBB plays a major role as a transport and metabolic barrier which is constituted by the neurovascular unit (NVU) with microvascular endothelial cells (ECs) as the main cellular component whose function is supported by other cell types such as pericytes, astrocytes, neurons and microglia.15–18 However, the highly specific and regulated nature of the NVU allows for transport of essential nutrients across the BBB. Although the BBB protects the CNS, it presents a challenge for transport of therapeutics in neurological diseases targeting parenchymal cells. 19 In GBM and brain metastasis, NVU dysfunction and BBB breakdown is manifested by edema formation that results in increased ICP limiting local perfusion and a transport barrier leading to hypoxia and ensuing pathologic tumor angiogenesis.1,3,4 The disrupted barrier also poses serious challenges for drug delivery due to increased ICP and disorganized and poorly perfused angiogenic vasculature. 19 Several studies demonstrated the negative effects of peritumoral edema in GBM progression. Patients with minor edema on preoperative cranial MRI scans were shown to exhibit longer survival compared to major peritumoral edema. 5 Several mechanisms were hypothesized for this finding, for example edematous brain-tumor tissue enabling enhanced GBM cell invasion and preservation. GBM stem-cells were also suspected to persist in this edematous area, escaping cytotoxic treatment and promoting tumor progression. Based on these findings, reduction of edema by restoring NVU function represents an important approach to GBM therapy. Dexamethasone, the synthetic, potent version of endogenous cortisol is widely used in this regard; however, the mechanisms by which DEX controls brain edema at the level of endothelial and tumor cells is still unclear. Furthermore, there are numerous side effects of DEX that demand for novel therapeutics to restore NVU function and BBB integrity in GBM. 6
Effects of dexamethasone in GBM therapy
Systemic adverse effects of DEX treatment for peritumoral edema in GBM and brain metastasis have been reported in up to 50% of the patients including hyperglycemia, cushingoid appearance and psychiatric symptoms among the top three.6,20 A retrospective multivariate analysis of 73 GBM patients revealed that concurrent DEX administration to radio-chemotherapy was a significant risk factor for poor overall survival (OS) (12.7 vs. 22.6 months, p = 0.004) and progression free survival (PFS) (3.6 vs. 8.4 months, p<0.0001). 21 These observations were supported by a retrospective multivariate analysis of more than 2000 GBM patients, providing evidence that DEX administration was an independent negative prognostic factor when adjusting for extent of resection, initial treatment, age and Karnofsky Performance Score (OS 12 vs. 17 months, p = 0.001). 22 Wong et al. studied 119 patients with recurrent GBM receiving either tumor-treating alternating electric fields (TTFields) or chemotherapy. In both groups DEX administration of >4.1 mg per day was associated with reduced OS (4.8 vs. 11.0 months, p = 0.0001 in the TTFields cohort and 6.0 vs. 8.9 months in the chemotherapy group, p = 0.0009). 23 A single center retrospective analysis showed that patients with dexamethasone-induced leukocytosis (DIL) had reduced OS and PFS (15 vs. 32 months for OS and 7 vs. 15 months for PFS, p = 0.009). 24 Furthermore, reduced CD3 lymphocyte and CD15 granulocyte infiltration in the tumor parenchyma of DIL positive patients (p<0.05) was demonstrated. This suggests that DEX might negatively interfere with immunotherapy as also demonstrated by Giles et al. (see below). 25 Beneficial effects of DEX-sparing has been assessed in combination with corticorelin acetate (CrA), 28 a synthetic formulation identical to the endogenous corticotropin-releasing factor/hormone which led to reduced vascular permeability. 26 The study demonstrated a significant reduction in DEX dose in patients that correlated with better neurological scores. 27 DEX is also administered in patients with brain metastasis that display severe vasogenic edema and ICP (reviewed in Ryken et al. 4 ), although the benefit has not been clearly demonstrated. In breast cancer patients, it has been reported that the incidence of metastasis increases with administration of DEX mediated by activation of glucocorticoid receptor leading to increased colonization in distant sites. 28 Dexamethasone is also used as an adjuvant for edema management in bacterial meningitis; however, its benefit for patient survival or neurological performance has not been unequivocally demonstrated.29–32 Overall, the negative influence of DEX on the therapeutic outcome warrant alternative treatment options for edema management.
