
Abstract
Previous data suggest a possible link between multidrug resistance-associated protein 1 (ABCC1) and brain clearance of beta-amyloid (Aβ). We used PET with 6-bromo-7-[11C]methylpurine ([11C]BMP) to measure cerebral ABCC1 transport activity in a beta-amyloidosis mouse model (APP/PS1-21) and in wild-type mice aged 50 and 170 days, without and with pretreatment with the ABCC1 inhibitor MK571. One hundred seventy days-old-animals additionally underwent [11C]PiB PET scans to measure Aβ load. While baseline [11C]BMP PET scans detected no differences in the elimination slope of radioactivity washout from the brain (kelim) between APP/PS1-21 and wild-type mice of both age groups, PET scans after MK571 pretreatment revealed significantly higher kelim values in APP/PS1-21 mice than in wild-type mice aged 170 days, suggesting increased ABCC1 activity. The observed increase in kelim occurred across all investigated brain regions and was independent of the presence of Aβ plaques measured with [11C]PiB. Western blot analysis revealed a trend towards increased whole brain ABCC1 levels in 170 days-old-APP/PS1-21 mice versus wild-type mice and a significant positive correlation between ABCC1 levels and kelim. Our data point to an upregulation of ABCC1 in APP/PS1-21 mice, which may be related to an induction of ABCC1 in astrocytes as a protective mechanism against oxidative stress.
Keywords
Introduction
Alzheimer's disease (AD) is by far the most prevalent type of dementia. AD is characterized by pathohistological hallmarks including the accumulation of beta-amyloid (Aβ) plaques and neurofibrillary tangles consisting of hyperphosphorylated tau protein in the brain as well as gliosis, which is marked by increases in activated microglia and reactive astrocytes near the sites of Aβ plaques. 1 In addition, oxidative stress (OS) plays an important role in the pathogenesis of AD and has been linked to decreased levels of glutathione (GSH), which is important for the detoxification of reactive oxygen species in the brain.2,3
The Aβ hypothesis points to the accumulation of Aβ peptides as the central event triggering neuronal degeneration. 4 It has been suggested that Aβ accumulation is caused by reduced Aβ clearance rather than increased Aβ production.4–6 Aβ clearance from brain is believed to occur via multiple pathways, i.e. active transport across the blood–brain barrier (BBB) into blood, enzymatic degradation in various cell types (i.e. endothelial cells, pericytes, astrocytes, neurons and microglia) and perivascular drainage of the extracellular fluid.7,8 Clearance across the BBB accounts for up to 70% of removal of Aβ from brain. Due to the fact that Aβ is a peptide which cannot cross the BBB by passive diffusion, its clearance across the BBB has to be facilitated involving receptors or transporters. There is evidence that the adenosine triphosphate-binding cassette (ABC) transporter P-glycoprotein (ABCB1) is involved in Aβ transport across the luminal membrane of brain capillary endothelial cells.9–12 Several lines of evidence suggested that ABCB1 expression and transport activity are impaired in AD, which may contribute to reduced Aβ clearance across the BBB.11,13–16
Multidrug resistance-associated protein 1 (ABCC1) is another member of the ABC transporter family which is expressed in the brain. ABCC1 is expressed in astrocytes, microglia, pericytes, neurons and in the basolateral membrane of choroid plexus cells.17–19 However, its localization at the BBB is controversial.20–23 This transporter plays an important role in protection against OS as well as in the cellular defense against xenobiotics and endogenous toxic metabolites. 24 It has been reported that the genetic knockout of Abcc1 in an AD mouse model of cerebral amyloidosis (APP/PS1-21) leads to 14-fold and 12-fold greater guanidine-soluble Aβ40 and Aβ42 levels, respectively, as compared with APP/PS1-21 control mice. 25 This suggests an inverse correlation of cerebral ABCC1 transport activity and the rate of Aβ accumulation with a possible link of ABCC1 actively exporting Aβ. 25 In line with this hypothesis, the treatment of AD mice with drugs which can activate ABCC1 in vitro was shown to significantly reduce cerebral Aβ load, especially the soluble Aβ fraction.25,26 Based on these results, these authors suggested that ABCC1 may play, besides ABCB1, a crucial role in mediating the clearance of Aβ from the brain and that cerebral ABCC1 transport activity may be impaired during the progression of AD.
Positron emission tomography (PET) with 6-bromo-7-[11C]methylpurine ([11C]BMP) has been proposed to visualize ABCC1 transport activity in the brain and lungs of mice.27–29 It is believed that this radiotracer enters cells as a prodrug by passive diffusion. Intracellularly, it is converted by glutathione-S-transferases into its GSH conjugate S-(6-(7-[11C]methylpurinyl))glutathione, which is exported from cells by ABCC1. 29
Aim of this study was to test if cerebral ABCC1 transport activity differs between APP/PS1-21 and wild-type control mice. We used [11C]BMP PET to measure ABCC1 transport activity and [11C]Pittsburgh compound B ([11C]PiB) to measure Aβ load at two different ages (50 and 170 days).
Material and methods
Chemicals
Unless otherwise stated, all chemicals were purchased from Sigma-Aldrich (Schnelldorf, Germany) or Merck (Darmstadt, Germany). The ABCC1 transporter inhibitor MK571 was obtained from Santa Cruz Biotechnology (Dallas, TX, USA). Before each administration, MK571 was dissolved in phosphate-buffered saline (PBS) at a concentration of 15 mg/mL and injected intraperitoneally (i.p.) into mice at a volume of 20 mL/kg body weight (300 mg/kg).
Radiotracer synthesis
[11C]BMP and [11C]PiB were synthesized as described before.27,30 For intravenous (i.v.) injection into animals, [11C]BMP was formulated in 0.9% (w/v) aqueous saline and [11C]PiB in 0.9% aqueous saline containing 0.1% (v/v) polysorbate 80. Radiochemical purity of both radiotracers was > 98% and molar activity at the time of injection into animals was 115 ± 118 GBq/µmol for [11C]BMP and 306 ± 187 GBq/µmol for [11C]PiB.
