
Abstract
Stroke is among the top 10 causes of death in children. The developmental stage of the brain is central to stroke pathophysiology. The incidence of childhood arterial ischemic stroke (CAIS) is lower than of perinatal arterial ischemic stroke but the rate of recurrence is strikingly high. Vascular inflammation is seen as major contributor to CAIS but the mechanisms that govern structural-functional basis of vascular abnormalities remain poorly understood. To identify the contribution of immune-neurovascular interactions to CAIS, we established stroke model in postnatal day 21 (P21) mice. We demonstrate acute functional deficits and histological injury and chronic MRI-identifiable injury, brain atrophy and marked derangements in the vascular network. In contrast to negligible albumin leakage and neutrophil infiltration following acute perinatal stroke, CAIS leads to significantly increased albumin leakage and neutrophil infiltration in injured regions of wild type mice and mice with functional CX3CR1-CCR2 receptors. In mice with dysfunctional CX3CR1-CCR2 signaling, extravascular albumin leakage is significantly attenuated, infiltration of injurious Ccr2+-monocytes essentially aborted, accumulation of Ly6G+ neutrophils reduced and acute injury attenuated. Unique identifiers of microglia and monocytes revealed phenotypic changes in each cell subtype of the monocyte lineage after CAIS. Taken together, CX3CR1-CCR2-dependent microglia-monocyte signaling contributes to cerebrovascular leakage, inflammation and CAIS injury.
Introduction
Stroke is among the top 10 causes of death in children 1 but has received disproportionally little attention. The developmental stage of the brain at the stroke onset plays key role in injury.2,3 In humans, the patterns of ischemic brain injury differ greatly between preterm and at-term neonates, 4 as well as between perinatal stroke and childhood arterial ischemic stroke (CAIS).5–7 Compared to perinatal stroke, the incidence of CAIS is considerably lower, whereas the rate of recurrence is markedly higher.8,9 Vascular damage associated with infection/inflammation was demonstrated to frequently precede CAIS, 10 and MRI used as surrogate measure of CAIS revealed abnormalities in arterial wall in pediatric patients, 11 increased tortuosity of cerebral arteries, 12 and linked vascular inflammation and arteriopathy. 5 Cumulatively, these data indicate the central role of interaction between the immune system and the vasculature that make children susceptible to CAIS and affect its progression but very little is known about the mechanisms that govern structural-functional basis of neurovascular abnormalities.
The multiple brain barriers, including the blood–brain barrier (BBB), blood–CSF barrier across choroid plexus (BCSFB) and the pia-arachnoid barrier provide essential physiological roles in the brain via efflux/influx mechanisms that control ionic, nutrient, glucose and other gradients and by limiting the exchange of solutes and various molecules between the brain and periphery. The concepts have emerged that BBB integrity is age-dependent but that integrity does not linearly decrease with postnatal age.13,14 In rodents, the decline of protein expression of several tight junction (TJ) and extracellular matrix (ECM) proteins during physiological brain maturation between postnatal day 7 (P7), P17 and adulthood, in parallel to increased expression of pericyte-associated proteins and the overall expansion of the vascular network during brain maturation, 14 could make BBB susceptible during particular “susceptibility windows” during postnatal brain development. In fact, under injurious conditions, extravascular albumin leakage through disrupted BBB occurs in adult rodents following transient middle cerebral artery occlusion (tMCAO) but leakage is strikingly lower following tMCAO in P7 rats and P9 mice (i.e. perinatal focal arterial stroke). 14 The pathophysiology of stroke, including the neuroinflammatory response, is also distinct between the two age groups.14,15 The responses of juvenile (P17–P25) brain to various excitotoxic and inflammatory conditions, including intracortical injection of IL-1β13,16 and brain trauma,17–19 are shown to differ from those in adult or newborn rodents, supporting the notion of existence of “susceptibility windows” to injurious stimuli during postnatal brain development. Neuroinflammation is a major contributor and modulator of brain injury after arterial stroke in the adult, 20 and following hypoxia–ischemia (H–I) and arterial focal stroke in neonates,2,3 but knowledge of the scope of differences and contribution of individual mechanisms in injured neonates, juvenile and adult rodents are insufficiently understood.
Microglial cells were viewed as purely injurious after stroke for a long time, in part due to production of inflammatory mediators, reactive oxidant species and other toxic molecules, 20 but role of these cells in injury is being reconsidered following discoveries of the heterogeneity of the microglial pool, 21 the demonstration that lack of microglial cells disrupts neuronal network activity and exacerbates injury after stroke, 22 and that some mediators produced by microglia and monocytes can play distinct, and even opposite, roles in stroke. 23 We previously reported that after perinatal stroke induced in P7–P9 rodents at least a subpopulation of microglial cells protects neurovascular integrity and attenuates injury and hemorrhagic transformation.14,24 Not only the vasculature, but microglial cells and leukocytes undergo major phenotypic changes during postnatal brain development, but effects of immune-neurovascular interactions in CAIS pathophysiology have never been studied in rodent models.
To understand the mechanisms of CAIS, we recently developed tMCAO model in P21 mice. We chose P21 to model CAIS because brain at this age is believed to represent brain development of a human toddler. 25 We demonstrate that tMCAO in P21 mice produces chronic MRI-identifiable injury and sustained vascular damage. Examination of the effects of CX3CR1-CCR2 microglial-monocyte signaling in mice with unperturbed or genetically ablated signaling shows that ablated CX3CR1-CCR2 signaling attenuates albumin leakage in the peri-focal ischemic region, essentially aborts monocyte infiltration, diminishes neutrophil adhesion and infiltration, alters microglial phenotypes and reduces injury.
Materials and methods
All research conducted on animals was approved by the University of California San Francisco Institutional Animal Care and Use Committee and by the Loma Linda University Animal Health and Safety Committee. All research was performed in accordance to the Guide for the Care and Use of Laboratory Animals (U.S. Department of Health and Human Services). Animals were given ad libitum access to food and water; housed with nesting material and shelters, and kept in rooms with temperature control and light/dark cycles.
The data reporting is in compliance with the ARRIVE guidelines (Animal Research: Reporting in Vivo Experiments). Two laboratory members who were unaware of animal identity analyzed data blindly. Randomization was used where possible.
tMCAO in P21 mice
To develop a CAIS model, we performed tMCAO in male and female P21 C57Bl/6 mice in a manner similar to what we previously detailed for MCAO surgery in P7 rats 26 and P9 mice 15 and varied suture characteristics and the length of suture advancement. Surgery was performed under 1.25–2% isoflurane anesthesia. A midline cervical incision was made exposing the common carotid artery (CCA). The skin and muscle were gently retracted with home built microretractors, exposing the internal carotid artery (ICA). A single thread from a 7–0 silk suture was used to tie a temporary knot at the origin of the ICA and pulled caudally, the long end clipped to the skin, whereas a second strand of 7–0 was looped 1 mm below the origin of the ICA, gently retracted cranially to prevent retrograde blood flow. A 0.21 mm thick 6–0 filament (Doccol Catalog# 602123PK5Re) with 2–3 mm long coating at the tip was inserted and the filament was advanced ∼6.5–7 mm past ICA/ECA bifurcation. The lower 7–0 suture was then gently tied with a releasable knot to keep the filament in place, and prevent retrograde bleeding from the arteriotomy. The skin was closed and the pups allowed to recover. Throughout the 7-min procedure, temperature of the pups was maintained by an overhead heat lamp or a heating pad below the animal. Reperfusion was achieved by re-anesthetizing the pups, removing a single skin suture to expose the cervical vessels, untying both 7–0 strands of silk, removing the occluding filament, and applying a small amount of collagen powder and pressure to the arteriotomy for 30 s, after which the skin was re-sutured.
We confirmed severe perfusion deficits during MCAO and recirculation upon suture removal using intra-jugular injection of FITC-isolectin B4 (n = 4–6/group). Histological outcome was determined on TTC and Nissl-stained brains 24 h after reperfusion (n = 34). There was no mortality associated with surgery and no mortality or bleeding associated with reperfusion. Within 72 h after reperfusion, mortality was <10%. Additional mice were subjected to longitudinal MRI during seven days after reperfusion (n = 11) followed by evaluation of vascular coverage/characteristics and histological (Nissl) outcomes, as described below. Two mice died after MRI scans at two and five days after MCAO and three mice were excluded due to very small or non-existent lesions after MRI at two days post MCAO. After characterizing the model in WT (C57Bl6) mice, tMCAO was performed on Cx3cr1GFP/+/Ccr2RFP/+ mice and Cx3cr1GFP/GFP/Ccr2RFP/RFP mice (C57Bl6). The latter mouse line was established at Dr. Charo’s laboratory at the Gladstone Institute at UCSF 27 ; founders for the colony were provided to us by Dr. Katerina Akassoglou (KA) at the Gladstone Institutes at UCSF. Histologic analysis was performed independently and blindly by two laboratory members who were unaware of animal identity.
Behavior tests
Inverted Wire grid hang and Open Field tests were performed 20–22 h after reperfusion.
For inverted Wire grid hang test, mice were placed with all four paws on a 24 cm diameter circular wire grid, the grid was gently rocked back and forth to encourage the mouse to grasp the wire with all paws and the grid carefully inverted so that the mouse is hanging over a padded surface. Latency to fall was recorded within 2 min. Measurements were repeated three times. Mice that were unable to grasp the wire for 5 s were excluded.
Open Field test was performed in a 30 × 30 cm box over a 2-min period. The overall activity, the number of grid crossings (6 × 6 cm 2 grid) and preferential circling behavior were video recorded and then analyzed.