Effects of dexamethasone in preclinical studies
Although unfavorable clinical effects of DEX in GBM patients are documented, preclinical studies revealed conflicting and in part opposing results. Beneficial results were observed in a rat permeability model where DEX led to reduced permeability of the brain ECs to tumor-derived VEGF and to reduced VEGF secretion by tumor cells.33,34 In addition, DEX was shown to decrease MMP-2 (matrix metalloprotease) secretion and cell invasiveness via MKP-1 (mitogen activated protein kinase phosphatase) induction in human glioma cells. 35 DEX also increased cell–cell adhesion and reorganized actin into stress fibers – resulting in reduced cell motility and invasion, which was also observed in human GBM neurospheres.36,37 Similar results were obtained in brain ECs where hydrocortisone treatment increased actin reorganization and reduced permeability. 38 Furthermore, DEX inhibited glioma cell proliferation in a dosage-dependent manner and even induced cell death. 39 On the counter side, an unfavorable interplay of DEX and alkylating agents was described. In primary GBM cell lines treated with TMZ, the addition of DEX counteracted TMZ-induced apoptosis and caused resistance to nutrient deprivation.40–42 Furthermore, in vitro DEX administration reduced the bystander effect of the thymidine kinase/ganciclovir system in suicide-gene therapy and reduced the antitumor effect of interleukin-4.43,44 The alkylating impact of TMZ targets the O6-alkylguanin in the carcinogenic lesion, which is reversed by the O6-methylguanine-DNA-methyltransferase (MGMT) repair enzyme. The human MGMT promotor harbors two glucocorticoid response elements and recent findings report the DEX-mediated induction of MGMT promotor as the most important factor of chemo-resistance. 45 Furthermore, mesenchymal glioma stem cells showed a significant upregulation of DEX-regulated gene network resulting in increased invasion, proliferation and angiogenesis in human-derived orthotropic tumors. 46 In the context of prevailing immunotherapies that are currently under way in GBM, further studies are needed to understand immune modulatory effects of DEX that include T lymphocyte depletion and suppression of co-stimulatory pathways.25,47
Anti-VEGF therapy for vasogenic peritumoral edema in GBM
Since the introduction of bevacizumab (BEV), a recombinant humanized monoclonal antibody against VEGF-A, for anti-angiogenic therapy in GBM, several clinical studies outlined an improved progression-free survival. However, the addition of BEV to standard radio-chemotherapy failed to show benefit in GBM OS.48,49 The chronic vascular hyperpermeability in GBM reflects the growth of structurally abnormal and immature vessels that have defective cell junctions and are deficient in pericytes. 50 The improper composition and functioning of the NVU in addition to high VEGF levels contribute to vascular permeability, which may lead to life-threatening edema.1–3 In fact, the permeability-inducing effects of VEGF were described before the effects on EC proliferation.51–53 Increased brain VEGF levels induced a 10-fold breakdown of the BBB. 54 In astrocyte-specific VEGF-A conditional knock-out mice, vascular leakage was prevented and brain edema reduced in a model of CNS inflammation. 55 Similarly, mechanistically blocking VEGF from reaching its capillary targets or blocking VEGFR2 function reduced capillary leakage and vasogenic edema.56,57
Almost two decades ago a correlation between the VEGF mRNA expression and the tissue plasma vascular volume and capillary permeability was described in human gliomas in vivo. 58 These findings supported the hypothesis that VEGF may play a pivotal role in peritumoral edema formation in addition to tumor angiogenesis in human gliomas. From those studies, it was concluded that inhibition of VEGF might be a useful approach to prevent edema formation in addition to tumor vascularization in vivo.58,59 Surprisingly, a clinical trial comparing DEX vs. VEGF inhibitors for edema treatment is still lacking, although several reports described a beneficial steroid-sparing effect in BEV-treated patients.6,60,61 In a murine GBM model, Pitter et al. reported benefit of anti-VEGF therapy compared to DEX and relate this finding to DEX-induced deceleration of cell cycle progression, via the induction of the cell cycle inhibitor p21, and thus elevated radio-resistance. 22 However, several adverse effects of anti-VEGF therapy limit its utility in perifocal edema therapy for GBM such as thromboembolic events, hemorrhage, surgical wound dehiscence, gastrointestinal perforation, hypertension and diarrhea.62,63 Besides these, the development of resistance mechanisms has been a major obstacle in anti-VEGF therapy. 64 VEGF/VEGFR inhibition failed to achieve durable responses in solid cancers due to acquired or tumor intrinsic resistance mechanisms which are not yet completely understood. 65 Although first-line BEV-treatment in GBM patients failed to show a survival benefit, it improved PFS and decreased the usage of steroids.48,49,64 Currently, BEV is commonly applied for edema management in recurrent GBM. 64 Resistance to anti-VEGF therapy in GBM includes the upregulation of additional growth factors such as Ang-2 as demonstrated in Scholz et al. 66 Furthermore, resistance is confined to innate immune cells which secrete a plethora of growth factors and cytokines that drive angiogenesis in GBM65,67 and negatively affect pre-clinical and clinical outcomes following anti-VEGF therapy.64,66,68,69 Consequently, alternative therapeutic options to treat peri-tumoral edema are warranted.
