
Abstract
Considerable controversy exists about whether postischemic hypothermia can permanently salvage hippocampal CA 1 neurons or just postpone injury. Studies of very brief cooling in rat have found transient benefit, whereas experiments in gerbil using protracted hypothermia report lasting protection. This discrepancy might be because of the greater efficacy of longer cooling or it might, for example, represent an important species difference. In the present study, a 48-hour period of mild hypothermia was induced starting 6 hours after 10 minutes of severe four-vessel occlusion ischemia in rats. Untreated normothermic ischemia resulted in total CA1 cell loss (99%), whereas delayed hypothermia treatment reduced neuronal loss to 14% at a 28-day survival. In unregulated rats, brain temperature spontaneously fell during ischemia, and stayed subnormal for an extended period after ischemia. This mild cooling resulted in more variable and less severe CA1 injury (75%). Finally, vertebral artery cauterization under halothane anesthesia caused an approximately 2°C drop in brain temperature for 1 hour, but prevention of this hypothermia did not significantly affect CA1 damage. In summary, protracted postischemic hypothermia provided robust and long-term CA 1 protection in rat. These results encourage the clinical assessment of prolonged hypothermia and its use as a model to understand ischemic CA1 injury.
Ischemic CA1 neurodegeneration is quite sensitive to alterations in brain temperature during the occlusion (for review see Ginsberg et al., 1992; Maher and Hachinski, 1993; Dietrich et al., 1996). In fact, even mild intraischemic hypothermia (i.e., 30°C to 32°C) can completely block CA1 damage (Green et al., 1992; Nurse and Corbett, 1994). Such findings led most investigators to regulate core temperature during ischemia. However, several groups have shown that rectal and even skull temperatures do not adequately approximate brain temperature during ischemia (Busto et al., 1987; Minamisawa et al., 1990; Colbourne et al., 1993; Miyazawa et al., 1993). Notably, brain temperature decreased by up to 5°C during four-vessel occlusion (4-VO) ischemia in rat even when core temperature was held at normothermia (Busto et al., 1987; Miyazawa et al., 1993).
Although the protective effects of intraischemic hypothermia have been known for decades, the efficacy of postischemic hypothermia has been vigorously debated (Colbourne et al., 1997). Most initial experiments in rodents found a beneficial effect with an early induction of mild and brief (<8 hours) hypothermia after ischemia in rats (Busto et al., 1989; Boris-Moller et al., 1989; Chopp et al., 1991; Coimbra and Wieloch, 1992, 1994) and gerbils (Coimbra and Cavalheiro, 1990; Buchan and Pulsinelli, 1990; Carroll and Beek, 1992). However, these studies used short survival times (e.g., <7 days), and more recent findings in rat (Dietrich et al., 1993, 1995) showed that neuroprotection with 3 hours of post-ischemic hypothermia was only transient. However, protracted (e.g., 24 hours), but not brief, durations of delayed (e.g., 1 hour) hypothermia do provide lasting (1 to 12 months) neuroprotection in young (Colbourne and Corbett, 1994, 1995) and old gerbils (Corbett et al., 1997) subjected to global ischemia. The discrepancy between studies in rat that did not find long-term CA1 protection and those in gerbil that found lasting protection may be related to assessment of different durations of hypothermia because briefer durations were used in the rat studies. However, it is also possible that gerbils are more easily protected than rats. If so, previous data in gerbil might have overestimated the true benefit.
The primary purpose of this study was to determine whether delayed, and prolonged, postischemic hypothermia would convey lasting CA1 neuroprotection in the rat 4-VO model. In the course of doing this we observed that both ischemia and the cauterization procedures caused mild hypothermia. Thus, we assessed the effects of these spontaneous temperature drops on CA1 neuronal injury. Finally, spontaneous movement activity was measured after ischemia and related to CA1 sector damage.
METHODS
Cannula implant surgery
Fifty-three adult male Wistar rats (150 to 175 g) were obtained from Charles River (Montreal, Quebec, Canada). Rats were first implanted (under halothane anesthesia) with a guide cannula that was placed at the dural surface 1 mm anterior and 1 mm lateral to bregma, as described previously (Colbourne et al., 1996). The cannula and a surrounding plastic shield were secured with dental cement. One day later telemetry brain temperature probes (XM-FH, Mini-Mitter, Co., Sunriver, OR, U.S.A.) were inserted to measure cortical temperature. Rats were placed in individual cages that rested on RLA-1020 receivers interfaced with a computer running DataQuest IV (DataSciences, Int., St. Paul, MN, U.S.A.). Temperature and activity data (movement of probe over receiver) were recorded every 30 seconds. Data from the following day (i.e., 1 day before cauterization surgery) served as a baseline. Temperature and activity data were analyzed by analysis of variance, or pooled or independent t tests.