High-resolution longitudinal MRI and unbiased MR data analysis
A subgroup of WT P21 mice with confirmed behavior deficits on the wire test 20–22 h after reperfusion was shipped to Dr. Obenaus’s laboratory for longitudinal MRI. Upon arrival, mice were allowed to recover till the next day and were imaged at two, three and seven days after tMCAO, as previously described for adult stroke model and H–I model in neonatal mice.28–31 Mice were anesthetized with isoflurane (3% induction, 1% maintenance) and placed in the coil. Body temperature was maintained at 37℃ by using a water-heated pad.
In vivo T2-weighted images (T2WI, TR/TE = 3000 ms/10 ms, 20 × 0.5 mm slices, matrix 192 × 192), diffusion-weighted images (DWI; TR/TE = 3000 ms/31.2 ms, 12 × 0.5 mm slices, matrix 128 × 128) and angiography (FLASH TOF 2D, TR/TE = 12 ms/3 ms, 30 × 0.4 mm slices, interval = 0.2 mm, matrix 256 × 256) were collected on a 11.7T Bruker Avance Instrument (Bruker Biospin, Billerica, MA). The total imaging time was 42 min for each animal at each time point. After imaging, mice were allowed to recover on a heated surface until ambulatory movements were apparent.
Unbiased MRI lesion analysis
MRI characteristics of the lesion at two-, three-, and seven-day post-injury were assessed using hierarchical regional splitting (HRS) computational method (US patent: 8731261; European patent: 11748009.5-1265) to quantify core, peri-focal, and total lesion volume, as we have previously reported.28,29 Each MRI was evaluated for T2 values within normal appearing brain matter (NABM) and within the lesion. Automated HRS threshold values were determined and HRS automatically extracted core, peri-focal and NABM features, including volumes and T2 values. Additionally, apparent diffusion coefficient (ADC) maps were generated using Jim analysis software (Xinapse Systems Ltd; West Bergholt, Essex; United Kingdom), in order to assess the extent of restricted diffusion exhibited by the lesion. The ADC lesion border was manually delineated using the Cheshire image processing software (version 4.3, Parexel, Waltham, MA) where hyper-intense tissue demonstrated restriction in days 2 and 3 post-injury, with a mixture of light and dark tissue types on the ADC lesion during the seven-day time point. Angiography slices were collapsed to generate a maximum intensity projection (MIP) using Image J (NIH) and then analyzed using the identical vessel features as described below for the vessel painting. Quantitative T2 and ADC values were extracted from the total lesion and exhibited similar temporal increases by 7 day after tMCAO (Figure 2(b)).
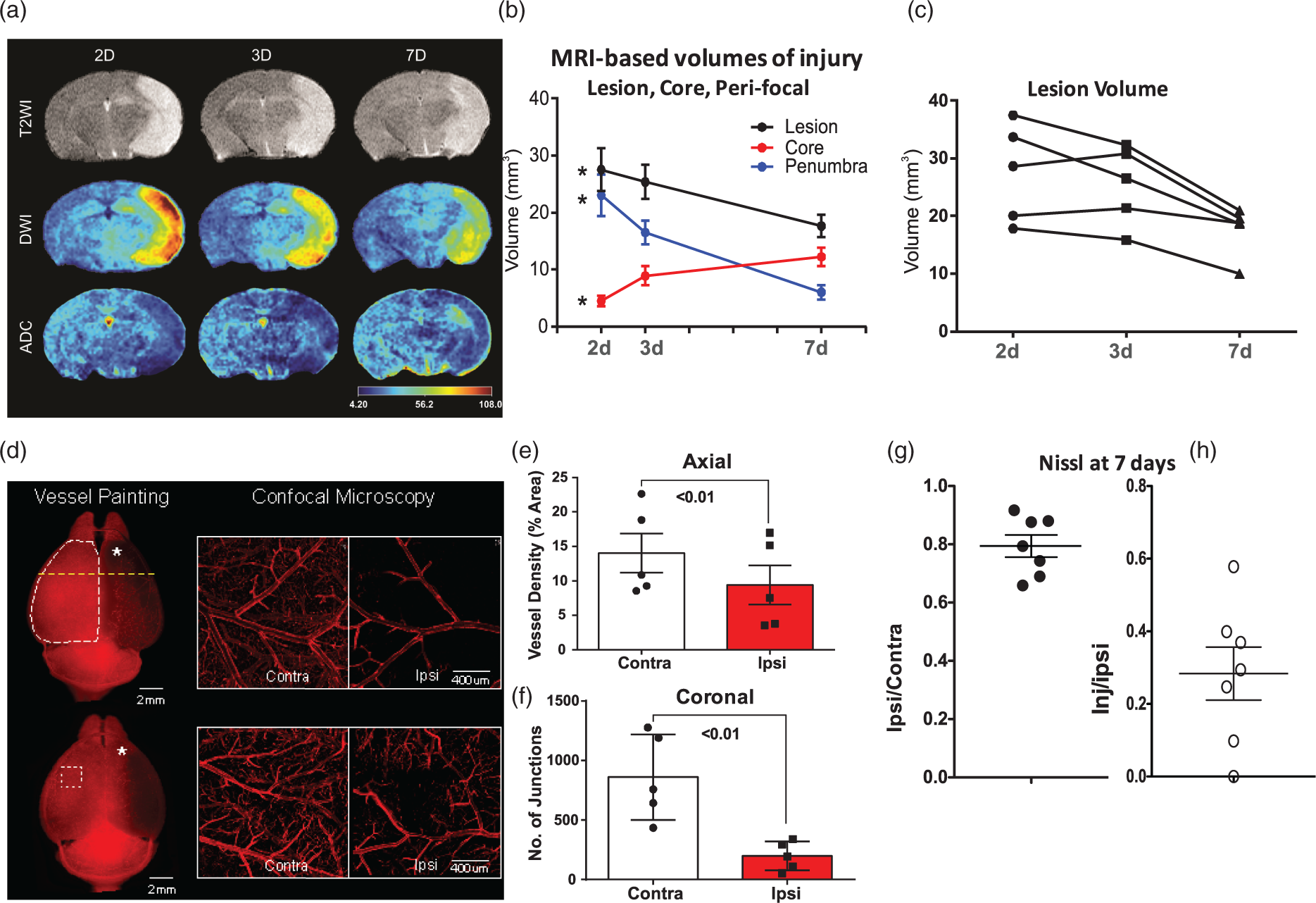
Brain injury 24 h after tMCAO in P21 mice with functional and disrupted CX3CR1-CCR2 signaling. (a) A representative set of four coronal TTC images 24 h after tMCAO, an injury pattern that we regarded as successful injury outcome during model development. (b) A representative set of five of eight coronal cresyl violet-stained sections used to quantify injury volume. (c) Percent volume of injured tissue in the ipsilateral hemisphere of cresyl violet-stained sections. (d– e) Functional deficits on Wire hanging test (d) and Open Field test (e) performed in the same mice before sacrifice. G/+/R/+ – double-heterozygote mice. G/G/R/R – double-homozygote mice. Dots represent data from individual animals. Significance levels as indicated on individual panels. tMCAO in P21 mice produces sustained MRI-identifiable injury and disruption of the vascular network during chronic injury phase. (a) Representative examples of evolution of MRI-identifiable lesion during seven days after tMCAO. (b) Quantification of percent of injured region within ipsilateral hemisphere based on the data shown in (a). Note the overall gradual reduction of lesion volume (black dots, *p = 0.01), reduced peri-focal volume (blue dots, *p = 0.005) and a parallel expansion of the ischemic core volume (red dots, *p = 0.01). (c) Evolution of MRI-defined injury in individual mice. (d–f) “Vessel Painting” method in vivo demarcates marked disruption of vascular network and vascular complexity seven days after injury. (d) Two representative examples of VP brains after wide-field epifluorescent microscopy illustrate the dramatic loss of vasculature (left column) that is apparent visually in the ipsilateral hemisphere (*). The white dotted line (top image, left) depicts the region of interest used for hemispheric (axial) vascular analysis. Yellow line depicts the location for subsequent coronal analysis. The white dotted square (bottom image, left) shows the approximate location for the confocal microscopy on the contra- and ipsilateral hemispheres. Confocal microscopy (right panel) delineates two examples of the loss of essentially all (top image, right) or most (bottom image, right) microvessels in the ipsilateral cortex. (e) Axial analysis of vessel density shows a significant decrease in vessel density in brains shown in (d). (f) Similarly, VP analysis of coronal sections shows a significant loss in the number of blood vessel junctions. (g–h) Quantification of injury volume seven days after tMCAO. Volume of the remaining injured hemisphere compared to contralateral hemisphere is shown in (g), whereas volume of injured region compared to volume of remaining tissue in ipsilateral hemisphere is shown in (h). Dots represent data from individual animals (**p < 0.01).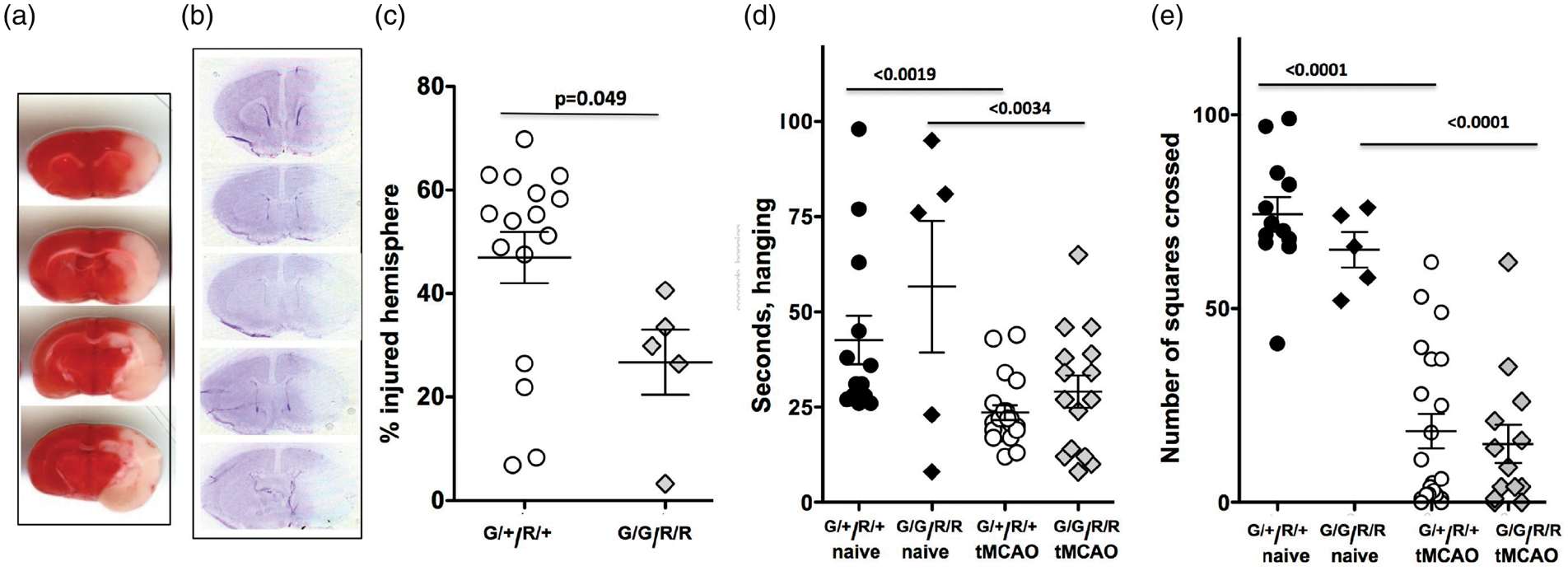
Vessel painting procedure, imaging and analysis
At the end of last MRI session, vascular structural-functional responses to CAIS were examined by vessel painting (VP), a method based on the ability of the fluorescent dye 1,1′-dioctadecyl-3,3,3′3′-tetramethylindocarbocyanine perchlorate (DiI) to bind to lipid membranes, as previously described.32,33 Mice were anesthetized with intraperitoneal injection of ketamine (90 mg/kg) and xylazine (10 mg/kg) and the following sequential injections were delivered: 30 µl SNP (Sodium Nitroprusside dehydrate + heparin; intracardiac injection into left ventricular over 10 s, 0.3 mg/ml DiI in PBS containing 4% dextrose, 250 µl, over 10 s), followed by 20 ml PBS perfusion and 40 ml of 4% PFA. The brains were extracted and post fixed in 4% PFA for 24 h, washed and stored in PBS at 4℃ until epi-fluorescent imaging. Successful VP was defined as uniform staining throughout the cerebrum; mice exhibiting patchy or non-uniform labeling were excluded from the study (1/6 mice). In vivo maps of the vascular bed were analyzed on both macroscopic and microscopic scales, as we previously described. 33 Images of the entire brains (axial and coronal view; 2 × magnification) were acquired using a fluorescence microscope (Keyence BZ-X700, Keyence Corp, Osaka, Japan) and reconstructed using the XY-stitching and Z-stack features. Then ipsilateral and contralateral MCA regions were imaged using confocal microscopy (5X magnification; Zeiss LSM 710 NLO, Zeiss, Jena, Germany). Classical vessel analysis, including vessel density, vessel length and number of junctions, was performed by using the AngioTool Software. 34