Animals
Overview of examined animal groups and numbers. a
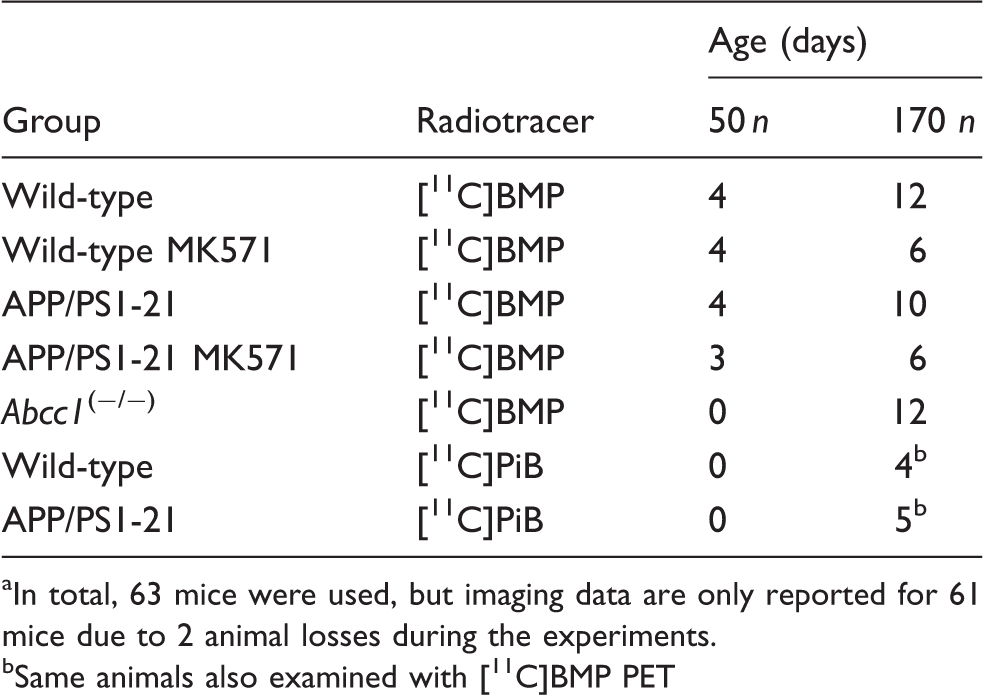
In total, 63 mice were used, but imaging data are only reported for 61 mice due to 2 animal losses during the experiments.
Same animals also examined with [11C]BMP PET
Experimental design
An overview of animal groups examined in this study is given in Table 1. Groups of wild-type, APP/PS1-21 and Abcc1(−/−) mice underwent a PET scan with [11C]BMP at 30 min after i.p. pretreatment with PBS (= vehicle used for administration of MK571). Selected groups of wild-type and APP/PS1-21 mice underwent a second PET scan with [11C]PiB at seven days after the [11C]BMP PET scan. Further groups of wild-type and APP/PS1-21 mice were scanned with [11C]BMP at 30 min after i.p. pretreatment with MK571 (300 mg/kg). The dosage of MK571 was selected based on previous work by Okamura et al. and by us.27,28
PET imaging
Imaging experiments were performed under isoflurane anesthesia (2.5–3.5% in air). Animals were warmed throughout the experiment and body temperature and respiratory rate were constantly monitored. Mice were placed in a custom-made imaging chamber and a lateral tail vein was cannulated for i.v. administration. A microPET Focus220 scanner (Siemens Medical Solutions, Knoxville, TN, USA) was used for PET imaging. Mice were i.p. injected under anesthesia at 30 min before start of the PET scan either with vehicle solution (PBS), or with MK571 (300 mg/kg). Subsequently, [11C]BMP (32 ± 8 MBq in a volume of 0.1 mL, corresponding to 0.64 ± 0.47 nmol of unlabeled compound) was administered as an i.v. bolus and a 90-min dynamic PET scan was initiated at the start of radiotracer injection. List-mode data were acquired with a timing window of 6 ns and an energy window of 250–750 keV. After seven days, anatomical magnetic resonance imaging (MRI) was performed on a 1 T benchtop MRI (ICON, Bruker BioSpin GmbH, Ettlingen, Germany) in selected mouse groups. Following the MRI, these mouse groups underwent an additional 90-min dynamic PET scan with [11C]PiB (28 ± 5 MBq in a volume of 0.1 mL corresponding to 0.18 ± 0.18 nmol of unlabeled compound) using identical acquisition parameters as for the [11C]BMP scans. At the end of the PET scan, a blood sample was collected from the retro-bulbar plexus and animals were killed by cervical dislocation while under deep anesthesia. Blood was centrifuged (13,000 × g, 4℃, 4 min) to obtain plasma. Whole brains were removed, divided into the hemispheres and shock frozen in liquid nitrogen. Aliquots of plasma were stored at −80℃ until analysis of MK571 concentrations by HPLC as described previously. 27
PET data analysis
The PET data were sorted into 25 time frames with a duration increasing from 5 s to 20 min to a total scan length of 90 min. PET images were reconstructed using Fourier re-binning of the three-dimensional sinograms followed by a two-dimensional filtered back-projection with a ramp filter giving a voxel size of 0.4 × 0.4 × 0.796 mm3. Using PMOD software (version 3.6, PMOD Technologies Ltd, Zurich, Switzerland), whole brain, cortex, hippocampus and cerebellum were outlined on the PET images using the Mirrione Mouse Atlas and guided by representative MRIs obtained in a few animals. Regions of interest were manually adjusted if necessary without modification in sizes to derive time-activity curves (TACs) expressed in units of standardized uptake value (SUV) ((radioactivity per g/injected radioactivity) × body weight).
From the log-transformed TACs in whole brain, cortex, hippocampus and cerebellum, the elimination slope of radioactivity washout (kelim, h−1) was determined by linear regression analysis of data from 17.5 to 80 min after radiotracer injection. 27 It should be noted that kelim is not identical with the efflux rate constant from brain into plasma k2 determined with compartmental modeling, which would require the consideration of an arterial plasma input function. For [11C]PiB scans, the ratio of radioactivity uptake in the hippocampus or cortex to that in the cerebellum as an Aβ-devoid reference region 13 was calculated at 25 min after radiotracer injection as an outcome parameter of Aβ load. 32
Assessment of conversion into glutathione conjugate
In separate groups of wild type (n = 3) and APP/PS1-21 mice (n = 3) aged 170 days, the percentage of unchanged [11C]BMP was assessed in plasma and brain at 15 min after radiotracer injection using radio-thin-layer chromatography (TLC) as described previously. 27 The employed TLC system comprised silica gel 60F 254 nm TLC plates which were developed in ethyl acetate/ethanol (9/1, v/v). On this TLC system, unchanged [11C]BMP had a retardation factor (Rf) of approximately 0.4, while the radiolabeled glutathione conjugate remained on the start.