Four-vessel occlusion preparative surgery
Severe forebrain ischemia was produced by a modified 4-VO surgery (Pulsinelli and Brierley, 1979; Pulsinelli and Buchan, 1988). Three days after cannula implantation, rats were anesthetized with halothane (3% induction with 1 % to 2% for maintenance in a 28% O2 and 70% air mixture), followed by electrocauterization of the vertebral arteries. In addition, 2-O silk was looped about both common carotid arteries. An 18-gauge needle was used to guide a 1-O silk suture through the neck and posterior to the trachea, esophagus, jugular veins, carotid arteries, and vagal nerves, but anterior to the cervical and paravertebral musculature. In the first experiment, brain temperature was measured during the cauterization procedure, but not controlled. In the second experiment, brain temperature was regulated (by infrared lamp) at normothermia in one group, but not in the other group in which brain temperature was allowed to fall spontaneously.
Ischemia
Experiment 1 The day after vertebral artery cauterization, rats were briefly anesthetized to allow opening of the neck wound and occlusion of both carotids with aneurysm clips while the 1-O silk thread was tightened. Some rats had their carotids manipulated but not occluded (SHAM; n = 5). Ischemia was produced for 10 minutes in the ISCH-REG (n = 10), ISCH-UNREG (n = 10), and ISCH+HYPO (n = 9; 2 others died during or after hypothermia) groups, described below. Three other rats were excluded because of seizures during ischemia. The ISCH-UNREG rats had their core temperature regulated at 37.5°C during ischemia, but their brain temperature was allowed to change. The ISCH-REG and ISCH+HYPO groups had their core temperature controlled and additionally had their brain temperature regulated at 36.5°C. After ischemia, rats were returned to their cages, where the ISCH-REG group had their brain temperature maintained at greater than or equal to 36.5°C for 70 hours (by use of a lamp). The ISCH+HYPO rats had their brain temperature maintained at greater than or equal to 36.5°C for 6 hours at which time they were slowly cooled to 32°C and kept at that temperature for 24 hours. They were then rewarmed to 34°C for an additional 24 hours. Finally, they were kept between 35°C and 36°C for 12 hours. Cooling and rewarming occurred at a slow 1.0°C/30 minutes because this is a clinically relevant rate. Temperature was precisely controlled (i.e., typically <1/3°C deviation from desired) in awake rats by a servocontrolled system that used infrared lamps (175 W), fans, and fine water misters (Colbourne et al., 1996). Four days after ischemia, rats were briefly anesthetized to remove the brain temperature probes.
Experiment 2 There were two groups of rats. One group had their brain temperature regulated near normothermia (≥36.5°C; CAUT-REG; n = 7) during the cauterization procedure and for 6 hours thereafter; the second group was allowed to become spontaneously hypothermic (CAUT-UNREG; n = 7). Subsequently, in both groups, ischemia and temperature procedures were identical to the ISCH-UNREG group of experiment 1 (i.e., only rectal normothermia during the 10 minutes of ischemia).
Histopathology
Rats were allowed to survive for 28 (experiment 1) or 7 days (experiment 2) after ischemia or sham operation. They were anesthetized with halothane and perfused with 250 mL of saline followed by 250 mL of 10% formalin. Coronal sections (7 µm) of the hippocampus at −3.3 mm (Paxinos and Watson, 1982) were taken and stained with hematoxylin and eosin. Viable CA1 neurons in the medial, middle, and lateral sectors were counted, averaged, and expressed as a percentage of SHAM values as previously described (Colbourne and Corbett, 1995). Data were statistically analyzed by analysis of variance, or pooled or independent t tests depending on the absence or presence of heterogeneity of variance, respectively.