Albumin leakage, histological analysis and immunofluorescence
Alexa-647 conjugated albumin (A34785, Life Technologies; 30 ul per 10 g mouse, 25 mg/ml in PBS injected over 30sec) was injected into the jugular vein 23 h after reperfusion followed by transcardial perfusion 1 h later, as we reported.
24
Perfusion-fixed, post-fixed, cryoprotected and flash frozen brains were sectioned on a cryostat (12um thick serial sections, 360 um apart) and injury determined in six coronal Nissl-stained sections. Sections adjacent to those used for histological analysis were immunostained and images captured in three fields of view (FOV) in the peri-focal and ischemic core regions in the caudate and the cortex and in the corresponding contralateral regions using an Axio Imager.Z2 microscope (Zeiss) equipped with Volocity Software (PerkinElmer), as we described.24,35 Peri-focal regions were defined based on the cumulative appearance of edge of morphologically transformed Iba1+ and fragmented DAPI+ nuclei adjacent to regions with regular morphology of DAPI+ nuclei and the layer of GFAP+ cells (as demonstrated in Figure 3(a) to (f)). The density, length, surface, volume of Glut-1+ vessels were determined using the automated protocols for signal intensity threshold. The number of GFP+ and RFP+ cells per FOV, their surface and volume characteristics and co-localization with Glut-1+ vessels were measured using custom-made thresholds and size-exclusion protocols created in Volocity software, as we described.24,35
Albumin leakage occurs in peri-focal regions of P21 Cx3cr1GFP/+/Ccr2RFP/+ mice but not Cx3cr1GFP/GFP/Ccr2RFP/RFP mice 24 h after tMCAO. (a–f) Representative images of ipsilateral hemisphere following intra-jugular injection of Alexa-647 albumin in wild type mice 24 h after tMCAO. (a) Low magnification image. (b–c) Higher magnification images acquired in boxed region in (a) demonstrate finer detail of albumin extravasation and neutrophil margination and infiltration. Thin yellow arrows point at areas of extravascular albumin leakage. Small thick arrows point to Ly6G+ neutrophils within the vessels and in the injured parenchyma. *demarcate uninjured region areas with normally appearing DAPI morphology. (d) Low magnification image. (e–f) Higher magnification images acquired in boxed region in (d) demonstrate extracellular albumin leakage in regions with irregular DAPI+ nuclei and activated Iba1+ cells. (g) Representative image of Iba1+ cells in cortex contralateral to MCAO. (h–i) Representative images of peri-focal region following intra-jugular injection of Alexa-647 albumin to Cx3cr1GFP/+/Ccr2RFP/+ mice 1 h before sacrifice at 24 h after reperfusion. White punctate curve demarcates a border of the peri-focal region. In (h), both intravascular and extravascular Alexa-647 albumin (turquoise) is observed. Thin yellow arrows point at extravascular Alexa-647 albumin. The presence of intravascular albumin (Glut1+/Alexa-647 albumin+) and extravascular albumin is visualized in (g). Cx3cr1GFP/+ microglia in injured cortex shows a variety of morphological phenotypes (b). The presence of Ccr2RFP/+ monocytes is evident in injured regions only (b). (j–l) Quantification of leakage in the peri-focal region (j), the caudate (k) and in ischemic core (l). (m) Quantification of Glut1+ vascular coverage in individual regions. G/+/R/+ – double-heterozygote mice. G/G/R/R – double-homozygote mice. Ipsi - Ipsilateral; C - Contralateral. Dots represent data from individual animals. Significance levels as indicated on individual panels.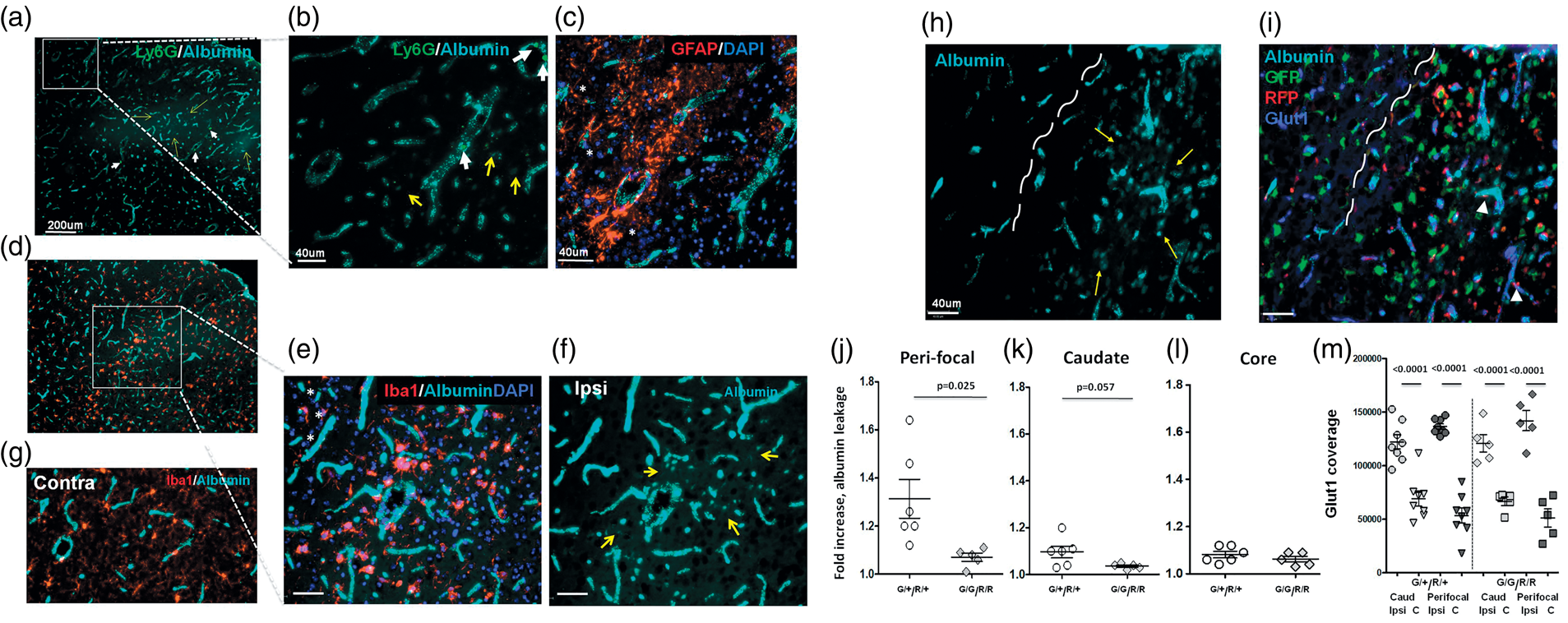
Cell isolation from injured and uninjured brain regions and multi-color flow cytometry
Samples for multi-color flow cytometry were prepared as we described. 35 Briefly, deeply anesthetized mice were transcardially perfused with 10 ml of Ca2+/Mg2+-free Hank’s balance salt solution (HBSS) to eliminate peripheral cells, meninges were removed, and the cortices of injured and matching contralateral regions were dissected on ice and enzymatically digested to obtain single brain cells using Papain-containing Neural Tissue Dissociation Kit (Miltenyi Biotec, Germany). Myelin was removed with myelin-conjugated magnetic beads (Miltenyi Biotec). 35 Isolated cells were plated at a density of 2 × 105 cells per 96-well, blocked for 15 min with CD16/32 (1:70; Biolegend) in FACS buffer containing 2% fetal bovine serum, washed once in FACS buffer, stained with a mixture of antibodies for 30 min on ice, and washed once with FACS buffer, as we described. 35 CD16/32 antibody blocking step was omitted in samples stained with unconjugated rat 4C12 monocyte antibody (specific to inflammatory monocytes 36 ), or unconjugated rat 4D4 monocyte antibody (specific for microglial cells), antibodies that were generated in the laboratory of Dr. Butovsky by vaccinating rats with splenic Ly6C+ monocytes (unpublished). The following conjugated antibodies were used: anti-CD11b-APC-Cy7 (1:2000; Biolegend); anti-CD45-Pacific Blue (1:2000; Biolegend); anti-Ly6C-PerCP5.5 (1:800; Biolegend); anti-Ly6G-AF700 (1:800; Biolegend). Samples were run on BD LSRII flow cytometer (BD Bioscience). The gating strategy was based on live single cells stained with Live/Dead Yellow reactive dye (Life Technologies). BD compensation beads (BD Bioscience) were used for compensation. Data analysis was performed in FlowJo software (Tree Star). All measurements to characterize antigen expression on microglial cells and infiltrated monocytes were performed on the population of SSC/CD11b+ cells, as indicated in individual figure legends. Ly6G+ cells were gated from single cell population.
Multiplex cytokine assay
Protein concentrations of IL-1α, IL-1β, IL-6, G-CSF, MCP-1, MIP1α, MIP1β, TNFα, LIF and KC were measured in whole brain homogenates from injured and matching contralateral regions, as we previously reported.15,35,37 Measurements were performed using Bio-Plex System (Bio-Rad) and StatLIA® software (Brendan Scientific) with a 5-parameter logistic curve fitting. The data were normalized to protein concentration in the same brain homogenate sample.