Western blot
Western blot analysis was performed on brains of MK571-pretreated animals collected at the end of the [ 11 C]BMP PET scans. Mice were killed by cervical dislocation while under deep anesthesia. After intracardial perfusion with PBS (10 mL), one hemisphere of each brain was snap-frozen in liquid nitrogen within 3 min after death and stored at −80℃ until use. The deep-frozen brains were soaked in RNAlater® for 1 h on ice. Hemispheres were homogenized using ceramic beads and a SpeedMill PLUS (analyticjena AG, Germany). Homogenized brain samples (20–30 mg) were thoroughly mixed with Lysis buffer 1 (100 mM Tris pH 7.4, 150 mM NaCl, 0.1% TritonX-100, DNAse, cOmplete™ mini protease inhibitor cocktail (Roche)). After 30 min incubation on ice, samples were centrifuged (13,000 r/min, 4℃, 90 min) and the pellets dissolved in membrane protein-lysis buffer. After 1–2 h incubation at 50℃, samples were centrifuged for clarification and protein concentration of the supernatant was determined using the BCA assay (Pierce™, Thermo Scientific, USA). Brain samples were adjusted to a protein concentration of 3 µg/µL using 4 × sample buffer and water. Brain samples (20 µL) were subjected to gel electrophoresis (7.5% TGX gel matrix, Bio-Rad, Germany). Protein transfer was performed onto PVDF membranes using a TransBlot Turbo (Bio-Rad). Membranes were blocked for 1 h at room temperature using 1.5% non-fat dry milk in PBS containing 0.05% Tween 20 followed by parallel anti-ABCC1 antibody ([MRPr1], abcam ab3368, 1:200) and anti-Actin antibody (AC-74, Sigma Aldrich, 1:5000) incubation at 4℃ over-night. HRP conjugated anti-rat antibody (Jackson Laboratories, 1:5000) was used first for detection of ABCC1 signals (Clarity ECL solution, Bio-Rad) which was followed by anti-mouse antibody (NordicBiosite, 1:10,000) after washing to detect actin signals as endogenous control. Band intensities were analyzed using Image Studio Lite Vers. 3.1 (LI-COR, Germany).
Statistical analysis
Statistical testing was performed using Prism 7.0 software (GraphPad, La Jolla, CA, USA). Differences between multiple groups were analyzed by one-way ANOVA followed by a Tukey’s multiple comparison test and differences between two groups were analyzed by a two-sided unpaired t-test. Pearson correlation coefficient r was calculated to assess correlations. The level of statistical significance was set to a p value of less than 0.05. All values are given as mean ± standard deviation (SD).
Results
We used [11C]BMP to measure cerebral ABCC1 transport activity in wild-type and APP/PS1-21 mice aged 50 or 170 days after pretreatment with vehicle or the non-subtype-selective ABCC transporter inhibitor MK571. As a control group, Abcc1(−/−) mice were studied. In addition, Aβ load in mouse brains was measured with [11C]PiB. In Table 1, an overview of all examined mouse groups and corresponding animal numbers is given. As an outcome parameter of cerebral ABCC1 transport activity, we determined the elimination slope for radioactivity washout from the brain from 17.5 to 80 min after radiotracer injection (kelim). Radio-TLC analysis revealed almost complete conversion (>95%) of [11C]BMP into polar radiolabeled species in plasma and brain samples of wild-type and APP/PS1-21 mice collected at 15 min after radiotracer injection.