RESULTS
Mean baseline brain temperature (±SD) and movement activity collected the day before vertebral artery cauterization
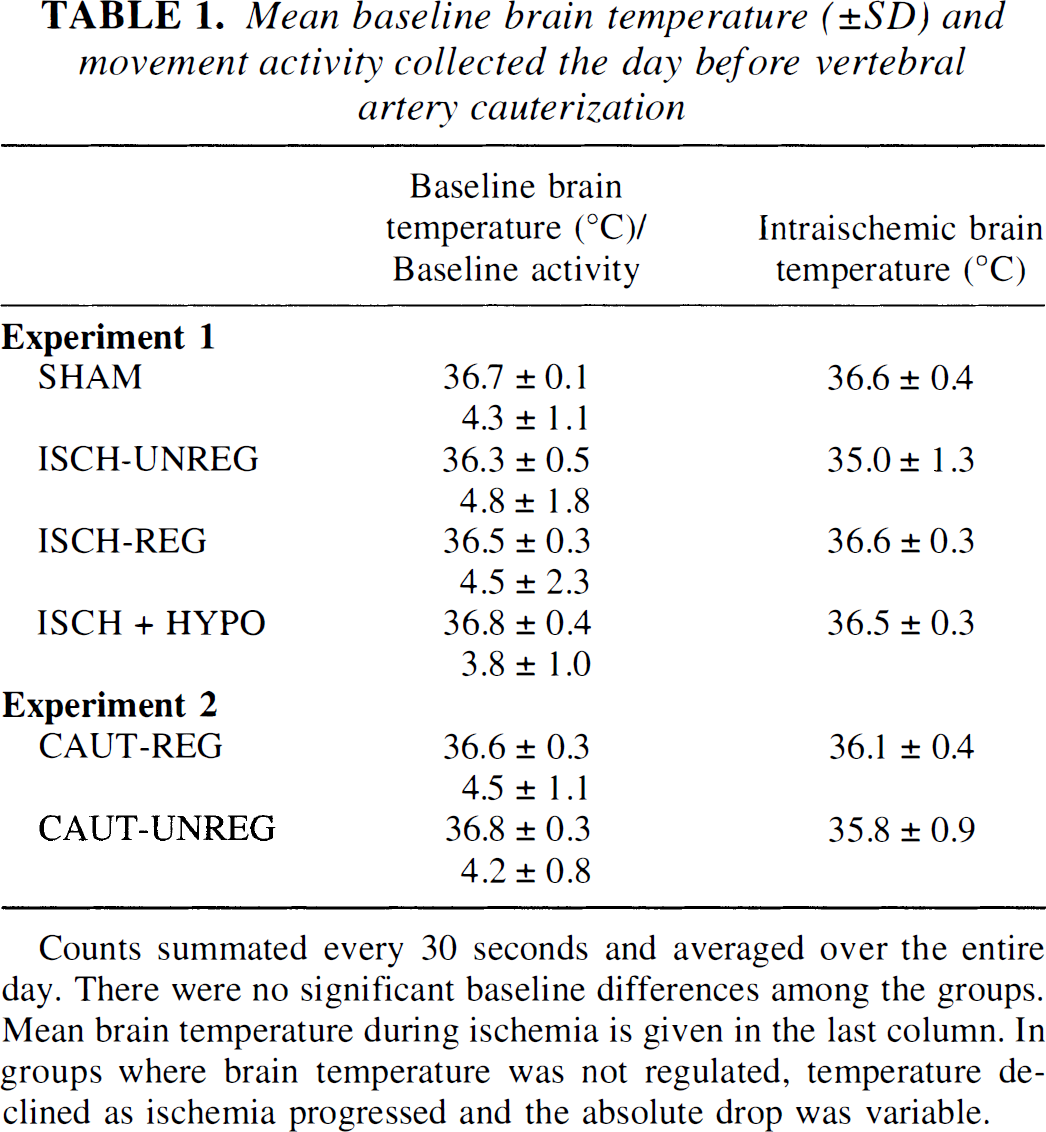
Counts summated every 30 seconds and averaged over the entire day. There were no significant baseline differences among the groups. Mean brain temperature during ischemia is given in the last column. In groups where brain temperature was not regulated, temperature declined as ischemia progressed and the absolute drop was variable.