Modifying Tie2 signaling to treat vascular leakage
Tie2 is a receptor tyrosine kinase predominantly expressed in vascular ECs that modulates vascular permeability via interaction with the Angiopoietin family of protein ligands12,70 (Figure 1). In the normal brain, Ang-1/Tie2 signaling promotes vascular stability via its effects on EC junctions. In the GBM microenvironment, increased Ang-2 limits Tie2 receptor tyrosine kinase phosphorylation leading to NVU dysfunction and vascular leakage.
3
VEGF promotes neovessel formation and BBB breakdown in conjunction with the Ang-2/Tie2 axis.3,12 It was previously shown that the combinatorial blockade of VEGF and Ang-2, an endogenous EC-derived antagonist for Tie2, leads to vascular normalization and decreased vascular permeability in mouse GBM models.66,68,69 In addition to Tie2 ligands, vascular endothelial protein tyrosine phosphatase (VE-PTP) is a modulator of Tie2 phosphorylation and vascular permeability, whose therapeutic targeting has been shown to improve EC barrier function in preclinical models for breast cancer/metastasis, brain ischemia, inflammation, renal and ocular diseases.71–74 The expression of VE-PTP is induced by hypoxia73,75 and upregulated within the hypoxic tumor vasculature.
76
The recent development of small-molecule VE-PTP inhibitors that restore Tie2 phosphorylation independent of ligand binding, and their efficacy in preclinical models of ischemic and VEGF-induced retinopathy14,75,77 resulted in Phase I/II clinical trials with AKB-9778, the lead VE-PTP inhibitor. AKB-9778 showed increased efficacy in combination with anti-VEGF therapy in the treatment of diabetic macular edema, a disease in which vascular leakage, similar to GBM, is driven by hypoxia (ClinicalTrials.gov: NCT02050828). The safety and efficacy of subcutaneously administered AKB-9778 in patients with moderate to severe non-proliferative diabetic retinopathy is currently being evaluated in a Phase IIb study (NCT03197870). Further clinical trials are similarly targeting the Angiopoietin/Tie2 pathway in conjunction with VEGF in diabetic macula edema using anti-Ang-2 and anti-VEGF (NCT02712008) or bi-specific Ang-2/VEGF antibody approaches (NCT02699450). In a mouse model of breast cancer metastasis, Tie2 activation by VE-PTP inhibition prevented vascular leakage through a normalized tumor vasculature, delayed tumor growth, metastatic progression, and enhanced response to chemo and radiation therapy.
74
Recent evidence from a preclinical brain ischemia model showed that neurovascular permeability can be successfully counteracted by restoring Tie2 phosphorylation via VE-PTP inhibition.
71
Together, these results provide a rationale in restoration of Tie2 signaling for edema management in GBM and brain metastases.
Interference of angiogenic growth factor signaling with junctional proteins and vascular leakage in GBM. (a) In the normal brain vasculature, paracrine Ang-1 signaling leads to the phosphorylation and subsequent translocation of Tie2 to endothelial cell junctions (EC junctions; upper right panel). VEGF receptor 2 (VEGFR2) is activated due to low amounts of the activating ligand VEGF, but a stable vasculature is maintained by concomitant Tie2 activation. In GBM, several mechanisms lead to the induction of vascular leakage: (1) Ang-2 is secreted from Weibel–Palade bodies and acts in an autocrine manner on Tie2, thereby blocking the Ang1-induced Tie2 phosphorylation and vessel stabilization (lower right panel). (2) Tie2 phosphorylation is further reduced by the activity of VE-PTP that dephosphorylates Tie2 at EC junctions, thereby promoting vascular permeability. VE-PTP expression is increased in hypoxic endothelial cells and in neovascular endothelial cells in mice with ischemic retinopathy suggesting increased expression in tumor vessels.