In Figure 1, [11C]BMP PET summation images (17.5–80 min) of the head region of wild-type and APP/PS1-21 mice aged 50 and 170 days pretreated with vehicle or MK571 are shown as well as PET images of Abcc1(−/−) mice. We outlined hippocampus and cortex as brain regions with substantial Aβ deposition and cerebellum as a control region with minimal Aβ load in all study groups (Supplementary Figure 1). In Figure 2, the corresponding TACs for these regions are shown. In all three regions, Abcc1(−/−) mice showed a markedly reduced washout of radioactivity as compared with the other groups (Figure 2(a) to (c)). MK571 pretreatment reduced the washout of radioactivity in all groups and brain regions as compared with the vehicle-treated groups (Figure 2(d) to (f)).
Representative sagittal PET summation images (17.5–80 min) of wild-type and APP/PS1-21 mice aged 50 and 170 days and Abcc1(−/−) mice pretreated with vehicle or MK571 (300 mg/kg, i.p.) at 30 min before the PET scan. Whole animal contour and whole brain region are marked with white broken lines. All images are scaled to the same intensity which is given in units of standardized uptake value (SUV). Time-activity curves (mean ± SD) in the cortex, hippocampus and cerebellum for vehicle (a–c) and MK571 pretreated (d–f) animals. Radioactivity is expressed as standardized uptake value (SUV). Please refer to Table 1 for number of animals in each group.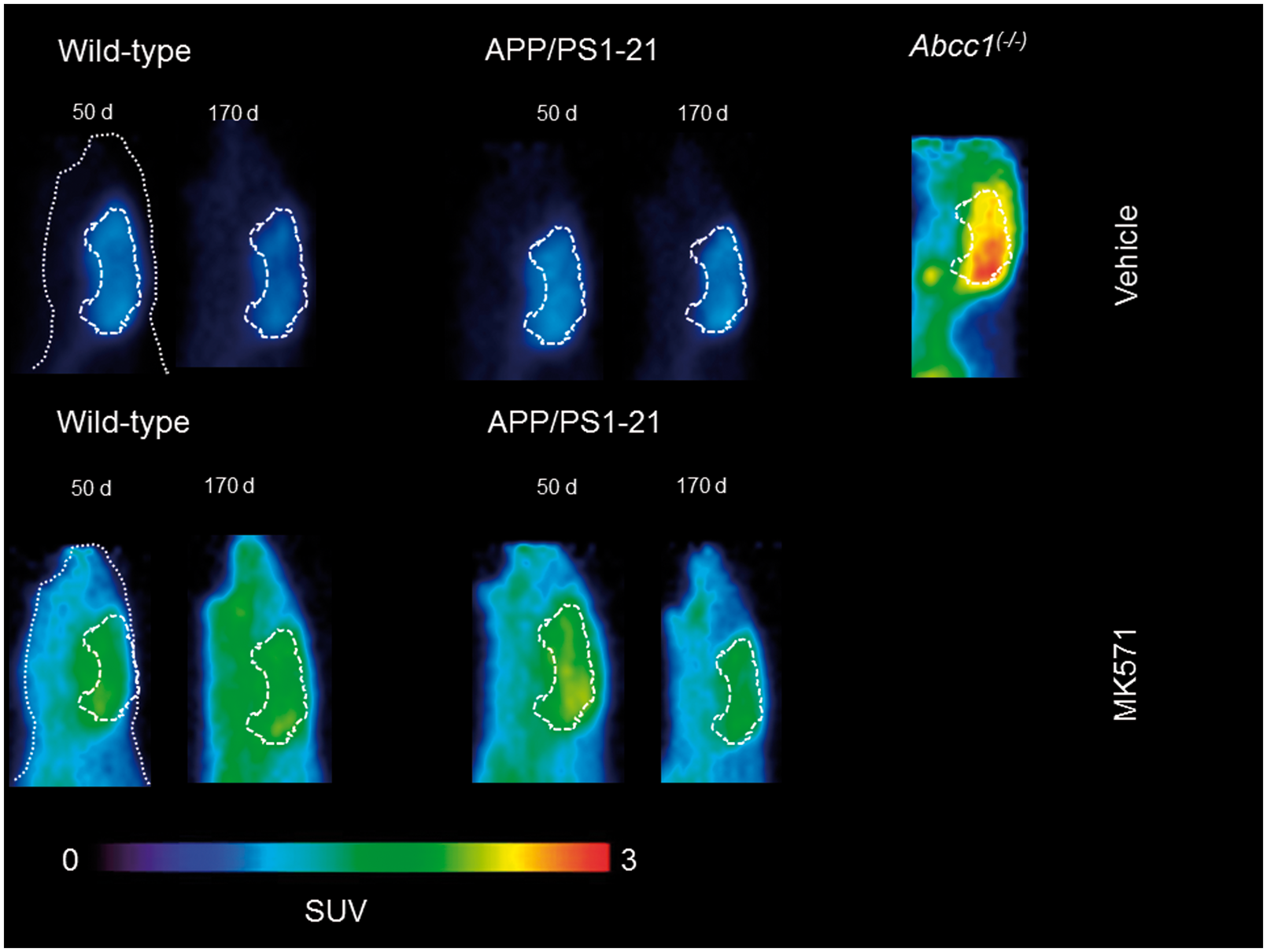
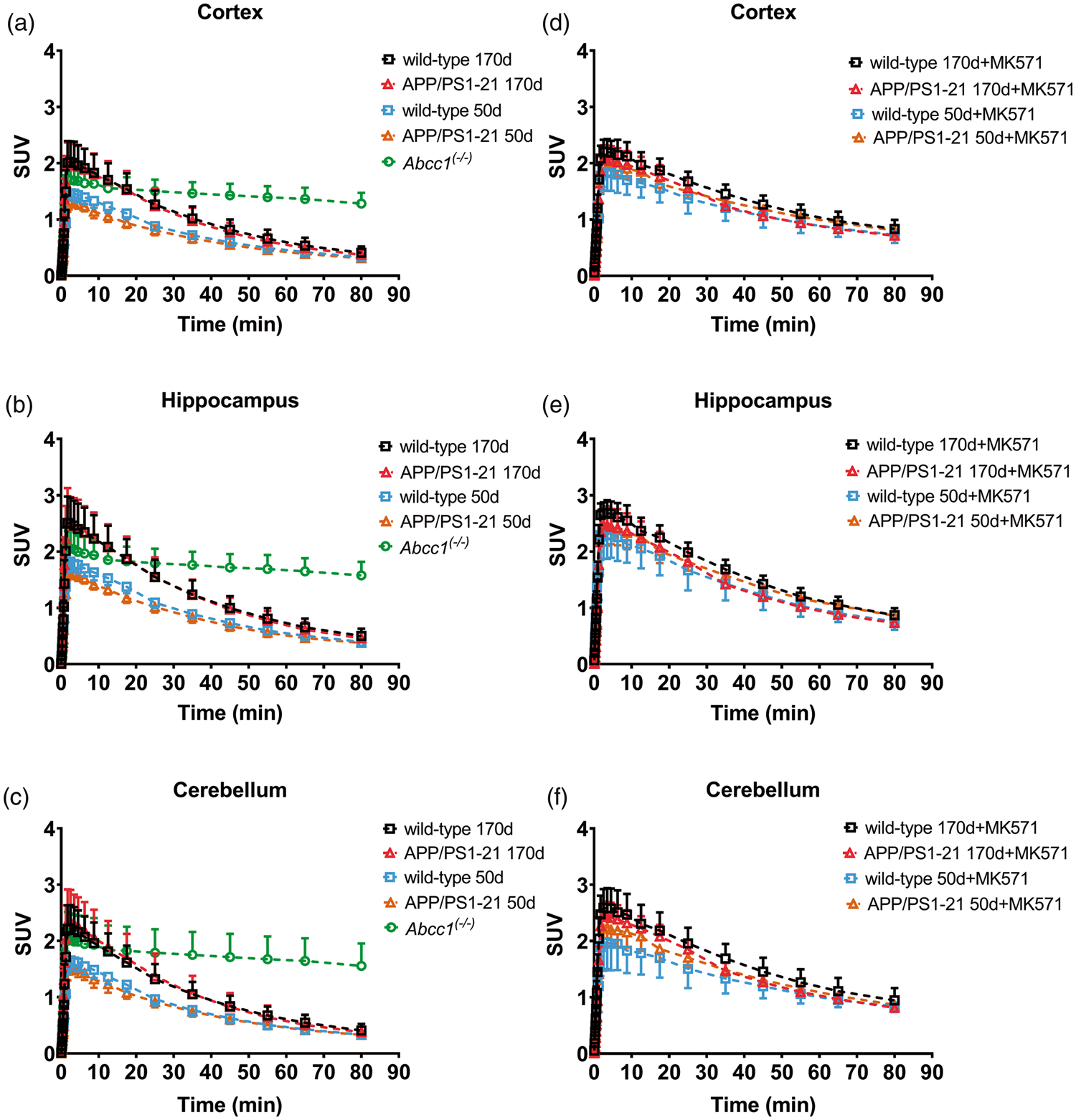
kelim values in different groups are shown in Figure 3. In the cortex (Figure 3(a)) and in the hippocampus (Figure 3(b)), kelim was significantly higher in vehicle pretreated wild-type and APP/PS1-21 mice aged 170 days than in the corresponding 50-days-old groups. In the cerebellum, significant differences in kelim values were only found between 170 - and 50-days-old vehicle pretreated APP/PS1-21 mice, but not between the wild-type groups (Figure 3(c)). After pretreatment with MK571, kelim was significantly decreased in all brain regions compared to the corresponding vehicle pretreated groups. Within the MK571 pretreated groups, we found in all brain regions of 170-days-old APP/PS1-21 mice significantly higher kelim values than in the corresponding wild-type group. Furthermore, in the cortex and the hippocampus of MK571 pretreated APP/PS1-21 mice, kelim was significantly higher in animals aged 170 days than in animals aged 50 days. We calculated the percentage of reduction in kelim after MK571 treatment relative to the mean of the corresponding vehicle group (Figure 3(d)). No differences in the percentage of kelim reduction were detected between wild-type and APP/PS1-21 mice in any brain region of the 50 days age group (data not shown). In the 170 days age groups, the percentage of kelim reduction was significantly higher in the cortex and hippocampus of wild-type mice (−45 ± 7% and −32 ± 5%, respectively) than in APP/PS1-21 mice (−36 ± 6% and −25 ± 2%, respectively), while no significant difference was observed in the cerebellum (Figure 3(d)). To exclude that the observed differences in the response to MK571 pretreatment were caused by differences in MK571 plasma concentrations, we determined MK571 concentrations in plasma samples collected at the end of the PET scan. MK571 plasma concentrations were not significantly different between 170-days-old wild-type mice and APP/PS1-21 mice (251 ± 63 µmol/L, n = 3 vs. 293 ± 41 µmol/L, n = 6).
