
Abstract
The incidence of perinatal stroke is high, similar to that in the elderly, and produces a significant morbidity and severe long-term neurologic and cognitive deficits, including cerebral palsy, epilepsy, neuropsychological impairments, and behavioral disorders. Emerging clinical data and data from experimental models of cerebral ischemia in neonatal rodents have shown that the pathophysiology of perinatal brain damage is multifactorial. These studies have revealed that, far from just being a smaller version of the adult brain, the neonatal brain is unique with a very particular and age-dependent responsiveness to hypoxia–ischemia and focal arterial stroke. In this review, we discuss fundamental clinical aspects of perinatal stroke as well as some of the most recent and relevant findings regarding the susceptibility of specific brain cell populations to injury, the dynamics and the mechanisms of neuronal cell death in injured neonates, the responses of neonatal blood-brain barrier to stroke in relation to systemic and local inflammation, and the long-term effects of stroke on angiogenesis and neurogenesis. Finally, we address translational strategies currently being considered for neonatal stroke as well as treatments that might effectively enhance repair later after injury.
INTRODUCTION
Perinatal arterial ischemic stroke is common and produces a significant morbidity and severe long-term neurologic and cognitive deficits, including cerebral palsy, epilepsy, neurodevelopmental disabilities, behavioral disorders, and impaired vision and language function.1–4 More than half of all children with cerebral palsy are born at term and in many instances the etiology is related to some form of cerebrovascular focal or global insult. 5 The pathophysiology of perinatal brain damage has proven to be multifactorial. 6 With the availability and safe use of magnetic resonance imaging (MRI) in newborns, older infants, and children over the past two decades, much has been learned about how the stroke phenotype differs in these age groups compared with adults, as well as the underlying causes and pathophysiology. From an experimental perspective, stroke models in neonatal animals are technically very challenging when one considers the surgical difficulties based on size (e.g., body weight of a postnatal day 7 (P7) rat pup is <15 g and of a P9 mouse pup is <5g). Therefore, the first and most commonly used model to mimic ischemic brain injury in term neonates is an hypoxia–ischemia (H–I) model in P7 to P12 rats and mice, which consists of ligation of the common carotid artery followed by systemic hypoxia. 7 This model more commonly is used to mimic ‘global’ hypoxic-ischemic encephalopathy (HIE) in term human babies although models of bilateral H–I may be more advantageous for this purpose. 8 More recently, age-appropriate models of focal arterial stroke involving middle cerebral artery (MCA) occlusion (MCAO) have been developed to mimic pediatric 9 and neonatal10–13 focal arterial stroke. We will briefly review some of the salient clinical aspects of neonatal stroke, describe some of the known differences compared with childhood stroke, and discuss in more detail current experimental knowledge regarding both distinct and common pathophysiologic mechanisms of perinatal stroke as compared with those in childhood and adult stroke. We will then discuss how maturational differences may affect ways to protect the neonatal brain and enhance long-term repair.
BRAIN IMMATURITY AT BIRTH AFFECTS THE PATTERNS OF ISCHEMIC INJURY
The maturational stage of the brain at the time of injury is a key factor in the pattern of brain damage, including regional and cell type-specific susceptibility. In preterm newborns, brain injury associated with hypoxia–ischemia generally coincides with a time window of high susceptibility of oligodendrocyte progenitors (OLPs) to excitotoxicity, oxidative stress, and inflammation,14,15 adversely affecting OLP differentiation into mature, myelinating oligodendrocytes. The arrest in OLP differentiation in turn predisposes the brain to defective white-matter tract development, including periventricular white-matter injury and cerebral palsy.16–18 Subplate neurons, which exist transiently during human fetal brain development, are involved in the formation of visual thalamocortical projections, and are another cell population selectively vulnerable to H–I in preterm babies that leads to long-term deficits. Several reviews have discussed the unique features of fetal brain neuroinflammation and injury in humans and in corresponding injury models.17,19,20
The patterns of ischemic injury in full-term newborns are different from those in preterm newborns; 6 injury is no longer diffuse and is manifested focally in gray-matter regions, most commonly in striatum, thalamus, and cortex.
ETIOLOGY OF PERINATAL STROKE
Perinatal stroke is defined as an insult occurring from 20 weeks gestational age, including in utero, to 28 days postnatally. 1 Subcategories within perinatal stroke include neonatal arterial ischemic stroke, neonatal cerebral sinovenous ischemic stroke, and presumed perinatal stroke. Perinatal arterial ischemic stroke, the primary focus of this review, has been defined more specifically by the NIH Workshop on Perinatal Stroke as a condition with acute encephalopathy, seizures, or neurologic deficit presenting in the term or preterm infant before the 29th postnatal day, with brain imaging confirming a parenchymal infarct in the appropriate arterial territory.4,21
Perinatal stroke occurs in 1 in 2,300 to 1 in 5,000 live infant births,1–4 and the estimated mortality rate of neonatal stroke is 3.49/100,000 annually. 2 These likely are underestimations, as to some degree neonatal brain imaging may be less commonly acquired in remote or low resource settings.
The maternal-fetal dyad, present in pregnancy and at delivery, gives rise to specific etiologic considerations with regard to neonatal stroke. Pathologic changes resulting in gestational diabetes, pregnancy-induced hypertension, and preeclampsia have long been linked to neonatal stroke. 22 Preeclampsia reduces placental blood flow, resulting in fetal cerebral hypoperfusion and the potential for emboli or global or focal ischemic injury. In case control studies, factors present at delivery including fetal bradycardia, fetal decelerations, prolonged rupture of membranes, or prolonged second stage of labor are associated with neonatal stroke, although it remains unclear whether these represent a causal relationship, or are reflective of intrauterine or ongoing stroke. Additional risk factors associated with difficult delivery such as vacuum delivery and emergency Caesarian section are associated with an increased risk of neonatal stroke. A recent study identified an Apgar score of <7 at 5 minutes, as well as maternal fever >38°C and hypoglycemia as independent risk factors for perinatal stroke.23,24
Increasing evidence suggests that the placenta has a significant role in neonatal neurologic disorders. Placental abruption with resultant decreased perfusion has been associated with neonatal stroke as well as HIE. A study of placental pathology in neonates with perinatal arterial ischemic stroke showed evidence of chronic poor perfusion of the placenta with acute changes at the time of delivery. 25 Additionally, placental chorioamnionitis even in the absence of maternal symptomatology is associated with neonatal stroke. Previous studies in which infants and children were diagnosed with cerebral palsy, including hemiplegic cerebral palsy, later in life and in whom the placenta was available for pathologic examination, often showed a high incidence of chronic infarctions, vascular pathology, anomalies of umbilical cord insertion, or the length of the umbilical cord or vessels.26,27
Pregnancy is considered to be a natural prothrombotic state. 4 Pregnancy is associated with changes in hemostasis due to evolutionary changes to protect the pregnant mother from fatal hemorrhage at the time of delivery. These changes predispose the mother to thromboembolism and place the fetus and the placenta at risk for thromboemboli. During pregnancy, coagulation factors V, VII, VIII, IX, X, XII, and von Willebrand factor and plasma fibrinogen concentrations significantly increase. 28 Concomitantly, fibrinolytic activity is decreased in pregnancy, and returns to normal within 1 hour after placental delivery. Tissue plasminogen activator decreases, while PAI-1 activity increases. 28 Additionally, consumption of cocaine during pregnancy places the fetus at risk for arterial stroke due to vasoconstriction and vasospasm. Vasospasm increases the risk of in utero stroke in the first and third trimester. 29
The presence of at least one prothrombotic factor substantially increased the incidence of stroke from 24% in neonates in the control group to 68% of 91 term neonates. 30 Increased lipoprotein (A) is considered as the most important prothrombotic risk factor in newborns. Other common prothrombotic factors associated with neonatal stroke include genetic polymorphisms of MTHFR (C677T), Factor V Leiden (G1691 A), and prothrombin (G20210A).30–34 Acquired prothrombotic states of anticardiolipin antibodies, activated protein C or protein S deficiency, or antithrombin deficiency also have been linked to neonatal stroke.31–34 These factors in combination with other triggering factors such as neonatal septicemia, perinatal asphyxia, or patent foramen ovale may potentiate the risk of stroke. Intracerebral hemorrhage accounts for 10% to 15% of stroke and is a particularly severe form of stroke. Mutations of COL4A1 have been shown to cause intracerebral hemorrhage in mice and humans.35,36 COL4A1 encodes the alphal chain of type IV collagen (Cola1), a major component of basement membrane. COL4A1 mutant mice and humans with COL4A1 mutations show increased sensitivity of brain vessels to antithrombotic agents and markedly increase the incidence of intracerebral hemorrhage that is associated with birth trauma.