Statistical analysis
Individual measurements were determined for each group and the Mean ± SD presented. Statistical analysis was performed on GraphPad Prism (GraphPad Software). One-way ANOVA with Bonferroni correction was performed to test for differences among groups and Student’s t test was used to examine for differences between two groups. Significant differences in MRI data were tested using repeated measures two-way ANOVA with post hoc testing.
Results
Transient MCAO in P21 mice produces acute behaviour deficits and sustained MRI-based and histologically defined injury
We developed the CAIS model in P21 mice, an age when mouse brain is thought to correspond to brain of a human toddler, 25 by modifying tMCAO surgical procedure we previously described for neonatal rats and mice15,26,38 by varying diameter of the filament, the length of its advancement and filament placement. tMCAO produced consistent histological injury 24 h after reperfusion (Figure 1).
We then used longitudinal multi-modal T2 and DWI to follow injury progression at two, three and seven days after tMCAO (Figure 2(a)). ADC-derived lesion volumes remained relatively constant over the seven-day period, while those from T2 decreased linearly and significantly over the seven days (p < 0.05). Core, peri-infarct and total lesion volumes from T2WI demonstrate significantly decreased peri-infarct volume temporally with an increasing core volume (Figure 2(b); p = 0.01 and p = 0.005, respectively, repeated measures two-way ANOVA; total lesion p = 0.01); total lesion was also reduced by 9.92 mm 3 from two to seven days (p = 0.012). 28 MR angiography (at two, three, seven days) to assess large vessels revealed no early significant differences but reduced vascular density (p = 0.048) and increased lacunarity (p = 0.02) at seven days.
Nissl analysis in the same brains showed tissue loss in all mice and injury in the ipsilateral hemisphere (Figure 2(g) and (h)). Large-to-medium size hemorrhages were observed in two mice.
tMCAO in P21 mice deteriorates vascular networks during chronic injury
To evaluate the pattern and the magnitude of changes in the vascular bed induced by CAIS, Dil was administered to all mice following MRI seven days after tMCAO. Analysis of the axial cerebral cortex revealed a dramatic loss of vasculature on the ipsilateral hemisphere (Figure 2(d)), ranging from partial (bottom panel, Figure 2(d)) to an almost complete (upper panel, Figure 2(d)) loss of microvessels on the ipsilateral cortical surface relative to the contralateral hemisphere. Loss of vessel density in the axial ipsilateral compared to the contralateral cortex was 9.40 ± 2.83% vs. 14.02 ± 2.82% (p = 0.008, t-test, Figure 2(e)). There were also decreased numbers of vessel junctions (p = 0.008, t-test) in the ipsilateral cortex (Figure 2(f)). Confocal microscopy showed a trend (ns) toward reduced vessel density within the ipsilateral MCA territory (axial view, Right; Figure 2(d); 15.15 ± 4.34% vs. 20.19 ± 4.50%), and decreased percentage of vessel area, number of junctions and average vessel length.
Genetic ablation of Cx3cr1 and Ccr2 significantly attenuates short-term histological outcome in a CAIS mouse model
To enable examination of CX3CR1-CCR2-dependent monocyte-microglial interactions to injury, we performed tMCAO in P21 mice with functional (Ccr2RFP/+/Cx3cr1GFP/+) and disabled (Ccr2RFP/RFP/Cx3cr1GFP/GFP) receptors due replacement of both gene copies with respective GFP and RFP. At 24 h after tMCAO, injury volume was 47.0 ± 5.0% of ipsilateral hemisphere in Cx3cr1GFP/+/Ccr2RFP/+ mice (Figure 1(c), n=16), with large injury involving both the caudate and the cortex observed in 75% mice, smaller size injury involving the caudate and the cortex in 12.5% mice, and injury limited to the caudate in 12.5% mice. Marked deficiency in paw strength (wire test, Figure 1(d)) and exploratory behavior were observed 24 h after tMCAO (open field test, Figure 1(e)).
Deficiency in CX3CR1-CCR2 signaling led to significantly smaller injury volume at 24 h, 26.7 ± 6.3% of ipsilateral hemisphere (Figure 1(c); n = 5, p < 0.05). Marked functional deficits were also evident in injured deficient mice on the Wire (Figure 1(d)) and Open Field (Figure 1(e)) tests, but the magnitude of deficits was similar in both groups. Therefore, while functional CX3CR1-CCR2 deficiency attenuated severity of histological outcome, it did not attenuate functional deficits after acute injury.
CX3CR1-CCR2 signaling contributes to albumin leakage through the BBB after acute CAIS
Knowing that albumin leakage into injured brain regions is minor 24 h following tMCAO in neonatal rats and mice, while albumin leakage is extensive in adult rats subjected to tMCAO, 14 here we asked if tMCAO in juvenile mice leads to albumin leakage. Albumin administered intra-jugular before sacrifice at 24 h remained within the vascular bed in contralateral hemisphere, whereas leakage was observed in the peri-focal region in the cortex and in the caudate of WT mice (Figure 3(a) to (f)) and Cx3cr1GFP/+/Ccr2RFP/+ mice (Figure 3(h) and (i)). Deficiency in CX3CR1-CCR2 signaling significantly reduced albumin leakage in the peri-focal region (Figure 3(j)) and tended to reduce leakage in the caudate (ns, Figure 3(k)). No leakage was observed in the ischemic core in the cortex (Figure 3(l)). Therefore, CX3CR1-CCR2 functionality modulates neurovascular integrity after CAIS. Compared to that in corresponding contralateral regions, volume of Glut1+- vessels was significantly but similarly increased in the peri-focal regions in the cortex and the caudate of both groups (Figure 3(m)), likely because of brain swelling after acute injury.
Deficiency in CX3CR1-CCR2 signaling aborts infiltration of Ccr2+-monocytes and affects neuroinflammation after CAIS
CAIS significantly increased the number of GFP+ cells and triggered their morphological transformation in the peri-focal region of Cx3cr1GFP/+/Ccr2RFP/+ mice at 24 h after tMCAO (Figure 4(a)). The number of GFP+ cells was reduced in the ischemic core compared to matching contralateral regions (Figure 4(b)). While volume of GFP+ cells was similar in contralateral hemisphere of mice with functional and deficient CX3CR1-CCR2 signaling (Figure 4(c)), the number of GFP+ cells tended to be lower in both the peri-focal region and the core of deficient mice (Figure 4(a) and (b)).