kelim values (mean h−1 ± SD) of radioactivity for the cortex (a), hippocampus (b) and cerebellum (c) of wild-type, APP/PS1-21 and Abcc1(−/−) mice pretreated at 30 min before PET either with vehicle or with MK571 (300 mg/kg, i.p.) (ns, not significant, *p < 0.05, **p < 0.01, and ***p < 0.001, one-way ANOVA followed by Tukey’s multiple comparison test). The mean ± SD percentage reduction in kelim in the cortex, hippocampus and cerebellum of individual MK571 pretreated wild-type and APP/PS1-21 mice aged 170 days relative to the mean of the kelim of the corresponding vehicle group is shown in d (ns, not significant, *p < 0.05, **p < 0.01, two-sided unpaired t-test). Please refer to Table 1 for number of animals in each group.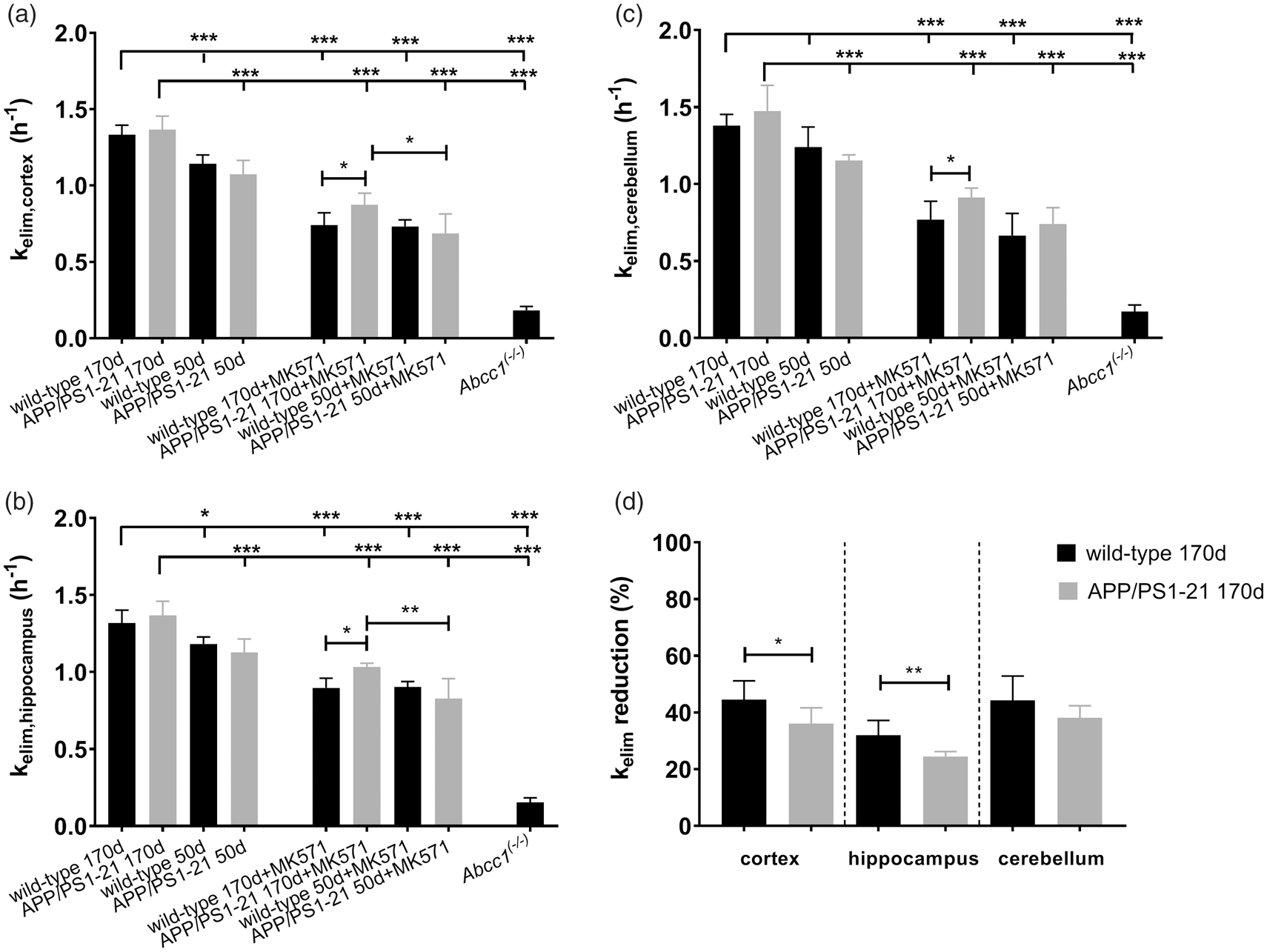
In MK571 pretreated APP/PS1-21 mice and wild-type mice aged 170 days, whole brain samples were collected after the PET scans and examined for ABCC1 levels by Western blot analysis (Figure 4(a)). There was a trend towards higher whole brain ABCC1 expression in APP/PS1-21 mice compared to wild-type mice, but statistical significance was not reached (Figure 4(b)). We found a significant positive correlation between kelim and ABCC1 levels in the whole brain (Figure 4(c)).