Infants with congenital heart disease are at a higher risk for perinatal stroke although the incidence is not clearly known. 37 Additionally, there is an increased incidence of stroke in the setting of cardiac surgery. With the incidence of 1 in 125 newborns having congenital heart disease, cardioembolic events resulting in stroke are another potential etiology. In contrast, one case control study with a relatively small number of newborns with stroke found that none had associated congenital heart disease. 5 There is currently no consensus regarding the evaluation of neonates suspected of having a stroke. Recent reviews and workshops have suggested various diagnostic approaches.4,38,39
The rate of neonatal stroke recurrence is low (i.e., <3%), and recurrence typically occurs in neonates who have prothrombotic factors or congenital heart disease. 40 Recurrence rates are higher in neonates with congenital heart disease, presumably due to a combination of factors including the type of congenital heart disease, the peri-operative cardiovascular status of the neonate, and the potential risk of complication from the needed surgical interventions such as cardiac reconstruction or cardiac bypass.
CLINICAL PRESENTATION OF PERINATAL STROKE
Most babies with neonatal stroke present during the first 3 days of life with symptoms including focal seizures, apnea, chewing or bicycling movements, and persistent feeding difficulties. Many newborns will have no lateralizing findings on examination. A variable percentage of neonates will have focal findings such as tone differences or an asymmetric Moro response. Hemiplegia is rare, and evolves over time. This has been attributed to the structural and functional changes in the ipsilateral corticospinal tract in the immature brain at the time of injury. Studies utilizing transcranial magnetic stimulation in infants with hemiplegic cerebral palsy have found that the ipsilateral corticospinal tract hypertrophies, when corticospinal tract injury is present in the contralateral hemisphere. 41
Stroke more frequently affects the left hemisphere and often involves the MCA territory. The International Pediatric Stroke Study has shown that perinatal stroke occurs in the anterior circulation 70% of the time and in 73% of newborns the left hemisphere was affected. 6 It has been suggested that the origin of the left carotid artery from the aorta allows a more direct vascular route to the brain as a corridor for cardiac emboli.42,43 Other considerations take into account the fetal circulatory system or the presence of a patent ductus arteriosus or patent foramen ovale.
Perinatal stroke may be missed initially if findings on examination are subtle or absent, but if new symptoms such as pathologic early handedness, hemiplegia, neurodevelopmental delay, or spasticity appear later in the first year of life, the condition will be described as ‘presumed perinatal ischemic stroke’. Newborns who present with seizures secondary to stroke may be nonencephalopathic and appear normal in the hours before the onset of seizures.
Epilepsy or motor impairment occurs in approximately one-half to two-thirds of injured neonates.4,22,23,44–47 Recent reports on the occurrence of recurrent seizures after perinatal arterial ischemic stroke has ranged between one-quarter and two-thirds of patients, with variable responses to therapy,47–49 with larger stroke size associated with the likelihood of seizures beyond the acute period. 49 Later development of infantile spasms is also an important complication of epilepsy due to perinatal stroke, accounting for 5% to 8% of cases.50,51
Motor outcomes in neonates with stroke are largely dependent on the location of injury. The internal capsule as well as the deep gray matter involving the basal ganglia more often is associated with neuromotor disability.52,53 Cortical involvement in the MCA distribution with involvement of the basal ganglia and the internal capsule is associated with hemiplegia, while those with basal ganglia and internal capsule without cortical involvement also may have motor coordination difficulties. 54
Magnetic resonance imaging has played a pivotal role in detecting stroke in term neonates.6,55 Diffusion-weighted MRI early after injury has identified the presence of severely injured regions as well as regions with more modest diffusion restriction, ‘the ischemic penumbra’, with higher likelihood of recovery in the penumbra (Figure 1A). Advanced MRI methods have been developed to quantitatively measure several aspects of brain development. For example, diffusion tensor imaging has become a standard technique to measure the direction and magnitude of water movement, which becomes more restrictive with increasing development of brain microstructure such as neuronal dendritic structure, axonal connections, and myelination. 57 Diffusion tensor imaging fiber tractography has enabled delineation of abnormalities in the white-matter fiber tracts of injured newborns.58,59 Optical imaging is an emerging method that allows mapping of the structural connectivity of the developing human brain (brain ‘connectome’). 60