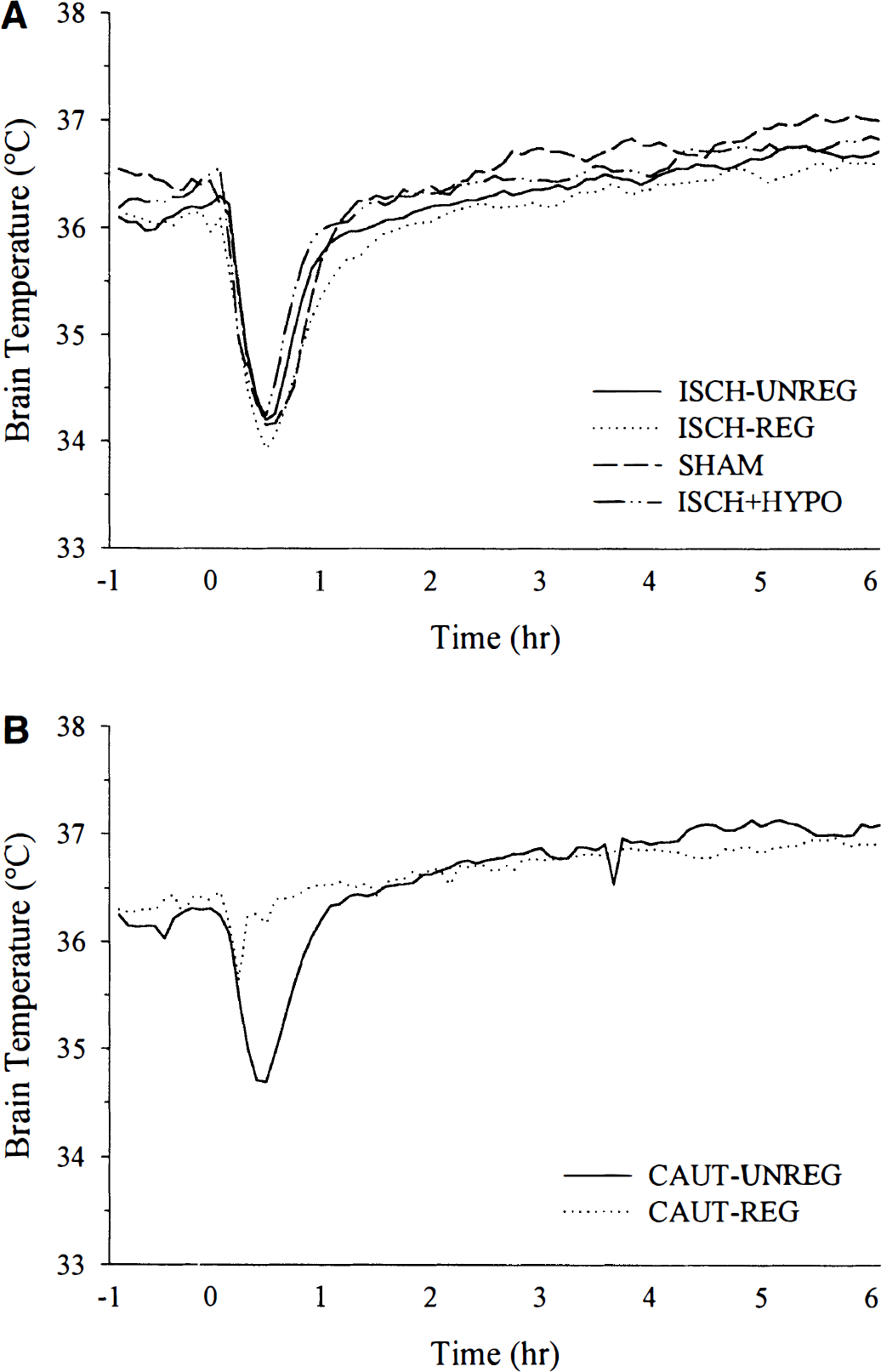
Brain temperature (°C) from approximately 1 hour before anesthetizing for the vertebral artery cauterization (at 0 hours) in experiments 1
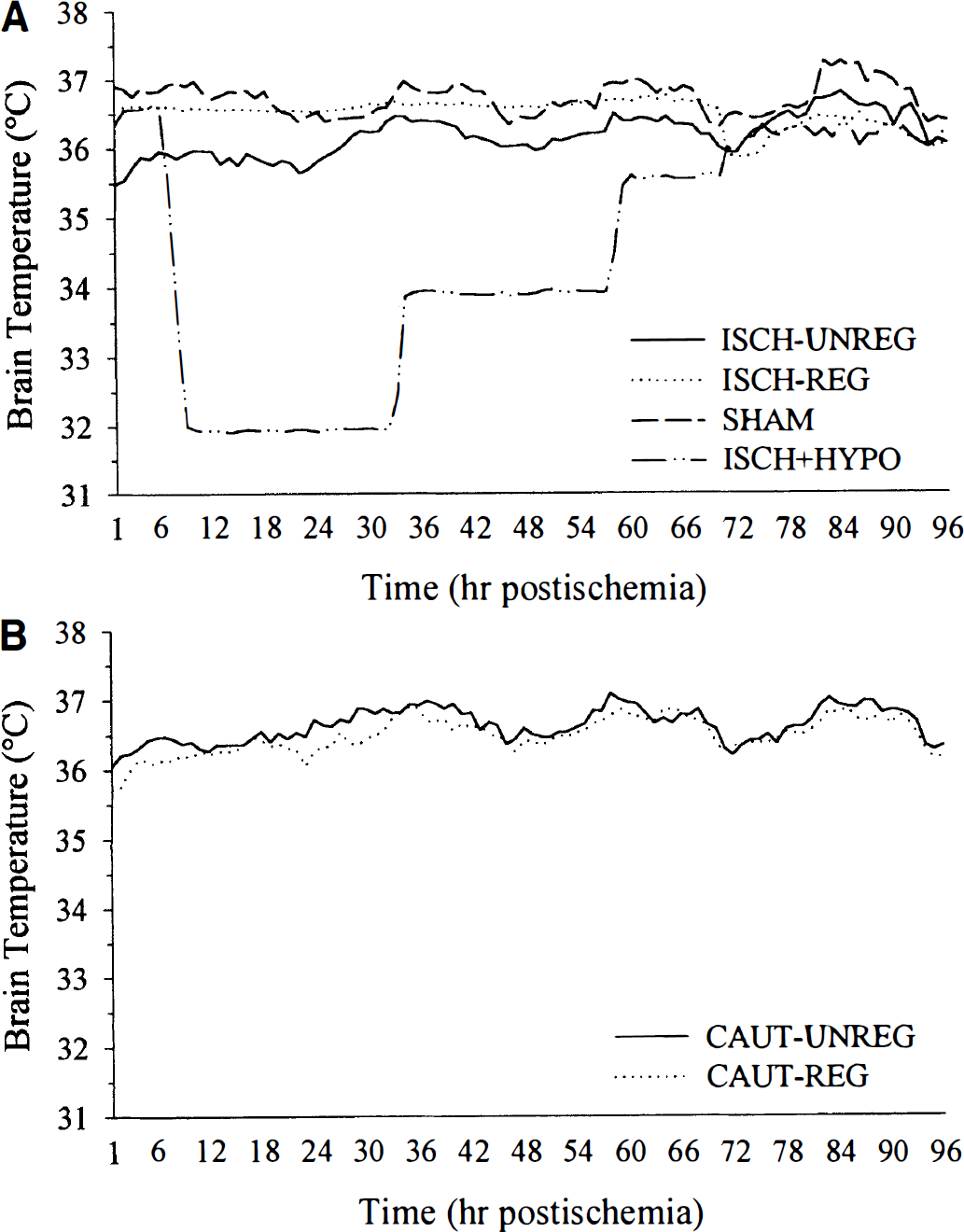
Brain temperature (°C) averaged every hour (sampled every 30 seconds) from immediately after ischemia or sham operation to 4 days later in experiments 1