(a) Representative whole brain Western blot of ABCC1 in wild-type and APP/PS1-21 mice aged 170 days. (b) Analysis of the ABCC1 protein bands detected by the anti-ABCC1 antibody in whole brain samples of wild-type and APP/PS1-21 mice. Relative abundance of the 190 kDa ABCC1 bands was determined from the total surface area of the respective bands normalized to β-actin as loading control (wild-type n = 3; APP/PS1-21 n = 5; ns, not significant, two-sided unpaired t-test). (c) Correlation between whole brain kelim and the ABCC1 levels determined by Western blot analysis (r = Pearson correlation coefficient). Shown data are from three wild-type mice (open squares) and five APP/PS1-21 mice (filled triangles) after MK571 pretreatment.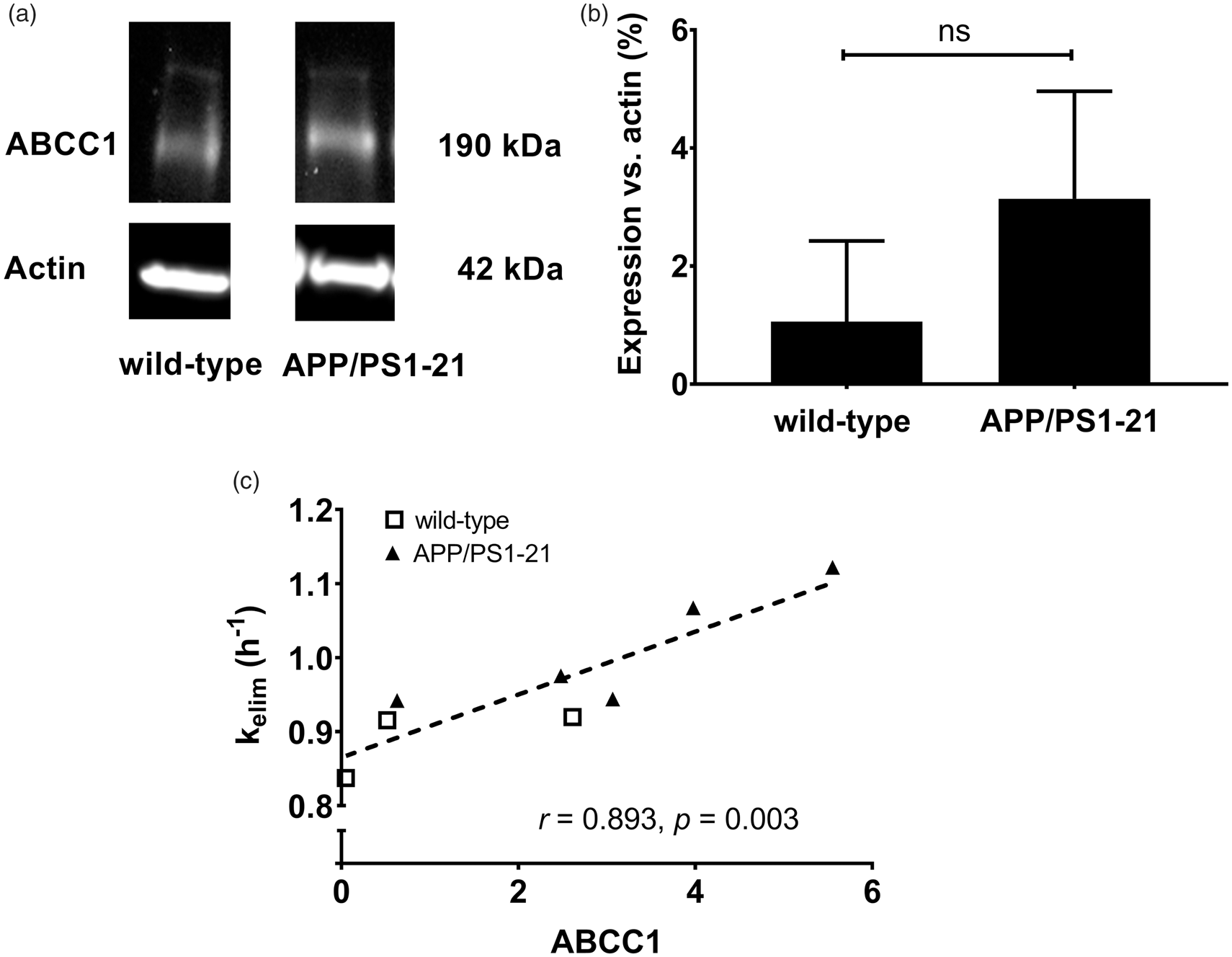
For the [11C]PiB scans, the cortex-to-cerebellum and the hippocampus-to-cerebellum ratios of radioactivity over time in 170-days-old APP/PS1-21 and wild-type mice are shown in Supplementary Figure 2, which revealed slightly increased ratios in APP/PS1-21 versus wild-type mice. At 25 min after radiotracer injection, the hippocampus-to-cerebellum radioactivity ratio (Figure 5(b)) was significantly higher in APP/PS1-21 versus wild-type mice, while there was only a trend towards higher cortex-to-cerebellum ratios in APP/PS1-21 mice (Figure 5(a)). We correlated regional outcome parameters from the [11C]BMP scans with those of the [11C]PiB scans in vehicle pretreated APP/PS1-21 mice aged 170 days (Supplementary Figure 3). No correlations were found between kelim,cortex of [11C]BMP and the cortex-to-cerebellum ratio of [11C]PiB or kelim,hippocampus of [11C]BMP and the hippocampus-to-cerebellum ratio of [11C]PiB.
Cortex-to-cerebellum (a) and hippocampus-to-cerebellum (b) radioactivity ratio at 25 min after injection of [11C]PiB into wild-type and APP/PS1-21 mice aged 170 days (ns, not significant, *p < 0.05, two-sided unpaired t-test). Please refer to Table 1 for number of animals in each group.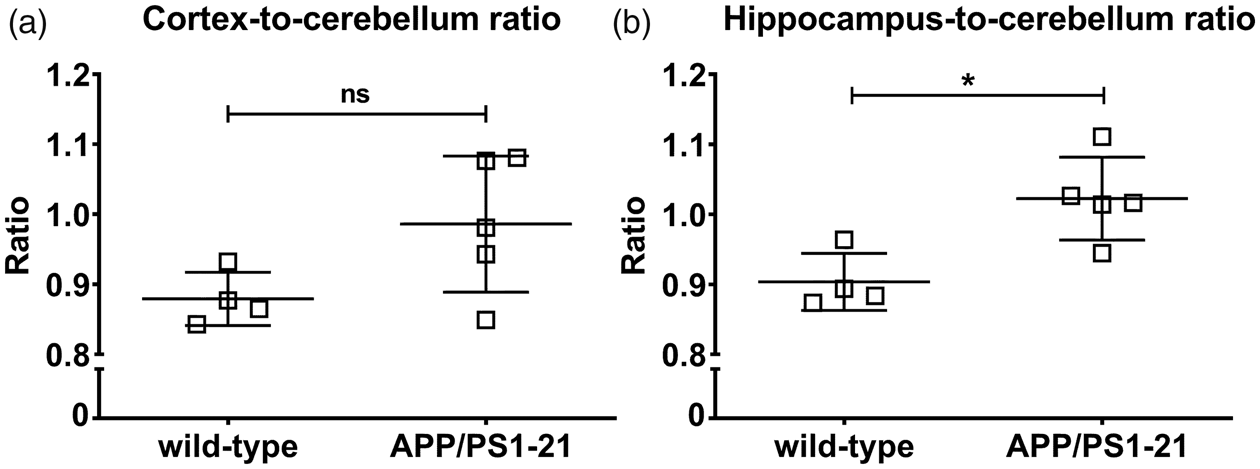
Discussion
In the present study, we used [11C]BMP, a validated ABCC1 PET probe,27,29,33,34 to assess cerebral ABCC1 transport activity in APP/PS1-21 mice 31 and in wild-type control mice at the age of 50 and 170 days, respectively. The age of 50 days corresponds to the onset of Aβ plaque deposition in this mouse model, while at the age of 170 days, substantial Aβ plaque deposition occurs, as shown in our previous study, in which we performed immunohistochemical Aβ staining of brain slices of APP/PS1-21 mice obtained from both age groups. 13 Moreover, we measured in the present study cerebral Aβ plaques with [11C]PiB, which has been shown to be suitable to visualize Aβ plaques in this mouse model. 35
[11C]BMP is rapidly (within 15 min after radiotracer injection) converted in the mouse brain into its GSH conjugate S-(6 -(7-[11C]methylpurinyl))glutathione, presumably by cytosolic glutathione-S-transferases within brain parenchymal cells (i.e. astrocytes, neurons).27,29,34 The GSH conjugate is assumed to be exported from parenchymal cells by ABCC1 followed by clearance across the BBB by other anionic transporters (i.e. organic anion transporter 3, SLC22A8 and multidrug resistance-associated protein 4, ABCC4). 33 Previous experiments showed that kelim of radioactivity from the brain following i.v. injection of [11C]BMP was almost reduced to zero in Abcc1(−/−) mice as compared with wild-type mice, which suggested that efflux transport by ABCC1 is a rate-limiting step in the elimination of [11C]BMP-derived radioactivity from the brain.27,29 However, direct intracerebral injection of S-(6 -(7-[11C]methylpurinyl))glutathione failed to reveal differences in kelim between wild-type and Abcc1(−/−) mice, indicating that ABCC1 does not contribute to clearance of radioactivity across the BBB. 33 In mice knocked out for the Abcc4 or Slc22a8 genes, a significant decrease in kelim was observed, which indicated that SLC22A8 and ABCC4 mediated transport of [11C]BMP-derived radioactivity from brain into blood. 33 Previous work has shown that kelim can be used as a quantitative parameter of efflux transport activity in the mouse brain without the need to consider an arterial blood input function, because the radiolabeled glutathione conjugate cannot enter the brain from the blood side. 33