76
(3) Local VEGF levels are increased leading to vascular endothelial cadherin (VE-cadherin) phosphorylation and internalization in an SRC kinase-dependent manner. Dexamethasone interferes with and prevents vascular leakage by a yet unknown mechanism that may include inhibition/downregulation of VEGF, whereas anti-VEGF treatment (BEV) neutralizes excess VEGF, and VE-PTP inhibition (AKB-9778) reduces the catalytic activity of VE-PTP to restore Tie2 activation to prevent vascular leakage. Illustration: Visual Science Communication. (b) Illustration of brain edema in GBM patients. Upon DEX treatment (left), the T2 weighted cMRI signal shows the anti-edema effect of DEX when compared with the DEX naive scenario in a GBM patient with perifocal edema (encircled in blue) in the left parietooccipital lobe. Upon BEV (anti-VEGF) therapy of another GBM patient (right), a weighted cMRI axial section shows decreased T2 signal enhancement demonstrating the anti-edema effect of BEV compared to progressive disease and perifocal edema (encircled in blue) of the corpus calosum before treatment.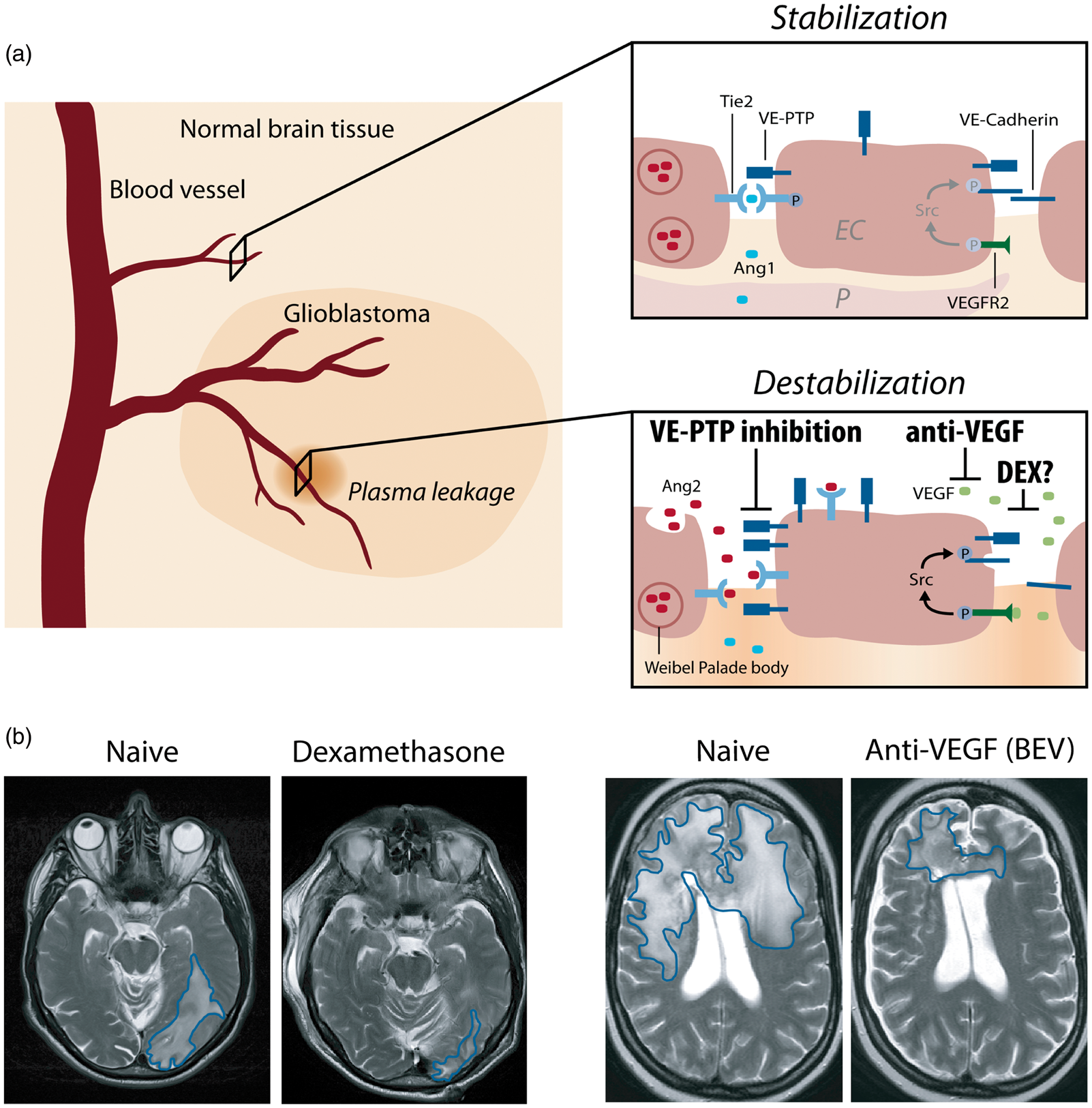
In Figure 2, a BBB-tightening effect of DEX is illustrated in transendothelial electrical resistance (TEER) measurements in vitro using primary porcine brain microvascular ECs as an in vitro BBB model similar to previous reports.9,78 DEX is able to rescue the VEGF-mediated endothelial permeability in this BBB model (Figure 2). However, the mechanisms responsible for reduced endothelial permeability by DEX are poorly understood and given the unfavorable immunosuppressive effects of DEX administration, there is a need for novel candidates to improve NVU function and BBB integrity in tumors. In this regard, targeting the Tie2 pathway via VE-PTP inhibition represents an attractive approach. Underscoring this possibility, a potent VE-PTP inhibitor is able to reduce endothelial permeability in the in vitro BBB model and is able to rescue VEGF-mediated permeability to a level comparable to DEX (Figure 2).