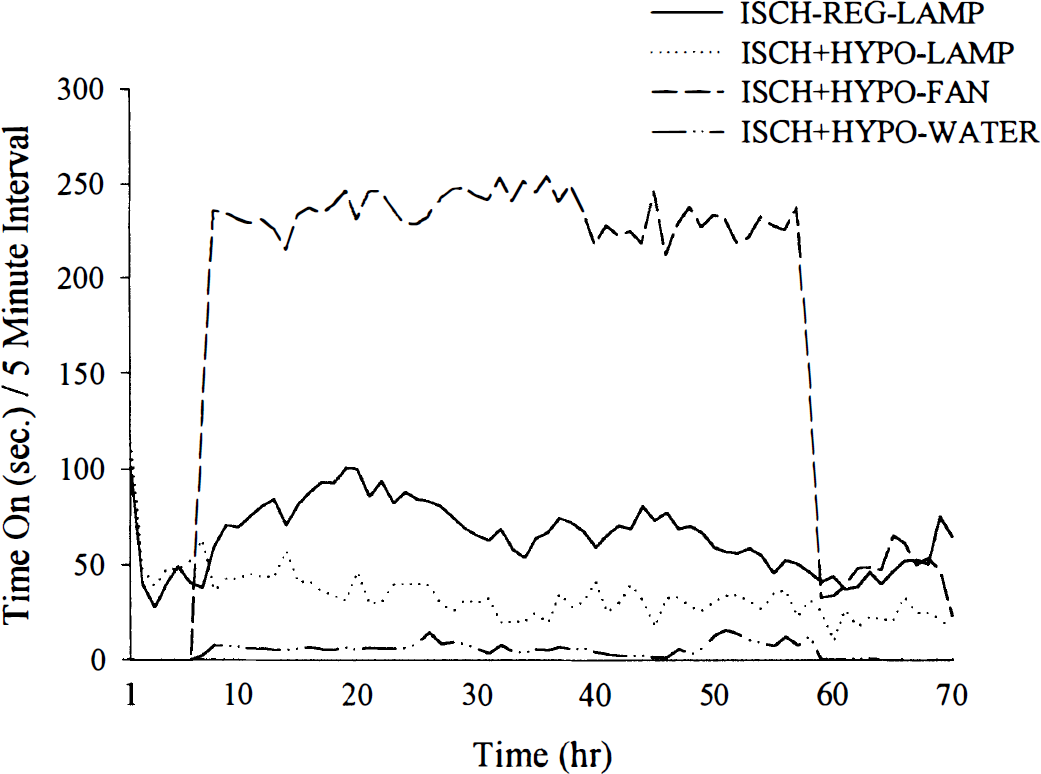
Lamp, fan, and water spray usage (per 5-minute interval) in ISCH-REG and ISCH+HYPO groups (data averaged every hour). The ISCH-REG group required a substantial and persistent amount of heating (time that lamp was on) to maintain normothermia. Although temperature rarely exceeded 36.5°C in the ISCH-REG group (Fig. 2), this was not prevented from happening.