We have previously performed [11C]BMP PET scans in heterozygous Abcc1 knockout mice (Abcc1(+/−)), which are assumed to have a reduction in cerebral ABCC1 levels as compared with wild-type mice. 27 These experiments revealed only a moderate reduction in kelim of [11C]BMP-derived radioactivity in the brain relative to wild-type mice. This may be attributed to the high transport capacity of ABCC1 in the mouse brain, which may compensate a reduction in ABCC1 expression. This suggested that [11C]BMP has only a limited sensitivity to detect moderate changes in cerebral ABCC1 expression/activity. In analogy to previous experiments with the ABCB1 probe substrate (R)-[11C]verapamil, 13 we therefore followed in the present study a strategy to partially inhibit cerebral ABCC1 to decrease the transport capacity of ABCC1 and thereby increase the sensitivity of [11C]BMP to detect small alterations in cerebral ABCC1 transport activity. As an ABCC1 inhibitor, we employed MK571, which is commonly used as a prototypical transporter inhibitor for in vitro experiments.36–38 We have shown before that pretreatment of mice with MK571 (300 mg/kg, i.p.) significantly decreased kelim of [11C]BMP-derived radioactivity in the brain, 27 suggesting that MK571 can cross the mouse BBB and reach sufficiently high concentrations in brain parenchyma to partially inhibit ABCC1 expressed in membranes of brain parenchymal cells.
In accordance with the results by Okamura et al., 29 we found in the present study a pronounced reduction of kelim in Abcc1(−/−) mice as compared with wild-type mice (Figure 3), which supported that ABCC1 transport activity plays a key role in brain clearance of [11C]BMP-derived radioactivity. Moreover, we found a significant positive correlation between kelim values from whole brain after MK571 treatment and whole brain ABCC1 levels measured with Western blot (Figure 4(c)), which further supported that [11C]BMP PET can be used to measure ABCC1 transport activity in the mouse brain.
In baseline scans without MK571 administration, we detected no significant differences in kelim values between APP/PS1-21 mice and control mice of both age groups (Figure 3). However, in both mouse strains, a significant age-associated increase in kelim values was observed. In MK571-pretreated animals, significantly higher kelim values were observed in APP/PS1-21 mice compared to wild-type mice at the age of 170 days, both in the Aβ-rich brain regions cortex and hippocampus as well as in the cerebellum, which is almost devoid of Aβ plaques (Figure 3(a) to (c)). In accordance with the PET results, Western blot analysis revealed a trend towards higher whole brain ABCC1 levels in APP/PS1-21 mice compared to wild-type mice at the age of 170 days (Figure 4(a) and (b)). In line with their increased cerebral ABCC1 levels, there was an attenuated response to ABCC1 inhibition with MK571, in terms of a kelim reduction, in APP/PS1-21 mice (Figure 3(d)). Our results suggested that [11C]BMP PET without administration of MK571 shows limited sensitivity to detect moderate alterations in ABCC1 transport activity in the mouse brain. Comparable results have been obtained with (R)-[11C]verapamil, which could only detect significant differences in ABCB1 transport activity in the brains of APP/PS1-21 mice versus wild-type mice after partial ABCB1 inhibition with tariquidar. 13 In an attempt to correlate regional ABCC1 transport activity with regional Aβ load, we performed [11C]PiB PET scans in the same animals that had undergone [11C]BMP PET scans (without MK571 pretreatment). In the [11C]PiB scans, hippocampus-to-cerebellum radioactivity ratios were significantly higher in 170-days-old APP/PS1-21 mice as compared with control mice, confirming Aβ deposition in APP/PS1-21 mice at the studied age. 13 The differences in [11C]PiB binding between APP/PS1-21 and wild-type mice were of similar magnitude as reported in literature for the same mouse model. 35 No correlation was found in our study between Aβ load and ABCC1 transport activity in the hippocampus and cortex (Supplementary Figure 3). This could be due to the limited sensitivity of baseline [11C]BMP PET scans to measure small variations in cerebral ABCC1 transport activity, but may also suggest that the increase in ABCC1 transport activity observed in APP/PS1-21 mice is independent of the presence of Aβ plaques.