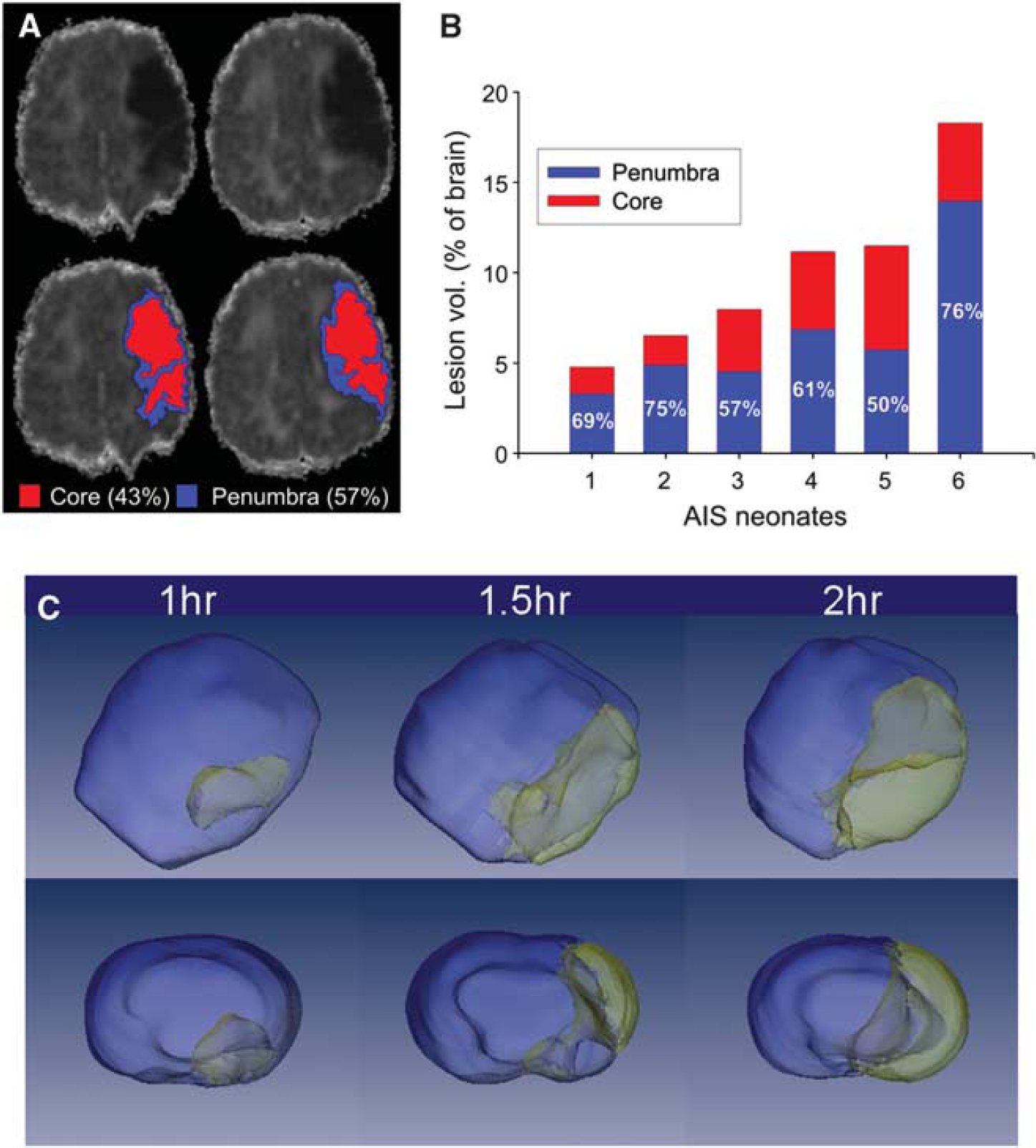
Magnetic resonance imaging (MRI)-based delineation of the ischemic core and penumbra. (
ETIOLOGY AND PATTERNS OF PEDIATRIC STROKE DIFFER FROM THOSE OF PERINATAL STROKE
Epidemiologic data from the International Pediatric Stroke Study show that the incidence of pediatric stroke is ~1 in 50,000 61 , that ~50% of strokes in children are hemorrhagic, that the majority of the remaining strokes are arterial ischemic strokes, 55 that only 24% of children are neurologically normal after a stroke, and that 20% to 30% have recurrent strokes.
In contrast to neonates, children with stroke present with the acute onset of weakness or altered mentation; only one-fourth will present with seizures.62–64 In comparison, the incidence of seizures as a presenting symptom is ~3% in adults with stroke 65 While the recurrence risk in neonatal strokes is low, the risk of recurrence for older children is nearly 20%. 66 Stroke etiology in children differs from strokes in adults or in neonates, in part owing to a higher incidence of certain arteriopathies. 67 Vascular damage associated with infection/inflammation is an additional risk factor. 68 Pediatric stroke may result from arteriopathies due to arterial dissection, moyamoya disease, as well as focal cerebral arteriopathies. Ongoing research continues to explore the role of inflammatory or infectious causes such as varicella or meningitis in pediatric arteriopathies. Moyamoya disease and infarction due to sickle cell disease occur in pediatric but not in neonatal stroke. Additional etiologies, similar to those found in neonates with stroke, include inherited and acquired thrombophilias and various subtypes of congenital heart disease. The Canadian Pediatric Stroke Registry found that 25% of children with stroke had underlying cardiac involvement. 6
Neurodevelopmental outcomes in pediatric and neonatal stroke are distinguished not only by the differences in motor outcomes, but also by cognitive outcomes such as perceptual reasoning, working memory, processing speed, and intellectual ability. In a study of 145 children, cognitive outcomes were compared in children with perinatal, early childhood, and later childhood onset arterial stroke. 69 The perinatal subset had poorer neurodevelopmental outcomes compared to children with later stroke onset regardless of lesion location; neonates with subcortical lesions performed significantly worse than those with childhood onset stoke, while conversely, cortical lesions led to greater cognitive impairment in childhood onset stroke compared with perinatal stroke. 69
In a prospective study comparing MRI and subsequent motor function, hemiparesis was found in 24% of neonatal and 56% of pediatric stroke patients, with comparable distribution of strokes. 70 The explanation for the different types of motor disability has been suggested to be related to brain/vascular maturational differences at the time of injury. In normal infants without stroke, transcranial magnetic stimulation of the corticospinal tract resulting in contralateral and ipsilateral responses, typically are extinguished by 18 months of age. In infants with neonatal stroke, this ipsilateral response may remain in an attenuated state, suggesting cortical reorganization.70,71 As this ipsilateral response extinguishes with normal development, subsequent brain injury after childhood stroke may not allow reorganization in such a robust manner.
ANIMAL MODELS AND THE UNDERLYING MECHANISMS OF PERINATAL ARTERIAL STROKE
Numerous models have been developed in fetal and neonatal nonhuman primate, sheep, lambs, piglets, rabbits, and rodents to elucidate the pathophysiology of brain damage relevant to term and preterm infants. 72 Although human physiology is more closely related to that of larger animals than to rodents, brain maturation at birth in most of these species corresponds to the developmental stage of the fetal human brain. 72 This limitation, along with practical issues, has limited the use of larger species for studying mechanisms of neonatal stroke.
Models of permanent MCA ligation 11 and transient MCAO (tMCAO) induced in P7 to P10 rats10,12,73 accomplished by a transient insertion of a suture filament enabled studies of perinatal arterial stroke. Varying the length of tMCAO has allowed production of injuries of different severity.73–75 The presence of recirculation in the tMCAO model76,77 also serves to mimic reperfusion, which frequently occurs in arterial stroke in term babies.