Rescue of VEGF-mediated permeability by activating Tie2 pathway comparable to dexamethasone in porcine brain endothelial cells in vitro. (a) Primary porcine brain endothelial cells isolated and cultured as described previously
71
were subjected to TEER measurements in cellZscope device (Nanoanalytics).
80
Expression of EC marker (VE-Cadherin/green, Claudin-5/red, DAPI/blue) indicate purity. VEGF (10 ng/ml), DEX (100 nM) and AKB 9785 (1 µm) treatments were performed in basal serum-free MCDB-131 medium upon reaching a plateau in TEER indicating confluency and barrier maturation. (b) Dexamethasone treatment resulted in high TEER values almost three-fold compared to controls and furthermore was able to rescue VEGF-mediated BBB breakdown. (c) AKB 9785 treatment was also able to tighten the BBB to similar levels as DEX and rescued VEGF-mediated permeability in this model.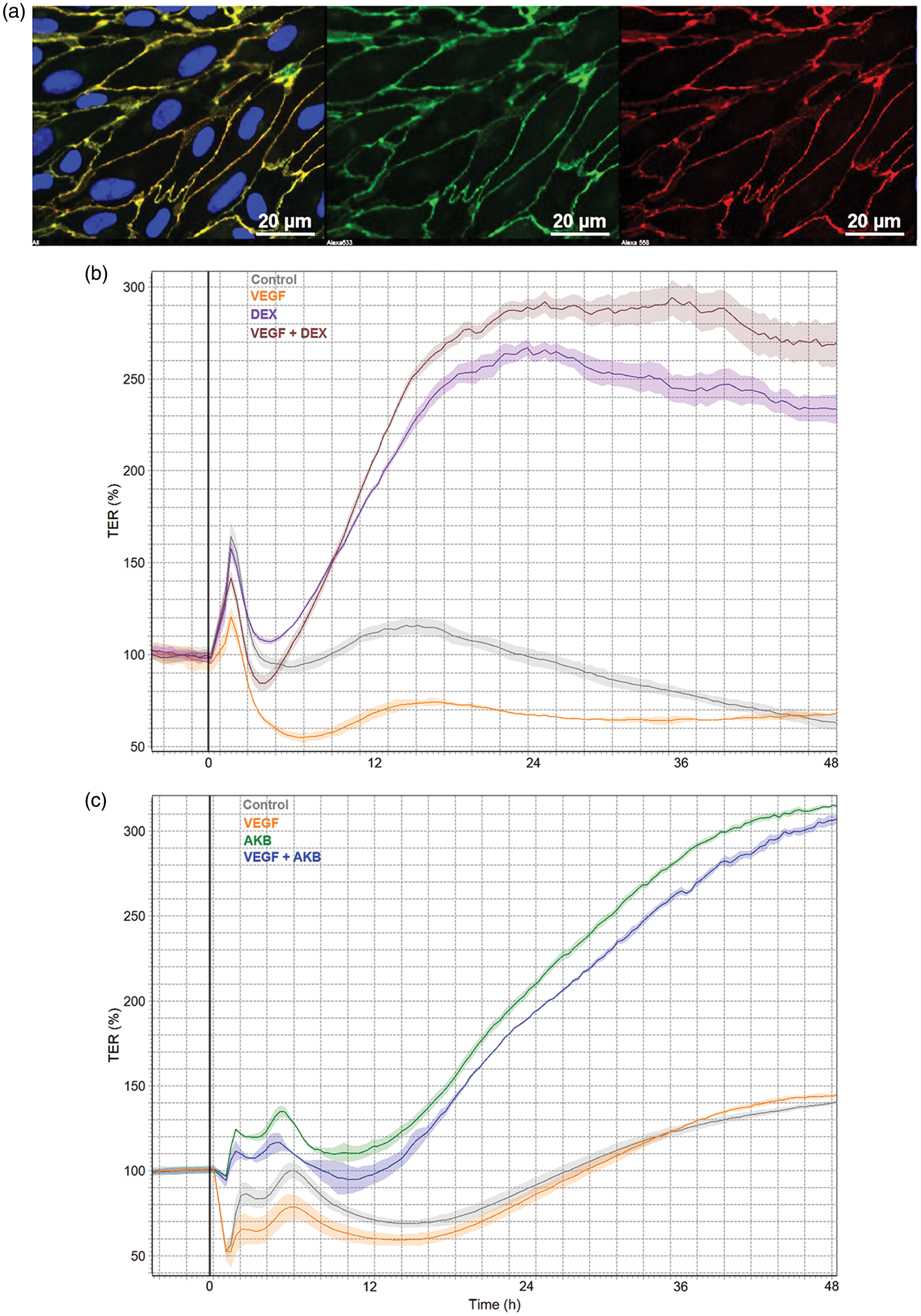
Conclusion
In conclusion, DEX, although highly effective in edema resolution in GBM, has a profound profile of adverse effects including T-cell-mediated immunosuppression. DEX application may even be related to reduced OS, although the mechanism of action remains to be elucidated. Increasing evidence supports the notion that DEX-sparing therapies might be warranted, particularly in light of upcoming immunotherapy trials (NCT02287428; J Clin Oncol 36, 2018, suppl; abstr 2020). Of note, DEX has recently been shown to negatively impact the clinical outcome after PD-1 checkpoint blockade in patients with lung cancer, 79 whereas GBM patients requiring DEX exceeding physiological replacement dosing have been excluded from immunotherapy trials (NCT02311920). In patients suffering from macula edema, VEGF blockade by BEV, ranibizumab or other inhibitors of VEGF signaling has been highly successful. 