Effects of CX3CR1-CCR2 deficiency on microglial activation and infiltration of Ccr2+-monocytes after CAIS. (a–b) The number of GFP+ cells in the cortical peri-focal (a) and the ischemic core (b) and in the matching contralateral regions in mice with functional (G/+/R/+) and dysfunctional (G/G/R/R) CX3CR1-CCR2 signaling, as determined by immunofluorescence. (c) Mean volume of GFP+ microglia in mice with functional (G/+/R/+) and dysfunctional (G/G/R/R) CX3CR1-CCR2 signaling. (d–e) The overall number of Ccr2+-monocytes in the peri-focal (b) and ischemic core (c) regions of mice with functional (G/+/R/+) and dysfunctional (G/G/R/R) CX3CR1-CCR2 signaling. (f) The overall number of infiltrated Ccr2+-monocytes in the peri-focal of mice with functional (G/+/R/+) and dysfunctional (G/G/R/R) CX3CR1-CCR2 signaling. Dots represent data from individual mice. Ipsi - Ipsilateral; C - Contralateral. Significance levels as indicated on individual panels, ns: non-significant (ANOVA with Bonferroni post hoc test).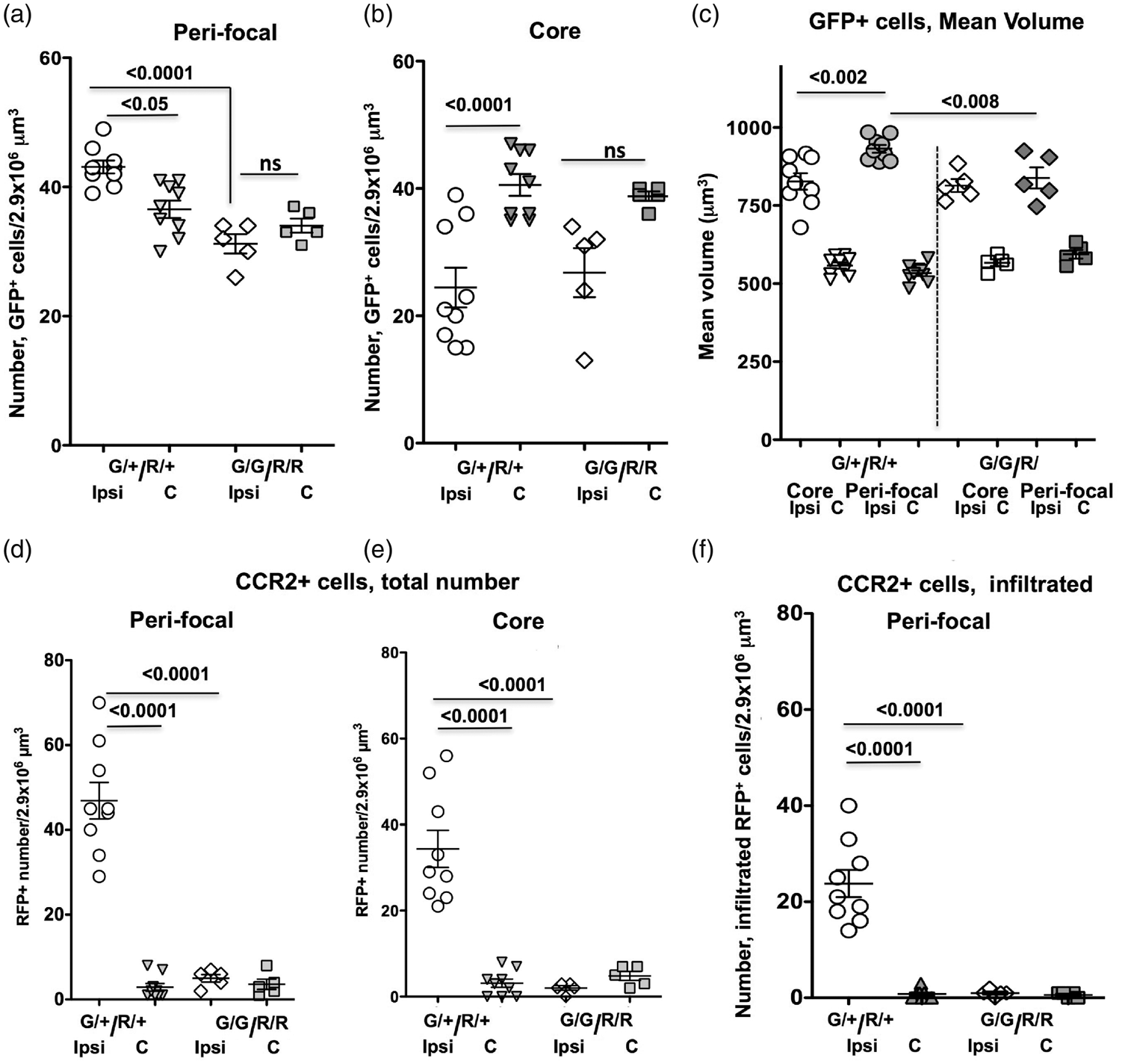
As expected, essentially no Ccr2RFP/+ monocytes were observed in contralateral hemisphere and in the cingulate in the ipsilateral hemisphere, while adhesion and transmigration of Ccr2 RFP/+-monocytes were triggered in the peri-focal and in core regions, as evident from -localization of Ccr2RFP/+ cells with Glut1+–vessels. Adherence (Figure 3(i) and Figure 4(d) and (e)) and transmigration (Figure 4(f)) of CCR2+ monocytes into injured tissue were essentially abolished in mice with disrupted CX3CR1-CCR2 signaling.
In mice with functional CX3CR1-CCR2 signaling, injury markedly increased the levels of leukocyte chemoattractants KC, MCP-1, MIP-1α and MIP-1β and cytokines IL-6, TNFα and G-CSF as compared to levels in matching contralateral regions, whereas the levels of IL-1β, IL-1α, IL-10 or IL-4 were not significantly affected by injury (Figure 5). In injured regions of deficient mice, the levels of KC, MCP-1 and MIP-1α, IL-6, TNFα and G-CSF were also significantly increased, whereas the levels of IL-1α, IL-10 or IL-4 were significantly elevated compared to injured regions of mice with functional CX3CR1-CCR2 signaling (Figure 5). The magnitude of increased levels of neutrophil chemoattractant KC was significantly lower in injured regions of mice with dysfunctional CX3CR1-CCR2 signaling (Figure 5). Therefore, the patterns of changes in the levels of individual inflammatory and anti-inflammatory mediators were affected by functional deficiency in CX3CR1-CCR2 signaling in injured juvenile brain.
Accumulation of cytokines and chemokines is altered by CX3CR1 and CCR2 deficiency in injured regions 24 h after tMCAO. Cytokine 14-multiplex showed marked upregulation of a subset of cytokines and chemokines by tMCAO, but unchanged levels of IL-1β. CX3CR1-CCR2 deficiency affected the levels of only some of measured chemokines and cytokines in injured regions. Dots represent data from individual mice. Ipsi - Ipsilateral; C - Contralateral. Significance levels as indicated on individual panels, ns: non-significant.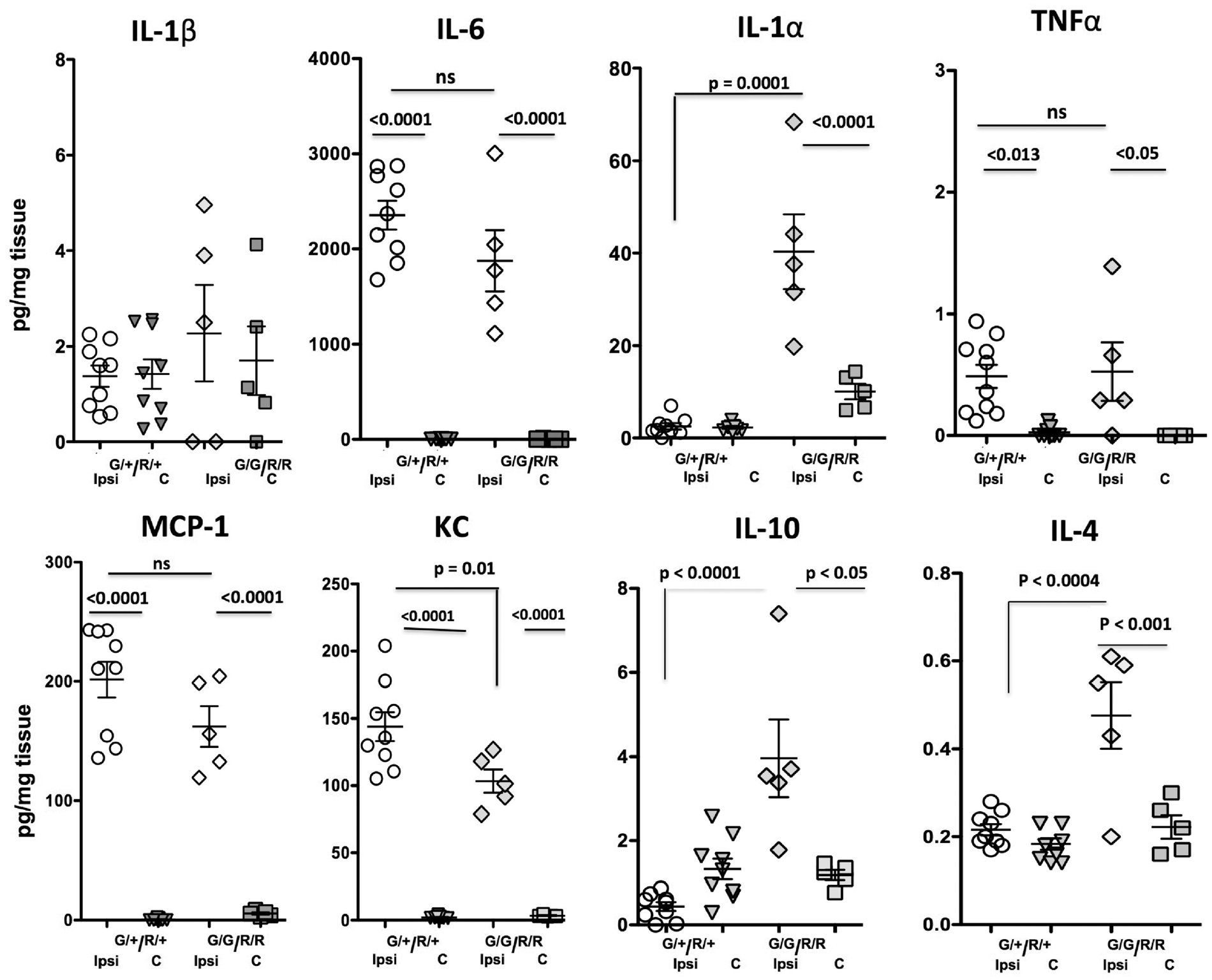
Disrupted CX3CR1-CCR2 signaling alters microglial and leukocyte phenotypes after CAIS
Considering that microglial cells, circulating monocytes and monocytes that differentiate into macrophages upon infiltration into injured regions share many surface antigens, we characterized the microglial and monocyte phenotypes by multi-color flow cytometry using multiple markers and unique cell-subtype identifiers. We applied gating strategy shown in Figure 6(a) (i.e. first gating on live single cells and then selecting CD11b+ cells using SSC/CD11b+ gate) to cells obtained from contralateral and injured regions. Consistent with immunofluorescence results (Figure 3), the patterns of GFP and RFP expression (Figure 6(b) and (c)) and number of GFP+/CD45low microglial cells were reduced in injured regions of both groups (Figure 6(d)), more so in deficient mice. The number of Ccr2+-monocytes was significantly increased in injured regions of Cx3cr1GFP/+/Ccr2RFP/+ mice (Figure 6(b)) but was aborted in injured regions of deficient mice (Figure 6(c)).