Diffusion-weighted MRI has been instrumental in delineating early injury, in experimental neonatal models of focal stroke, including the ischemic core and penumbra.13,74,78,79 Figure 1B shows that diffusion-weighted MRI depicts expansion of tissue ‘at risk’ associated with longer MCAO duration. Other MRI modalities have been proven useful in determining the extent of micro-circulatory disturbances during MCAO and after reperfusion (perfusion-sensitive MRI), 76 disruption of the blood-brain barrier (BBB) (Gd-enhanced T1W imaging), 76 elucidation of the evolving injury (T2W imaging),56,73,80 and changes in brain connectivity after injury during the neonatal period (diffusion tensor imaging). 81
Studies in tMCAO and H–I models in neonatal rodents have shown that although the components of injury that induce cell death and brain injury are similar to those occurring in experimental models of preterm injury—excitotoxic, oxidant, and inflammatory components—the targeted cell populations and regions affected by cerebral hypoxia and ischemia are different. Excitotoxicity, triggered by accumulation of glutamate and other excitotoxic molecules in extracellular spaces after neuronal membrane depolarization, glutamate efflux, and failure of its uptake, is a common initial damaging process caused by focal or global H–I. 82 The neonatal brain is more excitable and prone to oxidative stress than the adult brain due to higher levels of glutamate receptor expression83,84 and the different composition of individual NMDA receptor subunits, 85 and interaction with downstream signaling cascades. 86 Several other distinct characteristics of the immature brain, such as high oxygen consumption, higher iron levels, and lower expression of several endogenous antioxidant enzymes, contribute to the higher vulnerability of the immature brain due to the deleterious actions of free radicals. 87 However, overexpression of antioxidant enzymes does not necessarily protect the neonatal brain against H–I, as was shown for superoxide dismutase overexpressing pups, and is dependent on the balance of free radical scavenging pathways88,89 and cell types. 90 Inflammation, the third major contributor to neonatal brain injury can induce neuronal death by itself as well as by enhancing excitotoxicity and oxidative stress through the release of cytokines, free radicals, and other toxic products or trigger release of excitotoxic molecules, including glutamate. 91
MODES OF CELL DEATH IN THE INJURED NEONATAL BRAIN
Several concepts have emerged regarding the complexity of mechanisms underlying neuronal death in the injured immature brain. Neuronal apoptosis is commonly observed and may be quite severe after global 92 or focal ischemia-reperfusion 74 and declines with maturation. 93 Apoptotic pathways are more readily activated in the immature brain in part due to the high expression of many of the key components in apoptotic pathways that counteract neuronal overproduction during early brain development (programmed neuronal death). Although necrosis has been typically considered as the predominant early neuronal death mechanism in the ischemic core, and apoptosis and autophagy have been related more to delayed cell death in the ischemic penumbra, 94 apoptosis is an important contributing neuronal death mechanism in the core after neonatal focal stroke. 74 While pharmacological inhibition of caspase-3 protects the neonatal brain against H–I, 92 a lack of caspase-3 exacerbates injury via amplification of necrosis and caspase-3-independent injury, 95 suggesting that complete abolishment of caspase-3 activity can be injurious rather than beneficial. The modes and timing of cell death may depend on the cell type. For example, death of OLPs via caspase-3-dependent mechanisms is relatively low early after H–I, whereas a more delayed and sustained increase in OLP cell death is largely caspase-3 dependent. 96
Mitochondria have a central role in modes of cell death and the ability to attenuate mitochondrial responses by counteracting oxidative stress via modulation of expression of proteins within the apoptotic pathways has been shown.97,98 Both the extrinsic (cell death receptor dependent) and intrinsic (mitochondria dependent) apoptotic pathways are activated in response to neonatal brain injury. Inhibition of apoptotic effectors downstream from death receptors, such as caspases, has resulted only in partial protection.99–101 It has been suggested that autophagy may precede apoptosis after neonatal brain ischemia. 102 Importantly, failure to complete apoptosis results in programmed cell necrosis (also known as necroptosis), an intermediate form of cell death that exhibits features of both necrosis and apoptosis.94,103 Coexistence of several cell death mechanisms (i.e., necrosis, apoptosis, and autophagy) in the injured neonatal brain leads to the presence of a ‘continuum’ of hybrid cell death states and is thought to be unique to the newborn. 94
GENDER AND PERINATAL STROKE
Many central nervous system diseases display sexual dimorphism. Stroke, cerebral palsy, and related developmental disorders are more common in males than in females,104,105 but the reasons for these differences remain uncertain. Sex hormones can provide protection against ischemic injury, but the neonatal brain may not be as influenced by these hormones as the adult brain. Gender predominance in the mechanisms of caspase-dependent and -independent apoptotic death has been shown after neonatal H–I106,107 and focal ischemia. 108 While pharmacological inhibition of poly ADP ribose polymerase prevents neuronal death after H-l preferentially in male pups, 106 inhibition of caspase-3 primarily protects female pups, 109 There is also increasing evidence that the response to therapy may be gender dependent, as has been shown for protection of female but not male neonates against H–I by an inducible nitric oxide synthase inhibitor 2-iminobiotin. 110 A recent study did not observe gender differences in short-term injury outcome in tMCAO in a P20–P25 mouse model but did show that exogenously administered estrogen preferentially protected female but not males, 111 pointing out that gender and sex hormones may have independent effects on stroke in adults as well as in children.112,113 The existence of intrinsic gender-specific differences in cell death pathways in the neonatal period warrants further study to better understand the need for gender-specific therapies in neonatal and childhood stroke. A possible role of gender in stem cell treatment for neonatal HIE and stroke is being investigated. 114
NEUROINFLAMMATION AND INJURY
Clinical and translational studies have shown that inflammatory responses after stroke are different in neonates compared with adults.91,115 For example, underdeveloped nonclassical pathways of complement activation are present in term infants as well as in rat pups116,117 likely have important age-dependent effects. Genetic deletion of various major mediators of inflammation, such as NADPH oxidase, 118 interleukin 1β (IL-1β), IL-1α, or both IL-1αβ 119 , is not neuroprotective in the neonatal brain, an effect opposite to that observed in adults. Inhibition of NFkB activation produces opposite effects after H–I, depending on the duration and timing of inhibition. 120 While data on the injurious role of toll-like receptor activation on the development of the immature brain are emerging, 121 the extent to which long-lasting injury depends on receptor upregulation as opposed to the expression of appropriate ligands during early postnatal brain development has not yet been established.
Microglial activation has been traditionally considered as injurious in the ischemic brain due to their production of inflammatory mediators, free radicals, and other toxic molecules. 122 This classic view of activated ‘cytotoxic’ microglia/macrophages, the ‘bad guys’ in the ischemic brain, is supported by numerous studies showing that inhibition of inflammatory pathways, which are typically activated in microglia, results in neuroprotection in adult stroke and neonatal H–I animal models.91,122 However, more recent data suggest that microglia may have a dual role after stroke, injurious or beneficial, depending on the heterogeneity of the macrophage population, the origin of these, resident microglia versus infiltrated monocytes, as well as their phenotypes. Microglia also produce mediators that can initially be harmful but later enhance repair through remodeling of the extracellular matrix during the chronic recovery phase after stroke. 123 Although it is well established that the number of macrophages is markedly increased 1 to 4 weeks after H–I, little is known about the effects of individual microglial phenotypes and the relative contribution of microglia and invading differentiated monocytes after neonatal brain injury. One study showed that the macrophage population predominantly consists of activated microglia, not invading peripheral monocytes, within the first 24 hours after tMCAO in neonatal rats. 124 Selective depletion of microglia before induction of tMCAO in neonatal rats revealed that, far from reducing injury, the absence of microglia leads to increased brain levels of inflammatory mediators (Figure 2) and larger infarct size. 90 This study also showed that when microglia are absent, cytokine and chemokine synthesis and release are increased in other brain cell types. These data suggest that at least a subpopulation of activated microglia acts as a ‘buffering’ component in regulating the ‘pro’ and ‘anti’ neuroinflammatory response induced by neonatal stroke. Removal of apoptotic debris after neonatal stroke is another potential beneficial effect of microglia. Depletion of these cells or genetic deletion of the scavenger receptor CD36, a receptor that participates in the recognition and engulfment of apoptotic cells by microglia and macrophages, worsens injury and leads to accumulation of apoptotic neurons that die by caspase-3-dependent mechanisms.13,90 The distinct patterns of superoxide accumulation in microglia/macrophages after acute adult 125 and neonatal13,90 stroke may be central to the age-related differences in stroke mechanisms.