13 However, although effective in vasogenic edema management, anti-VEGF therapeutics have their limitations and contraindications in GBM patients. The successful inhibition of vascular leakage by inhibiting VE-PTP function in mouse models of macula edema, sepsis, stroke, inflammation and cancer12,14,71,72,74,75 resulted in promising clinical trials for ocular disease (currently Phase IIb, NCT03197870). In light of these recent developments, clinical trials evaluating DEX-sparing substances such as VEGF or VE-PTP inhibitors are highly warranted, in particular in GBM patients receiving immunotherapy.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: DD is a recipient of an Else-Kröner Foundation Research College fellowship. KHP and YR are supported by the Clinical Translation Platform of the Frankfurt Cancer Institute and the German Consortium for Translational Cancer Research (DKTK), partner site Frankfurt/Mainz, Germany. KD, YR and KHP are supported by the LOEWE Center for Personalized Translational Epilepsy Research (CePTER).
Acknowledgements
DD is a recipient of an Else-Kröner Foundation Research College fellowship. KHP and YR are supported by the Clinical Translation Platform of the Frankfurt Cancer Institute and the German Consortium for Translational Cancer Research (DKTK), partner site Frankfurt/Mainz, Germany. KD, YR and KHP are supported by the LOEWE Center for Personalized Translational Epilepsy Research (CePTER).
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: KGP is an employee of Aerpio Pharmaceuticals.