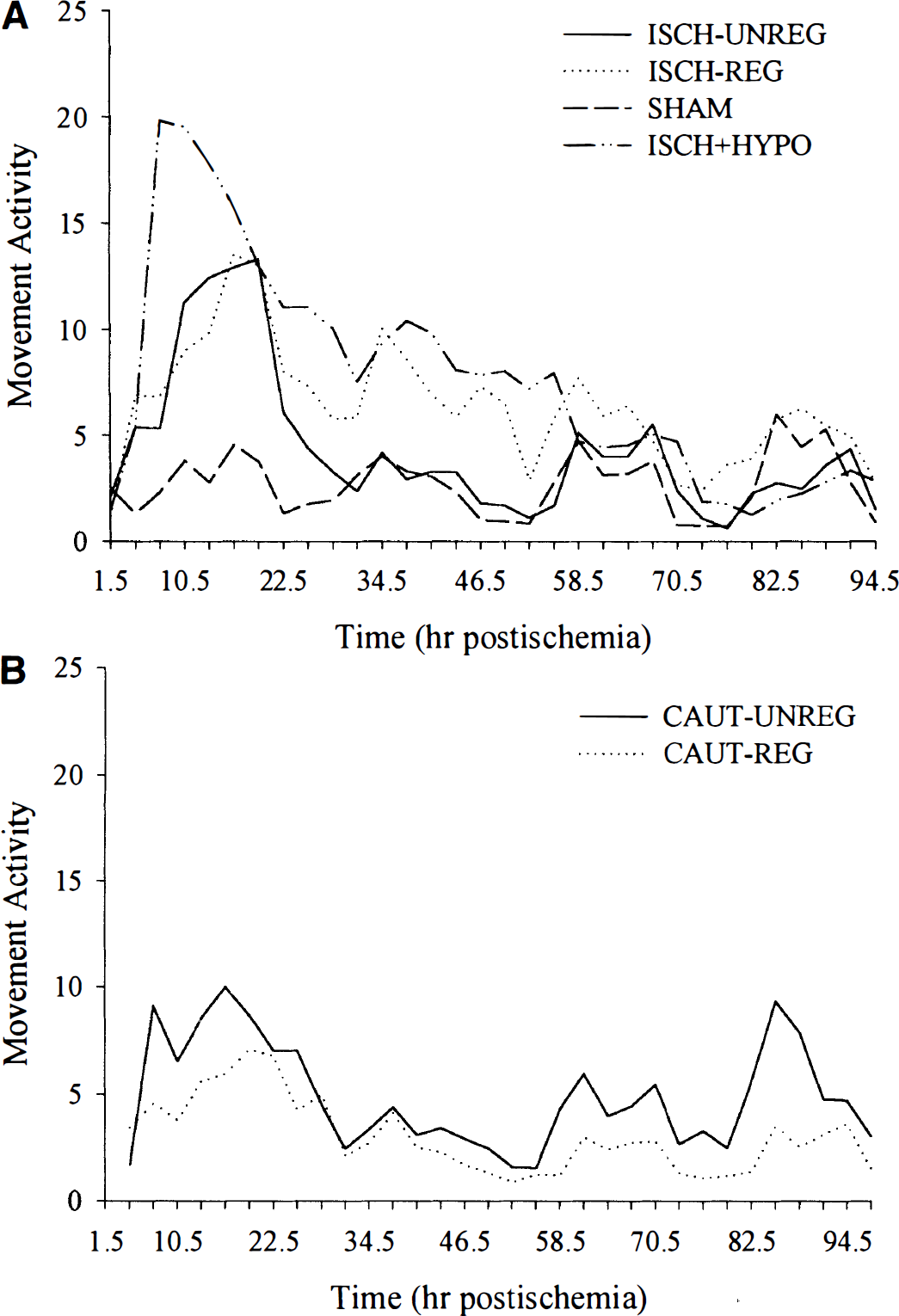
In the first experiment, all ischemic groups showed significant hyperactivity, which began soon after ischemia and returned to normal by the second or third day (Fig. 4). The ISCH-REG (P = 0.0018), ISCH-UNREG (P = 0.0032), and ISCH+HYPO (P < 0.0001) groups were significantly hyperactive (versus SHAM) on the first day (24-hour means), whereas only the ISCH-REG (P = 0.0012) and ISCH+HYPO (P = 0.0006) groups were significantly hyperactive on the second day.