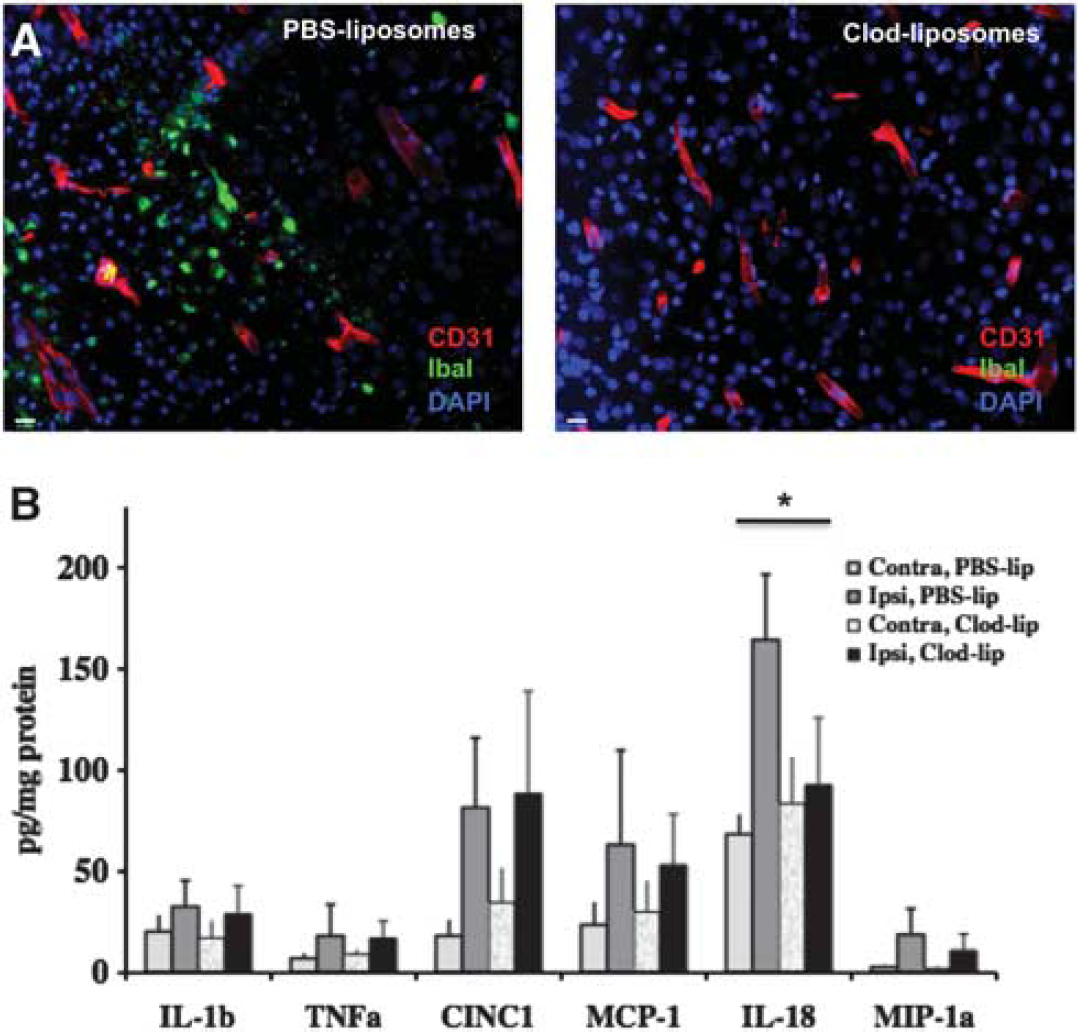
Depletion of microglial cells before a transient 3 hour middle cerebral artery occlusion (MCAO) in postnatal day 7 (P7) rats does not reduce the levels of several cytokines and chemokines elevated by injury induction. (
As a rapid responder to alterations in the local microenvironment, microglia ‘monitor’ the changes in BBB homeostasis and leakage of plasma components into the brain, preserving vascular integrity, as was shown in adult models.126,127 Macrophages also promote vasculogenesis during brain development, 128 but the role of microglia in neurovascular function after neonatal stroke is unknown.
SYSTEMIC INFLAMMATION AND BLOOD-BRAIN BARRIER INTEGRITY
Epidemiologic studies have shown a strong association between fetal infection and inflammation (e.g., chorioamnionitis) with brain damage in the newborn or neurologic impairments in survivors.129,130 The sensitizing effect of systemic inflammation on perinatal brain lesions induced by H–I or excitotoxic insults has been shown but the mechanisms underlying these effects are not yet fully understood, particularly as to how BBB function is altered.
In the adult, disruption of the BBB has a key role in injury after stroke due to entrance of unwanted molecules and cells from the peripheral circulation. However, many drugs that cannot access the brain through the intact BBB are able to enter through disrupted BBB and act centrally. Emerging evidence suggests that the early postnatal BBB is not as permeable as once thought. Mechanisms of BBB function in the fetus are different from those in the adult. 131 Tight junctions are present early in embryonic development, restricting entrance of proteins into the brain, while influx and efflux transporters are present during midgestation. At birth, the BBB is functional with no fenestrations in most brain regions. 132 Blood-brain barrier endothelial proteins undergo major changes in expression from the postnatal period to adulthood. 77 As pericyte and astrocyte coverage increases with brain maturation, their control of neurovascular integrity in the normal brain increases. 133 However, BBB permeability after injury does not decrease linearly with age, as was shown by the finding that BBB disruption is much higher after a local inflammatory challenge (i.e., intrastriatal IL-1β injection) in P21 rats than in 2-hour-old rat pups. 134
A recent comparative study provided direct evidence for the different functional BBB response to acute experimental stroke between neonates and adults. 77 Extravasation of albumin that circulated for 20 hours starting at 2 hours after reperfusion was shown to be increased in the injured adult rat brain (5- to 25-fold), but, in stark contrast, only a 2-fold increase occurred in the injured cortex and caudate in the neonate. 77 Blood-brain barrier permeability to intravascular tracers of various sizes, from large (~65 kDa) to relatively small (560 Da), also remained low in the cortex and caudate of neonates after acute injury, while leakage and brain accumulation of tracers are significantly increased in these regions in adults. 77 The transcriptional profiles in endothelial cells isolated from injured and noninjured adult and neonatal tissue in part explain the observed age differences in BBB susceptibility to stroke by showing that the patterns of upregulated and downregulated endothelial genes are largely nonoverlapping between the two ages (Figure 3), and that gene and protein expression of several tight junction components and basement membrane components (collagen IV and laminin) is better preserved in neonates. 77 Other studies observed increased BBB permeability during a subchronic injury phase after tMCAO in P7 rats 77 and P10 rats. 78 The affected region was larger in spontaneously hypertensive pups 78 than in normotensive pups of the same age. 135 As expected, permanent MCAO in P7 rats resulted in rapid BBB disruption and leukocyte extravasation, 136 suggesting that persistent lack of cerebral microcirculation contributes to the BBB collapse. Together, these data suggest that intrinsic developmental differences in basement membrane and extracellular matrix formation may contribute to a better preserved BBB integrity after acute neonatal arterial stroke.