The phenotypes of microglial cells and phenotypes of monocytes and their infiltration induced by CAIS depend on CX3CR1-CCR2 signaling 24 h after tMCAO. (a) An example of gating strategy used in flow cytometry data analysis to first choose viable and then single cell populations (insert in a, top left) and CD11b+ population in contralateral and injured regions of mice. All results in Figure 6 were obtained in CD11b+-gated cells. (b–c) Representative examples of GFP+ and RFP+ cell populations in contralateral and injured regions of mice with functional (b) and dysfunctional (c) CX3CR1-CCR2 signaling. (d) Quantification of morphological transformation of GFPlow microglia to acquire GFPmed/high in injured regions. (e–f) Representative plots of the number of CD11b+/Ly6C+ cells in mice with functional (e) and dysfunctional (f) CX3CR1-CCR2 signaling. Note that CX3CR1-CCR2 deficiency reduces the overall number of CD11b+/Ly6Chigh and CD11b+/Ly6Clow cells and essentially abolishes CD11b+/Ly6Clow cell population. (g–h) Quantification of infiltrating CD11b+/Ly6C+/Ccr2+ monocytes demonstrates that tMCAO triggers infiltration of Ccr2+/Ly6C+ cells in Cx3cr1GFP/+/CCR2RFP/+ mice and that Ccr2+ cells are predominantly Ly6Chigh (g). Accumulation of both Ccr2+/Ly6Chigh and Ccr2+/Ly6Clow subpopulations is aborted in mice with dysfunctional CX3CR1-CCR2 signaling. (k–l) Quantification of the number of 4C12+/GFP+ cells. Note that infiltrating Cx3Cr1+ monocytes constitute only a minor subpopulation of 4C12+ monocytes in injured juvenile mice, i.e. less than 4% of Cx3cr1+/4C12+ cells in contralateral regions of both groups. Cx3cr1+/4C12+ cell population remains small in injured regions (ns). (m–n) Representative plots for CD11b+/Cx3Cr1+/4D4+ cells in contralateral and injured regions of mice with functional and dysfunctional CX3CR1-CCR2 signaling. Note broader-range GFP expression levels in mice with functional receptors. (o–p) A representative example of Cx3Cr1+/4D4+ cells. Note that all Cx3Cr1+ cells are positive for 4D4, whereas CCR2+ monocytes do not express 4D4. White arrowheads point to few CCR2+ monocytes engulfed by Cx3Cr1+/4D4+ microglia. Dots in (d), (g–h), (k–l) represent data from individual mice. Ipsi - Ipsilateral; C - Contralateral. Significance levels as indicated on individual panels.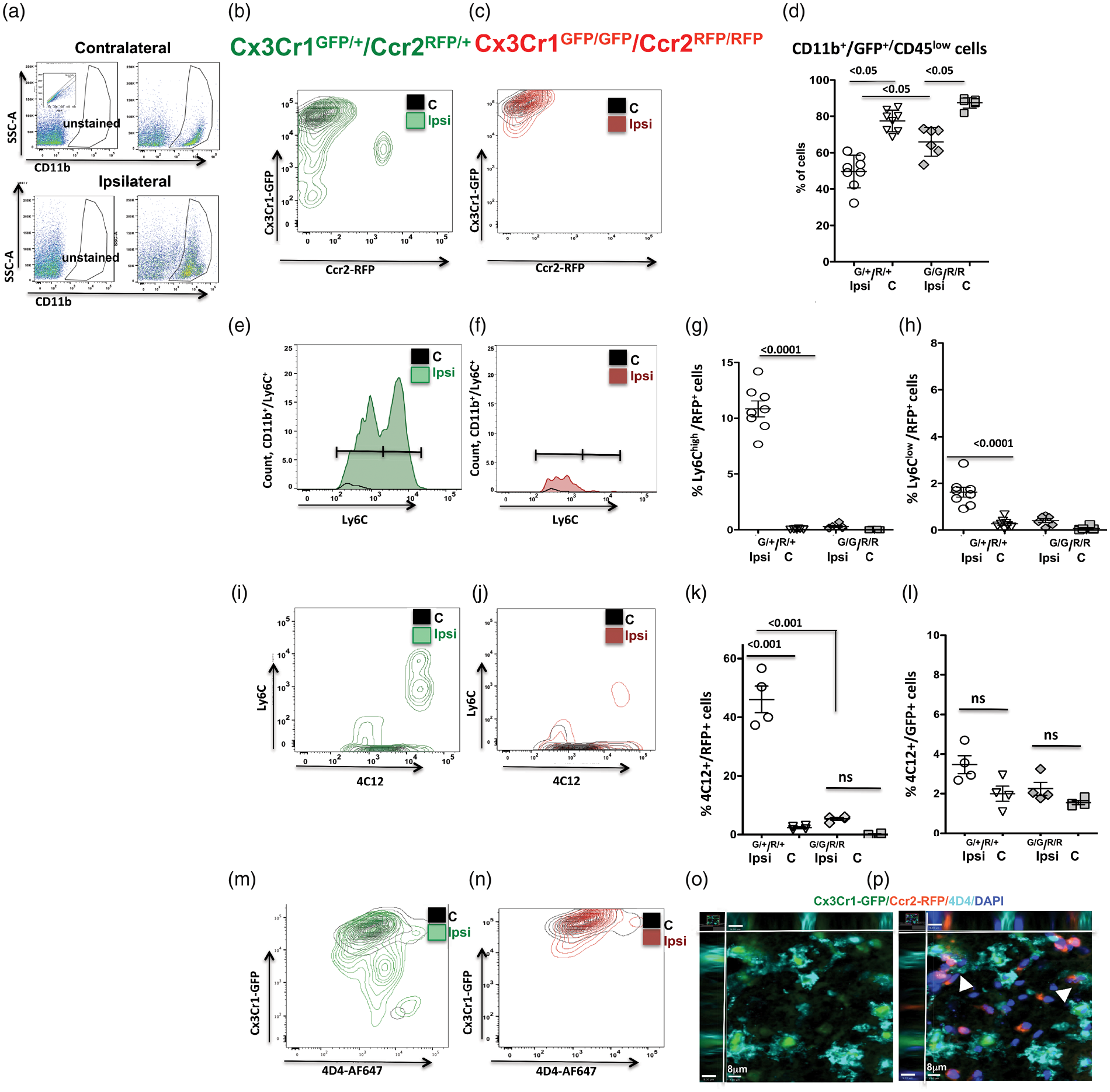
The Ly6Clow and Ly6Chigh subpopulations were shown to play distinct roles in models of stroke and hemorrhage in adult mice. 39 In CAIS, we observed increased number of Ly6C+/CD11b+ cells in injured regions in both groups (Figure 6(e) and (f)), but accumulation of Ly6C+ cells was significantly reduced in mice with dysfunctional CX3CR1-CCR2 signaling, including significant reduction in the number of Ly6Clow cells and an almost completely abolished accumulation of Ly6Chigh cells (Figure 6(e)). Quadrant analysis of CCR2+/Ly6C+ cells (Figure 6(g) and (h)) showed a much higher number of CCR2+/Ly6Chigh cells than CCR2+/Ly6Clow cells in mice with functional receptors and less than 1% of CCR2+/Ly6Chigh and CCR2+/Ly6Clow cells in mice with dysfunctional receptors. These results clearly demonstrate phenotypic changes in the cells of the monocyte lineage that stem from CX3CR1-CCR2 deficiency.