Significant (versus SHAM, P < 0.0001) CA1 sector neuronal loss occurred in the ISCH-UNREG (25.1% ± 27.9% of SHAM; ± SD) and ISCH-REG (1.1%) ± 0.7% of SHAM) groups at a 28-day survival (Figs. 5 and 6). Cell loss was significantly greater in the ISCH-REG group (P = 0.0236). Delayed postischemic hypothermia treatment significantly (P < 0.0001) and persistently reduced CA1 loss (86.0% ± 29.0% of SHAM) in the ISCH+HYPO group (versus ISCH-REG group) to an extent that injury was not significantly lower than SHAM counts (P = 0.3091). CA1 injury was also significantly greater in the ISCH-UNREG group than in the ISCH+HYPO group (P = 0.0002). There were no notable sector (medial, middle, or lateral) or side differences, and mortality was not significantly different among groups (P ≥ 0.4762, Fisher exact test).
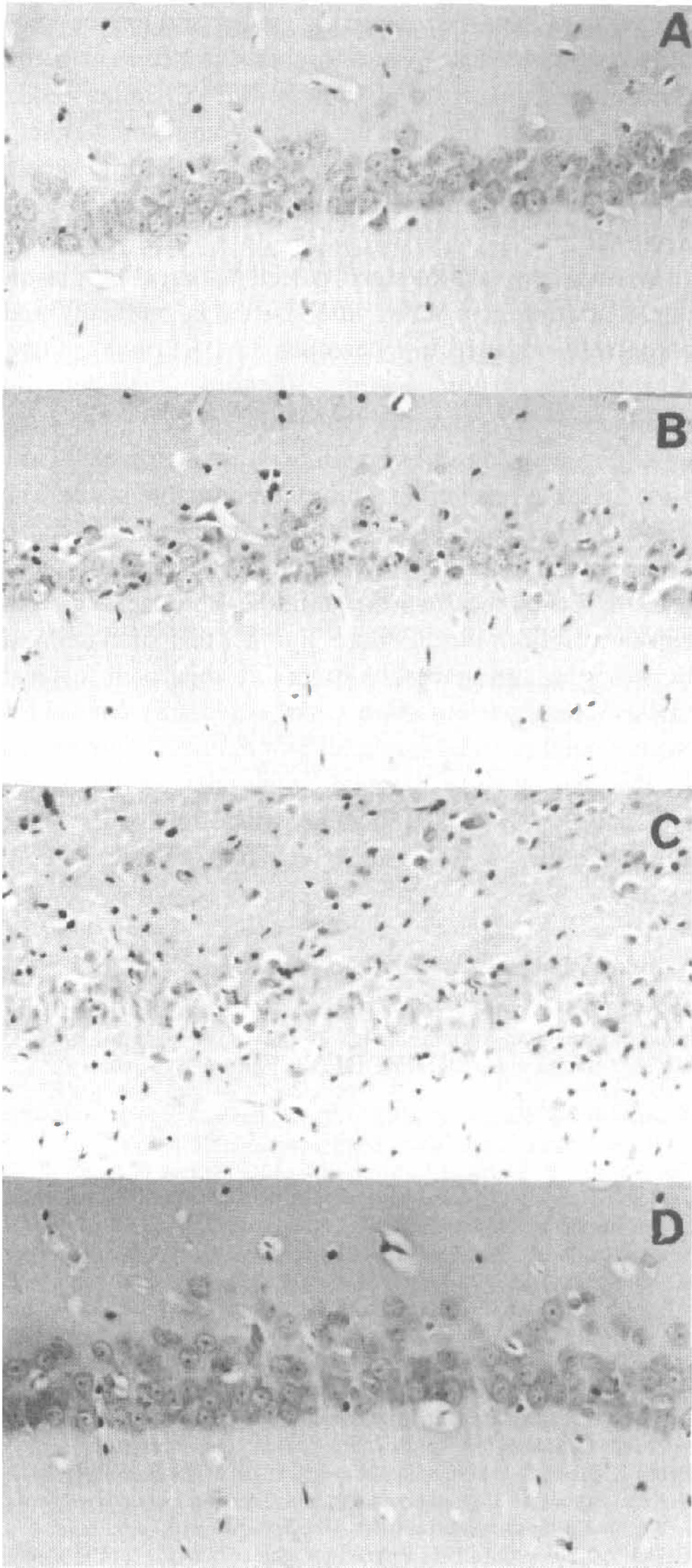
Hippocampal CA1 sector cell counts (% of SHAM) from experiments 1 and 2. Maintenance of normothermia during and after ischemia (ISCH-REG) resulted in a significantly more severe and consistent degree of CA1 sector injury. Six-hour delayed postischemic hypothermia largely attenuated this injury (versus ISCH-REG group) in all but one rat. There was no obvious reason for this outlier, but perhaps that rat had a carotid thrombus that embolized and exaggerated the ischemic insult. The mild spontaneous hypothermia that occurred during the vertebral artery cauterization did not affect subsequent ischemic CA1 damage (CAUT-UNREG versus CAUT-REG). Photomicrographs (original magnification ×160) of the middle CA1 zone from typical SHAM 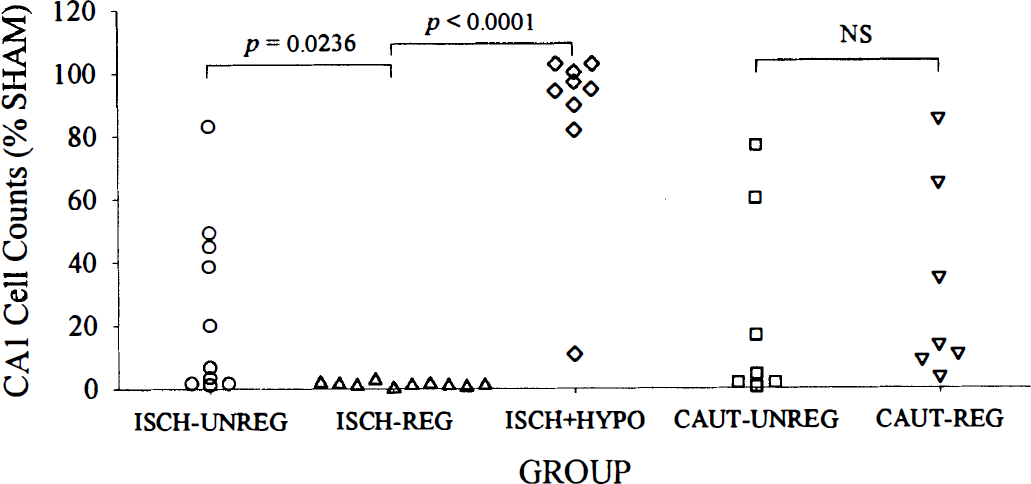