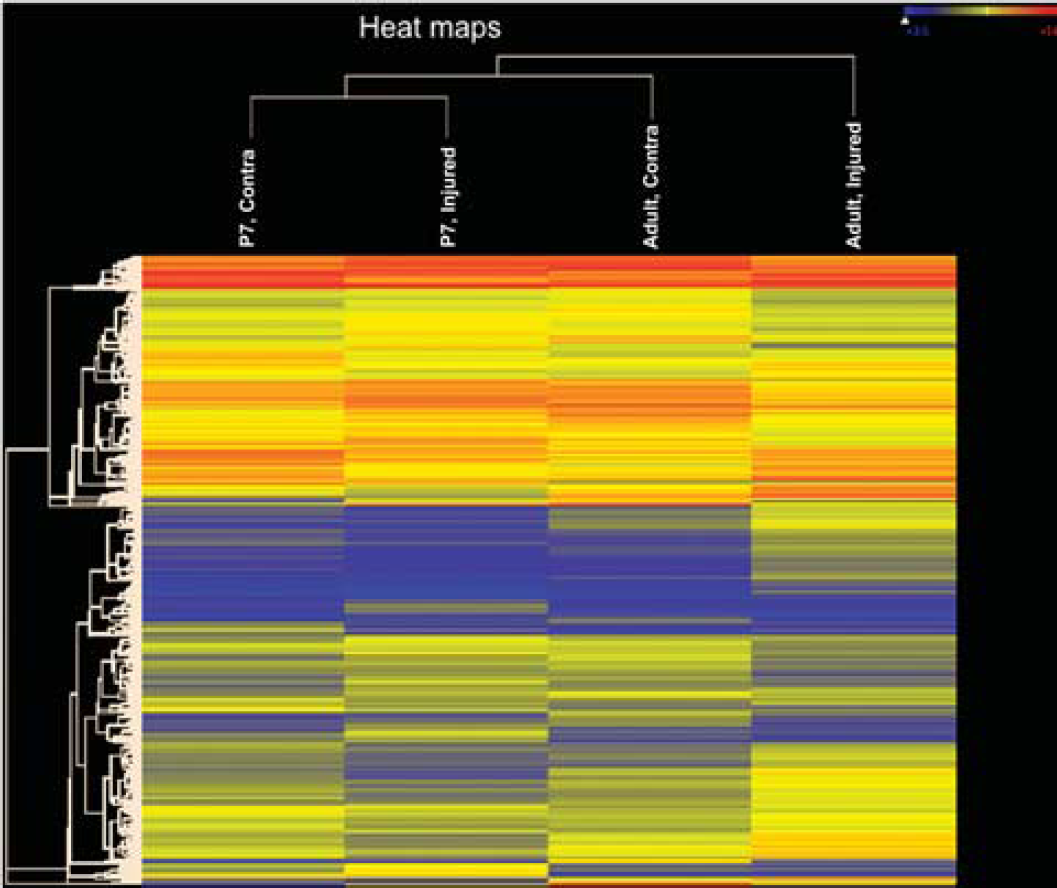
Stroke induces rapid nonoverlapping changes in gene expression in endothelial cells within injured regions in adults and neonates. Heatmaps obtained in endothelial cells isolated from injured adult and neonatal brains 24 hours after transient 3 hours MCAO, as described in Fernandez et al. 77 Heatmap visualization from a total of 31,042 probe sets shows that the expression levels of endothelial genes are markedly changed in injured regions compared with the corresponding contralateral anatomic regions in the same rats in each age group but that the patterns of changes are distinct after adult and neonatal stroke. The expression levels of genes are indexed by color.
Interaction of endothelial cells with peripheral leukocytes serves as an important contributor to BBB disruption (Figure 4). Both animal and human studies in the adult show that neutrophils infiltrate the brain and contribute to BBB disruption after transient cerebral ischemia (Figure 4B) and that neutropenia is neuroprotective.137,138 In contrast to adults, neutrophil infiltration in neonates is negligible after tMCAO 77 and is very limited 139 or brief 140 after H–I. Although the exact mechanisms that restrict neutrophil infiltration in the injured neonatal brain are yet to be understood, and it remains unclear whether the higher resistance of the neonatal BBB to stroke is a cause or a consequence of reduced leukocyte infiltration, the different patterns of expression of adhesion molecules and matrix metalloproteinases between neonatal and adult rodents, along with changing chemokine gradients between the brain and the blood, may limit neutrophil extravasation in the neonate. 77
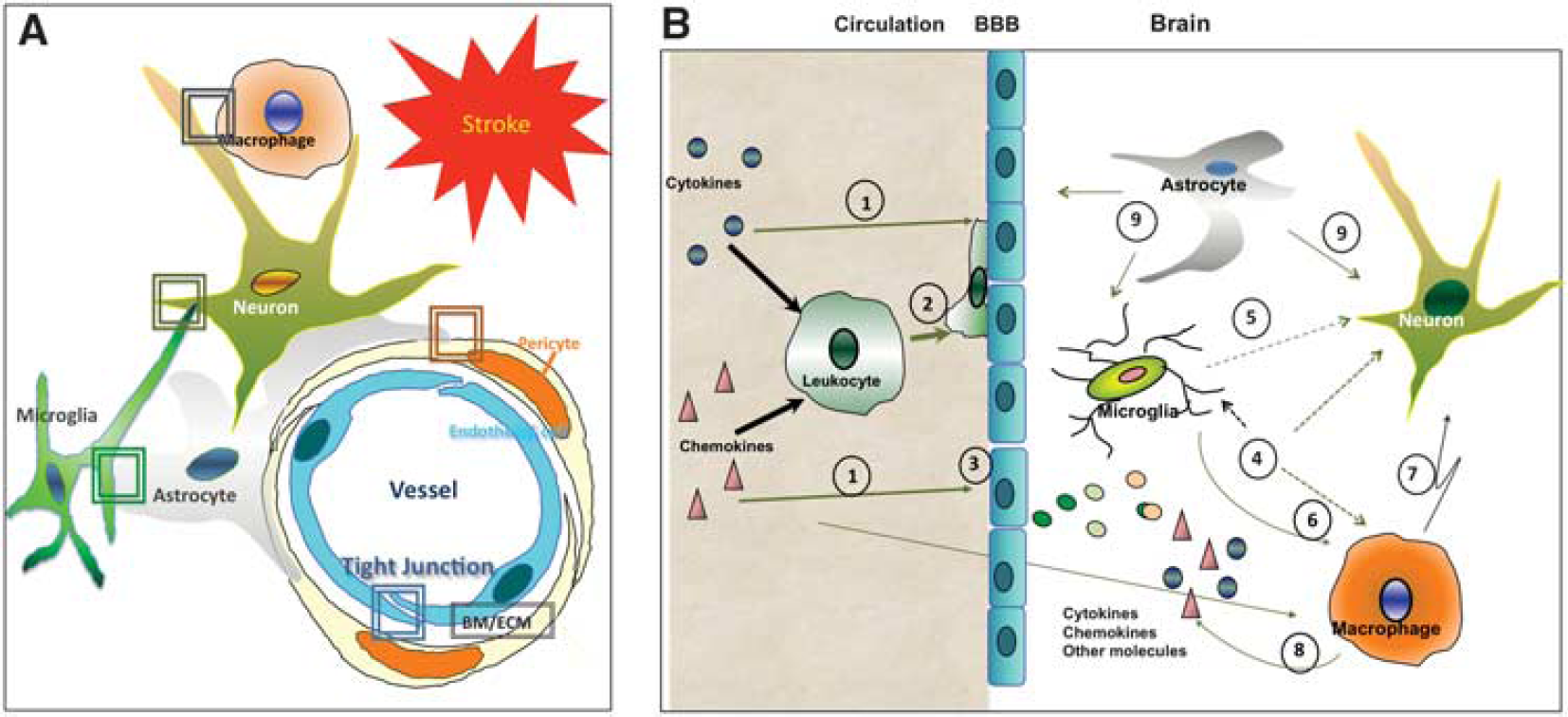
A schematic diagram that shows major differences in the response of the blood-brain barrier to acute stroke between adults and neonates. (
Astrocytes and pericytes, cells known to participate in the maintenance of vascular impermeability, may contribute differently to neonatal and adult stroke (Figure 4A). In adults, astrocyte swelling, mediated by water channels (aquaporin-4) present in astrocyte end feet, is one of the mechanisms involved in the generation of cytotoxic edema during the early phase after stroke. 141 Retraction of astrocyte end feet from the parenchymal basal lamina of vessels and cellular redistribution and/or degradation of aquaporin-4 at later stroke stages are involved in BBB leakage and formation of vasogenic edema. 142 While coverage of vessels with astrocytic end feet begins before birth, it continues to increase during the first postnatal week. 133 However, these particular mechanisms have not yet been specifically characterized after neonatal stroke. Pericytes are another important cell determining the adequate formation and maintenance of the BBB.133,143 Pericyte coverage continues to increase during postnatal brain maturation and may affect the interactions of these cells with brain vessels in distinct ways after neonatal versus adult stroke that likely differentially affect angiogenesis. Mast cells can be a source of toxicity. 144
As will be discussed later, emerging data on the relative intactness of the BBB after acute neonatal stroke may have broad implications for developing therapies for injured newborns as many classes of drugs may still be precluded from entering the injured brain, at least acutely.
NEURAL REPAIR RESPONSES AFTER NEONATAL STROKE
Compared with the body of knowledge accumulated from adult animal models of stroke, characterization of long-term neural repair responses, including angiogenesis, neurogenesis, oligodendrogenesis, and neural plasticity in experimental models of neonatal stroke is still in its infancy. Transient MCAO gives rise to cell proliferation in the subventricular zone (SVZ) in the adult 145 and neonatal146,147 brain. Neuroblasts migrate from the SVZ into the damaged striatum, a region in which neurogenesis does not occur in the intact brain, and begins to express phenotypic region-specific mature neuronal markers,148,149 and forms synapses. 150 Gain- and loss-of-function stroke studies in the adult have shown that neuroblast migration occurs in association with remodeling of blood vessels, 151 via a link between angiogenesis and neurogenesis within the ‘neurovascular niche’, and that blockage of angiogenesis abolishes neurogenesis after adult stroke. 151 Several recent studies have shown that as opposed to the common belief that the immature brain has greater capacity for endogenous neural repair after injury, repair responses in neonates are more limited and more delayed in time compared with adults.