We then confirmed the origin of cells of the monocyte lineage. While all microglial cells express Cx3cr1 in vivo under normal and injurious conditions, 40 recent studies have demonstrated that peripheral monocytes also express Cx3cr141 as are infiltrated monocytes that differentiate in injured regions after stroke, 42 thus precluding from definite distinction between microglia and monocytes. Ly6C+ was initially thought to be expressed by monocytes only, with the level of Ly6C+ expression defining toxic (Ly6Chigh) or beneficial (Ly6Clow) properties, 39 but microglia also express low levels of Ly6C in injured neonatal brain. 35 Given that neither Cx3cr1 nor Ly6C expression can be used as comprehensive discriminator between microglia and monocytes, but microglial cells uniquely express molecules not expressed by toxic monocytes 43 and monocytes express molecules not found in microglial cells, we used 4C12 antibody specific for monocytes and 4D4 antibody that uniquely identifies microglia. Both Ly6Chigh and Ly6Clow cells were 4C12+ (Figure 6(i) and (j)). Approximately half of 4C12+ cells were RFP+ in injured regions of mice with functional receptors (Figure 6(l)), whereas more than 97% of GFP+ cells were negative for 4C12 in contralateral regions in both groups and only a small increase in 4C12+/GFP+ cells was seen in injured regions of both groups (ns; Figure 6(l)). In parallel, essentially all GFP+ cells were 4D4+ and only few cells were either 4D4+/RFP+ cells or RFP+ cells engulfed by 4D4+/GFP+ cells (Figure 6(m) to (p)), consistent with low rate of Cx3cr1+ monocytes in the infiltrating monocyte population (white arrowheads, Figure 6(o) and (p)).
Disrupted CX3CR1-CCR2 signaling attenuates neutrophil recruitment after CAIS
To determine the presence of neutrophils in injured regions, we characterized the number of Ly6Ghigh/Ly6C− cells in single cell population. tMCAO triggered accumulation of Ly6Ghigh/Ly6C− neutrophils in injured regions in both groups (Figure 7(a) to (c)), but the number of Ly6Ghigh/Ly6C− neutrophils was significantly attenuated in injured regions of mice with deficient CX3CR1-CCR2 signaling.
Lack of CX3CR1-CCR2 signaling attenuates neutrophil accumulation in injured regions 24 h after tMCAO. (a–c) Disrupted CX3CR1-CCR2 signaling attenuates neutrophil accumulation in injured regions. Representative examples of Ly6G/Ly6C plots gated on single cells in Cx3cr1G/+/Ccr2R/+ mice (a) and Cx3cr1G/G/Ccr2R/R mice (b). The number of Ly6Ghigh/Ly6G+/Ly6C− neutrophils is significantly increased by injury in both groups but to a significantly lower extent in mice with disrupted CX3CR1-CCR2 signaling compared to mice with functional CX3CR1-CCR2 signaling (c). Dots in (c) represent data from individual mice. Ipsi - Ipsilateral; C - Contralateral. Significance levels, as indicated on individual panels.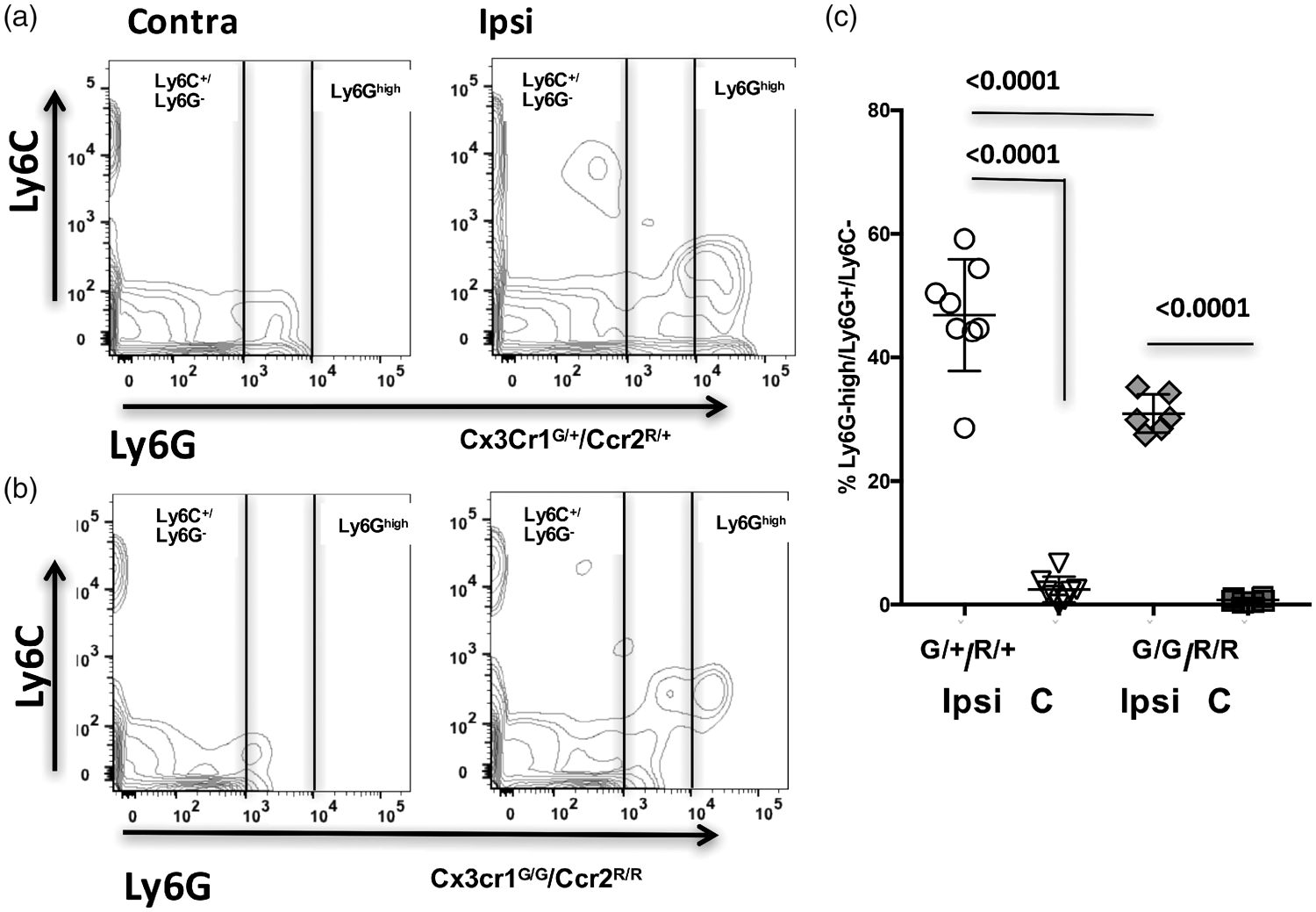
Taken together, these data demonstrate that CAIS triggers neutrophil accumulation in injured regions and that dysfunctional CX3CR1-CCR2 signaling markedly attenuates neutrophil recruitment.
Discussion
We demonstrate that the new age-appropriate mouse CAIS model produces functional deficits and histologically defined acute injury as well as chronic MRI-identifiable injury, brain atrophy and substantial loss and derangements in the vascular network. We then show that in contrast to previously established negligible albumin leakage and neutrophil infiltration in neonatal rodents after acute tMCAO, in juvenile mice, albumin leakage is significantly increased in the peri-focal region and neutrophils infiltrate into acutely injured regions, revealing that maturational step of postnatal brain development at the time of stroke induction affects neurovascular and immune responses. Furthermore, we demonstrate that disrupted CX3CR1-CCR2-dependent microglial/monocyte signaling contributes to albumin leakage, essentially aborts monocyte infiltration, alters microglial phenotypes, diminishes neutrophil adhesion/infiltration and reduces injury.
Epidemiologic data show that the incidence of CAIS is approximately 1 in 50,000, 8 that only 24% of children are neurologically normal after a stroke, and that 20–30% have recurrent strokes. Etiology and patterns of CAIS differ from those of perinatal stroke, in part owing to vascular damage. Inflammation is a major factor in stroke in all age groups, but inflammatory signatures depend on the maturational step of the brain.2,3 Increased blood levels of inflammatory biomarkers correlate with CAIS recurrence 44 and infection/inflammation frequently precedes CAIS. 10 MR angiography points to abnormalities in arterial wall in pediatric patients 11 and links inflammation and arteriopathy. 5 Vascular lesions in children are strongly predictive of CAIS occurrence and recurrence.5,6 We chose to mimic CAIS by inducing tMCAO in P21 mice, an age believed to represent brain development of a human toddler, 25 and to focus on the immune-neurovascular interactions as injury modifiers.