Our findings appear to be in good agreement with the results of Ye et al., 39 who reported a transient, up to 2-fold increase in cortical ABCC1 levels in 5 × FAD mice between an age of 1.5 and 3.5 months. This increase in cortical ABCC1 levels was attributed to an induction of the expression of the GSH transporter ABCC1 in astrocytes by monomeric Aβ as a protective mechanism against OS. At later time points (≥ 7 months), ABCC1 levels were decreased to sub-control levels in the 5 × FAD mouse model, presumably due to an inhibition of ABCC1 induction in astrocytes by aggregated Aβ. The 5 × FAD model represents a very fast amyloidosis model. Therefore, the age range of elevated ABCC1 levels observed in 5 × FAD mice can most likely not directly be compared with our APP/PS1-21 model. Our results are not in line with the hypothesis of Krohn et al. 25 that diminished ABCC1 transport activity may contribute to reduced Aβ clearance from the brain. Our data rather support that ABCC1 expression/activity was upregulated in our AD mouse model at the investigated age of 170 days. It should be noted, however, that Krohn et al. 25 used APP/PS1 mice with a different genetic background (FVB/N) and neither measured ABCC1 expression nor ABCC1 transport activity in FVB/N and APP/PS1-FVB/N mice. The genetic background is known to modulate the amount of Aβ in the mouse brain, which also depends on the mitochondrial genotype and ATP levels.40–42
The spatial resolution of PET is insufficient to distinguish different cell types which contributed to efflux of [11C]BMP-derived radioactivity from the mouse brain. Hence, it is not known in which cell type(s) the observed upregulation of ABCC1 transport activity occurred. While the limited spatial resolution of small-animal PET did not allow us to investigate the ventricular system and the choroid plexus in the mouse brain, the global increase in kelim values across all investigated brain regions argued against a selective upregulation of ABCC1 in the basolateral membrane of choroid plexus epithelial cells, where ABCC1 may promote transport of Aβ from cerebrospinal fluid into blood. It is tempting to speculate that astrocytes were involved in the observed kelim increases as these cells strongly express ABCC1, contain glutathione-S-transferases and are uniformly distributed throughout the brain.19,43–45 ABCC1 mediates export of the antioxidant GSH and its oxidized form glutathione disulfide (GSSG) from astrocytes, thereby maintaining cellular redox homeostasis. 24 The induction of OS in astrocyte cultures was shown to enhance efflux of GSH and GSSG, which was significantly reduced by the addition of MK571 supporting active transport by ABCC1.36,37 It has been suggested that Aβ could target astrocytes and that increased GSH and GSSG release from astrocytes could protect neurons against OS, by supplying them with precursors for GSH synthesis.39,46 This increased GSH efflux from astrocytes could contribute to a decrease in GSH levels in the AD brain.47,48 Astrocytes, which are surrounding Aβ plaques, become reactive in AD, contribute to the local inflammatory response and produce reactive oxygen species. 49 Reactive astrocytes can take up and degrade Aβ, but the exact mechanisms of this process are not fully understood.49,50 Provided that ABCC1 can transport Aβ, it can be speculated that increased expression of ABCC1 in astrocytes may decrease their intracellular Aβ content and thereby contribute to altered Aβ clearance. Astrocyte reactivity is an early feature of AD, potentially providing a promising target for early diagnosis and treatment. 49 As there is a lack of PET tracers to specifically image astrocyte function, 49 ABCC1 activity measured with [11C]BMP PET merits further investigation as a possible biomarker of astrocyte activation.
Conclusion
In this work, we showed that [11C]BMP PET in combination with the ABCC1 inhibitor MK571 can measure cerebral ABCC1 transport activity in the mouse brain. We found an age-associated increase in cerebral ABCC1 transport activity in both wild-type and APP/PS1-21 mice. Moreover, ABCC1 transport activity was higher in APP/PS1-21 mice relative to age-matched control mice, which appeared to be independent of Aβ plaque load measured in the same animals with [11C]PiB PET. The observed increase in ABCC1 transport activity may be mainly caused by an induction of ABCC1 expression in astrocytes to promote GSH release as a protective mechanism against OS, suggesting that [11C]BMP may be used in future studies as a marker of astrocyte function.
Supplemental Material
Supplemental material for Measurement of cerebral ABCC1 transport activity in wild-type and APP/PS1-21 mice with positron emission tomography
Supplemental Material for Measurement of cerebral ABCC1 transport activity in wild-type and APP/PS1-21 mice with positron emission tomography by Viktoria Zoufal, Severin Mairinger, Markus Krohn, Thomas Wanek, Thomas Filip, Michael Sauberer, Johann Stanek, Claudia Kuntner, Jens Pahnke and Oliver Langer in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Austrian Science Fund (FWF) [I 1609-B24, to O. Langer], the Deutsche Forschungsgemeinschaft (DFG) [DFG PA930/9-1, to J. Pahnke] and the Lower Austria Corporation for Research and Education (NFB) [LS14-008, to T. Wanek]. The work of J. Pahnke was further supported by the following grants: DFG [DFG PA930/12], Wirtschaftsministerium Sachen-Anhalt [EFRE, ZS/2016/05/78617], the Leibniz Association [Leibniz-Wettbewerb SAW-2015-IPB-2], the Latvian Council of Science/Latvia [lzp-2018/1-0275], Nasjonalforeningen [16154], HelseSØ/Norway [2016062, 2019054, 2019055], Norsk forskningsrådet/Norway [251290, 260786], Horizon 2020/European Union [643417, PROP-AD]. PROP-AD is an EU Joint program – Neurodegenerative Disease Research (JPND) project. The project is supported through the following funding organizations under the aegis of JPND – ![]() : AKA #301228 – Finland, BMBF #01ED1605 – Germany, CSO-MOH #30000-12631 – Israel, NFR #260786 – Norway, SRC #2015-06795 – Sweden. This project has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement #643417 (JPco-fuND).
: AKA #301228 – Finland, BMBF #01ED1605 – Germany, CSO-MOH #30000-12631 – Israel, NFR #260786 – Norway, SRC #2015-06795 – Sweden. This project has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement #643417 (JPco-fuND).
Acknowledgements
The authors wish to thank Mathilde Löbsch for help in conducting the PET experiments and Alexander Traxl for help with PET data analysis. Dr. Toshimitsu Okamura (National Institute of Radiological Sciences, Chiba, Japan) is gratefully acknowledged for providing the reference standards for the radiosynthesis of 6-bromo-7-[11C]methylpurine.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
Oliver Langer, Markus Krohn, Thomas Wanek and Jens Pahnke designed the research; Viktoria Zoufal, Severin Mairinger, Markus Krohn, Thomas Filip, Michael Sauberer, Johann Stanek and Claudia Kuntner performed the research; Viktoria Zoufal, Markus Krohn and Thomas Wanek analyzed the data; Viktoria Zoufal and Oliver Langer wrote the paper.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.