The low rate of stroke-induced endogenous neural repair observed in neonatal rodents may be related to insufficient induction of angiogenesis after focal ischemia compared with adults. In the adult brain, angiogenesis is a latent process and the turnover of endothelial cells is very slow, whereas the neonatal brain's vasculature is very dynamic, with the vascular network growing in complexity during the first three postnatal weeks, 152 along with the gradual increase in the brain's increased metabolic activity that results in increasing cerebral blood flow. 153 Stroke in adult rodents induces a relatively rapid angiogenic response, which starts within 24 hours of injury,154,155 while existing data on angiogenesis after neonatal stroke reveal surprisingly delayed induction of angiogenesis after neonatal focal stroke. 156 Vessel density and the number of proliferating endothelial cells are markedly reduced in the ischemic core and even in the ischemic penumbra, and the first signs of increased endothelial proliferation are not observed in the ischemic boundaries until 14 days after tMCAO in P7 rats. 156 Studies utilizing tMCAO in P10 rats also have shown a limited degree of angiogenesis 1 and 2 weeks later157,158 despite rapid induction of neuronal and then astrocytic vascular endothelial growth factor (VEGF). 12 Angiogenesis and neurogenesis are enhanced by delayed administration of recombinant human VEGF and disrupted by VEGFR2 inhibition, showing the role of VEGF signaling for the maintenance of angiogenesis and repair in the injured ischemic core.157,158 In this regard, therapies oriented to promote vascular remodeling after neonatal stroke may have a great impact on the long-term progression of brain injury and recovery.
Another factor limiting endogenous repair is the pattern of differentiation of cells that proliferate in the SVZ of the postnatal brain. The formation of GFAP+ cells,159,160 and not neurons or oligodendrocytes, contributes to astrogliosis and inhibits neural repair in the ischemic boundaries. Brain connectivity depends on white matter, and lack of OLP maturation adversely affects myelination of white-matter tracts19,96 and may be the underlying cause for some of the functional deficits in children with neonatal white-matter injury, 19 such as cerebral palsy and long-term cognitive impairment.17,61–63 Arrested differentiation and delayed cell death of OLPs limit the potentially regenerative effect of increased OLP proliferation observed after H–I. 96 While myelination is affected (both ipsilaterally and contralaterally) after neonatal H–I, it is not well understood whether the effect is due to dysregulation of myelination from regulators of myelination, such as Axin2, or altered individual pathways, such as the Wnt pathway. Along with alterations in OLP differentiation, axonal death, malfunction of the subpopulation of astrocytes promoting myelination, and disturbed cell-cell communications can also contribute to myelination defects. 163 While the effects of growth factors are viewed as purely positive, individual VEGF isoforms, for example, can differentially affect the proliferation and differentiation of SVZ progenitors, since VEGF-A promotes the production of astrocytes from SVZ glial progenitors while VEGF-C stimulates the proliferation of both early and late OLPs, cautioning interpretation of the relative isotype-specifk effects on myelination after perinatal brain injury. 164 While these phenomena have not been studied after neonatal stroke, the presence of injury in white-matter structures strongly suggests that these events could be extrapolated to neonatal focal stroke.