Literature has emerged that in normally developing rodent brain, BBB permeability does not linearly decrease with postnatal age13,14 and that unsynchronized changes in expression of several TJ proteins, continuously increasing astrocyte and pericyte coverage, evolving brain myelination and maturation of leukocytes and microglia during physiological postnatal brain development likely contribute to “windows of susceptibility” of the vasculature to injury during brain development.13,14 Comparison of BBB leakage between newborn, juvenile and adult rat brains following intra-cerebral IL-1β administration showed that BBB leakage in newborn brain is lower than in the adult and that BBB leakage in juvenile brain is strikingly higher than in both the newborn and adult brain. 13 Brain trauma or intra-striatal CINC-1 injection exert more severe brain edema, CBF dysregulation and injury outcome in juvenile than in adult rodents.17–19,45,46 Impaired vascular reactivity has been reported in pediatric brain trauma models in several species, 47 changes not necessarily associated with structural barrier breakdown. 19 In CAIS model, we demonstrate that monocyte/microglial CX3CR1-CCR2 signaling contributes to neurovascular disturbances and injury.
The relative contribution of monocytes and microglia in stroke pathophysiology remains unresolved, in part because of the lack of readily available experimental tools to discriminate these two cell subpopulations that share many similar pathways, 43 but are of different origin, 48 and, based on chimeric studies, may have distinct and even opposite effects in stroke. 23 While the dominant long-standing notion in the stroke field was that microglial cells are purely injurious, selective depletion of microglial cells was recently shown to exacerbate neuronal metabolism and injury in a murine adult stroke model. 22 Pharmacological depletion of the entire monocyte population, in turn, triggered hemorrhagic transformation and exacerbated injury. 49 Stroke studies in bone marrow (BM) chimeric mice deficient in inflammatory monocytes that infiltrate into brain post-stroke via CCR2-dependent mechanism and in BM chimeric mice that lack circulating CX3CR1+/Ly6Clow monocytes showed a complex step-wise temporal dynamics of monocyte recruitment into injured brain, with early infiltration of CCR2+/Ly6Chigh monocytes and transdifferentiation of these cells in the brain parenchyma over time. 50 The extent and the timing of recruitment of peripheral immune cells to the injured brain can play multifaceted role50,51 and greatly depend on spatio-temporal changes in brain cytokine and chemokine levels. Factors that induce IL-10 secretion by activated monocytes can attenuate integrin-mediated monocyte adhesion to endothelial cells and protect after acute stroke. 51
Given the demonstrated distinct roles of beneficial (Ly6G−/Ly6Clow) and toxic (Ly6G−/Ly6Chigh) monocytes, the pro-inflammatory nature of CCR2high monocytes, and a dynamic spectrum of injury-induced reprogramming of CCR2+ monocytes in the adult brain, 41 we characterized the presence of individual subtypes of cells of the monocyte lineage. In mice with functional CX3CR1 and CCR2 receptors, we observed significant accumulation of CCR2+/Ly6Chigh monocytes, monocytes involved in acute inflammation and injury in adult stroke models, and only a small fraction of CCR2+/Ly6Clow and 4C12+/CX3CR1+ “patrolling” monocytes, whereas inflammatory CCR2+/Ly6Chigh monocyte subpopulation was essentially abolished in deficient mice. Such distinct phenotypic makeup of monocyte subtypes would favor acute protection that we observed, especially in conjunction with increased levels of IL-10 and IL-4 in injured regions of deficient mice, cytokines known to ease injury and promote recovery. The effects of CX3CR1 and CCR2 deficiency on accumulation of other cytokines were rather specific, with a small but significant increase in cytokine TNFα, but not IL-1β, and markedly increased levels of several chemoattractants for monocytes and microglia, MCP-1 and MIP-1α, that were similar regardless of a CX3CR1-CCR2 function. Yet, several fold increase in monocyte chemoattractants did not induce infiltration of CCR2-deficient monocytes. The magnitude of increased neutrophil chemoattractant KC-1 in injured regions, in turn, was substantially lower in mice with deficient CX3CR1-CCR2 signaling, likely contributing to attenuated neutrophil infiltration and neurovascular leakage, and constituting another protective mechanism.
Further characterization of recruited monocytes using a unique monocyte-specific monoclonal antibody 4C12 demonstrated the presence of CCR2high4C12+ monocytes in injured brain regions of mice with functional CX3CR1-CCR2 signaling, but sparing of such cell population in injured brain regions of deficient mice. The contribution of CX3CR1high4C12+ monocytes is small (less than 4%) and is similar in injured regions of both groups. Monocytes infiltrated and differentiated into macrophages would be another cell population, but differentiation requires days42,50 and as such should be negligible within 24 h after reperfusion in our study. There were essentially no 4D4+/RFP+ cells. As expected, the cells were 4D4+/GFP+.
As immune cells in the brain, microglial cells have a broad scope of functions under physiological conditions, including brain surveillance, support of brain homeostasis and production of growth factors. They play unique physiological roles in the developing brain,52,53 including regulation of embryonic neural vasculogenesis and sprouting,54–56 controlling synaptic pruning and the formation of precise synaptic circuits,57–59 and supporting cortical layer formation via production of growth factors. 60 Microglial cells undergo marked maturational changes during postnatal brain development themselves, including reciprocal changes in expression of individual genes, such as decline of Apoe gene expression and increase in C1q, P2ry12, TGFβ1 and TGFbR1 gene expression between P4 and P21.43,61
CX3CR1 deficiency not only affects microglial function but also has direct implications for neuronal function in immature brain. In the early murine postnatal brain, CX3CR1 deficiency delays microglial recruitment, affecting maturation of postsynaptic glutamate receptors, 57 leading to increased dendritic spine density58,62 and defective distribution of particular subpopulations of interneurons in the developing brain. 63 In our study, the number and volume of GFP+ cells are unaffected by CX3CR1-CCR2 deficiency in the contralateral cortex and caudate at P22, whereas in the peri-focal cortical regions, the number of GFP+ cells is significantly increased compared to matching uninjured regions of Cx3cr1GFP/+ mice and to the number of GFP+ cells in injured regions of deficient mice, consistent with aborted Cx3cr1-dependant migration of microglial cells. In the ischemic core of Cx3cr1GFP/+ mice, the number of GFP+ cells is significantly reduced, while only a trend toward reduction is observed in deficient mice. Immunolabeling with a microglia-specific 4D4 antibody demonstrated that GFP+ population consists almost entirely of 4D4+ microglial cells (GFP+/4D4+) with a range of activation states.
Neutrophils are of vital importance for immunity against invading microorganisms and inflammation but can damage the brain, as demonstrated in adult experimental stroke64–67 and in humans. Yet, therapies targeting neutrophil egress have been largely ineffective.68,69 Neutrophils contribute to the age-dependent effects on BBB permeability, 13 and can modify injury in immature brain independently and in consort with monocytes. 3 In perinatal stroke, recruitment of neutrophils is low in part because of increased neutrophil chemoattractant levels in the systemic circulation. 14 In CAIS, we demonstrate marked neutrophil accumulation in mice with functional CX3CR1-CCR2 signaling but attenuated accumulation in mice with deficient CX3CR1-CCR2 signaling. The latter data support the notion of importance of neutrophils in stroke severity and of their interaction with peripheral monocytes. These data, however, do not allow discrimination of whether lessened neutrophil response is the direct consequence of attenuated monocyte activation/infiltration or because of diminished extent of elevation of neutrophil chemoattractant KC levels.
Many other factors can contribute to maturation-dependent BBB susceptibility to stroke injury and affect immune-neurovascular interactions and long-term CAIS outcomes. Our findings of the overall markedly reduced vascular coverage and preferential loss of smaller size vessels and capillaries in ischemic-reperfused regions are consistent with profound long-lasting vascular changes reported in a model of pediatric brain trauma.18,47 While we focus on consequences of disruption of particular pathways in microglia-monocyte function for BBB leakage and injury after acute CAIS, altered interactions will likely have broad range long-lasting effects. For example, microglia were demonstrated to regulate the density of oligodendrocytes precursor cells (OPCs) and myelination early postnatal 70 and possess “immune memory” of previous challenge.3,71,72 Injury could affect the renewal of microglial cells. While the majority of cortical microglia remains unchanged under homeostatic conditions, under injurious conditions, like unilateral facial nerve axotomy, clonal expansion of microglia with the formation and self-renewal of microglia can increase. 73 Some recent data revealed that while harmful role of Ly6Chigh and CCR2+ monocytes in adult stroke is well defined, immune preconditioning by low dose LPS protects adult mice from MCAO via induction of Ly6Chigh in monocytes and reprograming of gene expression in monocytes, with CCR2 playing an essential role in neuroprotection. 74 These data raise a possibility that the magnitude, the timing and the phenotypes of infiltrated and differentiated monocytes can be tuned to support long-term recovery but it is unknown whether similar scenario exists in injured juvenile brain. An improved understanding of leukocyte/neurovascular/microglial axis would shed light on how to enhance vascular remapping and brain connectivity after CAIS.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work was supported by RO1 NS44025 (ZV), RO1 NS76726 (ZV), R21 NS98514 (ZV), R01 NS103483 (ZV, AO).
Acknowledgements
We thank Dr. David Fernandez-Lopez for his contribution to earlier stages of the project and Ms. Elodie Blanchard and Ms. Shivani Mahuvakar for help in cutting the brains.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
JF, SC – performed experiments and data analysis, contributed to manuscript writing.
ND – established animal model and performed experiments and data analysis. Mr. Derugin died but his contribution was essential for the study.
AJ, MH, EH – performed experiments and data analysis, contributed to manuscript editing.
OB – created antibodies used in the study, discussed the results with Dr. Vexler and contributed to manuscript writing and editing.
AO – performed statistical data analysis of MRI data obtained in his laboratory, wrote portion of the manuscript and contributed to editing of the manuscript.
ZV – senior author. Designed, supervised/oversaw the entire study, performed statistical data, wrote manuscript.
*These authors contributed equally to the work.