TRANSLATIONAL ASPECTS AND TREATMENTS
Therapies available in clinical practice in the perinatal period are very limited and the most commonly used ones such as antiepileptic drugs have side effects that can affect recovery after brain injury. Studies in human neonates have shown that perinatal administration of phenobarbital and phenytoin is associated with reduced brain size and cognitive decline. 165 Consistent with this observation, in rats, a broad range of antiepileptic drugs with various mechanisms of action (GABAA mimetics or inhibitors of voltage-gated sodium channels) trigger neuronal apoptosis throughout the brain during synaptogenesis166–168 but not later in life. Over the past two decades, an array of therapeutic agents has been tested in neonatal ischemic brain injury models to target excitotoxic, oxidative, and inflammatory injury components, but, as in adult stroke, with limited success.
Currently, hypothermia is the only neuroprotective treatment for injury resulting from perinatal global HIE. Recent multicenter clinical trials show that when hypothermia is initiated within hours in neonates with moderate HIE, it can reduce the risk of major neurologic disabilities.169,170 A recent analysis of 11 randomized controlled trials revealed that therapeutic hypothermia is beneficial in term newborns with HIE and that cooling reduces mortality without increasing major disability in survivors. 171 Selective brain cooling has been shown to potentially induce antiinflammatory effects. 172 The beneficial effects of hypothermia seen in experimental models of H–I, from mice to sheep, may be the result of a wide range of biologic effects, including preservation of energy metabolism and reduction of reactive oxygen species production and neuroinflammation. Hypothermia may also widen the window for repair but the beneficial effects of hypothermia alone seem insufficient, suggesting that outcomes may improve when hypothermia is used in conjunction with other therapies.173–177
Considering that initial observations on a limited endogenous neural repair are not specially promising, as injury can adversely affect postnatal brain development and reprogram the neonatal brain after stroke, more recent studies have begun targeting repair as a way to improve long-term recovery after focal or global H–I injury during the newborn period. Growth factors may prove potent for perinatal stroke and HIE when given alone or as adjunctive therapies. Erythropoietin administration has been shown to enhance neurogenesis and promotes functional recovery after neonatal H–I 178 and MCAO179,180 in rodents, with long-lasting effects after multidose erythropoietin treatment. Currently, promising clinical studies are being conducted on the effects of hypothermia and erythropoietin after perinatal HIE 181 but much needs to be learned about the optimal conditions for these interventions and alternative means to target the severely injured neonatal brain, especially in the setting of stroke.
As in experimental adult stroke studies, transplantation of stem cells and cell-based therapies, including mesenchymal stem/progenitor cells, provide promising results in animal models of neonatal brain injury182–185 but several aspects of stem-cell therapy including stem-cell origin, number and location of the transplanted cells, and timing of transplantation after injury still need to be refined before considering their use in humans. Another aspect that has yet to be understood is whether engrafted cells themselves enhance the repair by replacement of dead neurons or by stimulating the microenvironment (gene expression of growth factors and inflammatory molecules), which, in turn, permits remodeling and improvement of neurologic function. Some studies have shown that engrafted cells survive for weeks 182 while other studies show that the survival of engrafted cells sharply decline even when the beneficial effects continue. 184
Human umbilical cord blood (HUCB) cells are a rich source of immature stem cells, which have the potential to repair the injured brain. Transplantation of HUCB can induce functional recovery in postischemic neonatal brain in multiple ways, including direct protection, reduced neuroinflammation, production of growth factors, angiogenesis, and neurogenesis, but the exact mechanisms are currently unknown. 186 Some studies suggest that particular subpopulations of HUCB such as endothelial progenitor cells isolated from HUCB may be even more potent and thus preferred. Endothelial progenitor cells, bone marrow-derived precursors are capable of proliferating, circulating, and differentiating into mature endothelial cells. Endothelial progenitor cells have been shown to have a critical role in endothelial repair and vasculogenesis associated with stroke. Their use may have benefits compared with the use of HUCB due to endothelial progenitor cells differentiation into mature endothelial cells and incorporation into vessels or indirectly by secreting factors to stimulate neovascularization. 187 Studies are needed to establish the safety and the temporal and spatial effects before considering clinical trials.
An ongoing preclinical effort to characterize the effects of transplanted cells is supported by MRI to track migration, proliferation, and spatial location and proliferation of neural stem cells implanted after focal stroke in neonatal rats. 182 The ability to monitor cell therapies noninvasively with three-dimensional MRI quantification of ferromagnetically labeled neural stem cells would be important in preclinical and clinical trials using neural stem cells.
CONCLUSIONS AND FUTURE DIRECTIONS
Considerable progress has been made in early stroke diagnosis and MRI-based outcome prognosis in humans and in delineating the complexity of cellular injury, including better understanding of the network of intracellular signaling pathways and the rationale for identification of therapeutic targets and development of effective therapies. Over the past two decades, a major milestone has been the acknowledgment that age at stroke onset is the key factor determining the mechanisms and progression of ischemic brain injury. Recognition of the dynamic nature of changes during brain maturation and the integrative nature of brain injury, which evolves via communication between different cell types and between regions that may reprogram injured immature brain, is another conceptual advance. While much is to be learned about the underlying mechanisms, the knowledge gained is important for advancement to clinical trials. Distinct age-related susceptibility of particular cell populations and mechanisms controlling local inflammation and immune cell infiltration may guide a time-dependent selection of therapeutic targets. In view of new knowledge of the relative impermeability of the BBB after acute neonatal stroke, the distribution and bioavailability of therapies must be carefully considered to ensure that agents that are expected to produce effects locally in the brain do in fact reach the brain.
Short-lived protection of acutely administered therapies, paradoxical responses to antiinflammatory and antioxidant drugs, and the plasticity of the immature brain suggest that one of the critical features to improve outcomes should be a focus on rebuilding the injured neonatal brain by enhancing repair. It is also likely that combinatorial therapies may be superior to single therapies but future research should address the optimal timing for modulating effects of interventions and the role of genetic factors. Among other unanswered questions are why pediatric stroke is recurrent while perinatal stroke is rarely recurrent. Also, the role of prothrombotic pathways after injury is unknown due to lack of neonatal or pediatric thrombotic stroke models.
Elucidating how to enhance the repair process, with a focus on angiogenesis, neurogenesis, and preservation of function will improve the opportunities to enhance recovery and improve outcomes. Emerging evidence also suggests that interventions may be gender specific. Studies are obviously needed to examine the relative risk and benefit of any treatment and to determine whether proposed interventions may adversely affect long-term development.
Footnotes
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
The authors thank Dr Donna Ferriero for useful discussions pertaining to this review.