In the second experiment, the mild hypothermia experienced in the CAUT-UNREG groups was prevented during and after vertebral artery cauterization in the CAUT-REG group (Fig. 1). These groups (Fig. 2) had similar (<0.5°C difference) brain temperatures during (P = 0.4723) and after ischemia (first day mean, P = 0.4722). In addition, these groups had similar postischemic activity profiles (Fig. 4), and the 24-hour means on the first and second days were not significantly different from each other (P ≥ 0.2227). CA1 neuronal loss at 7 days (Fig. 5) was also not significantly different (P = 0.6226) in the CAUT-REG (31.7% ± 31.7% of SHAM) and CAUT-UNREG groups (23.1% ± 31.7% of SHAM).
Spontaneous movement activity (Fig. 7) on the first day after ischemia (i.e., 24-hour activity mean) significantly predicted (r = −0.425, P = 0.0383) CA1 sector injury (i.e., very damaged rats tended to be more active) in rats in which brain temperature was not regulated during or after ischemia (i.e., ISCH-UNREG, CAUT-UNREG, and CAUT-REG groups combined). Movement on the second day did not significantly predict CA1 sector injury (r = 0.177, P = 0.4068), but this is expected because spontaneous activity returned to normal by this time.
Scatter plot of postischemic movement activity (average of first day) and CA1 sector neuronal counts in ISCH-UNREG, CAUT-UNREG, and CAUT-REG rats. Hyperactivity was significantly but weakly associated with increased CA1 neuronal injury.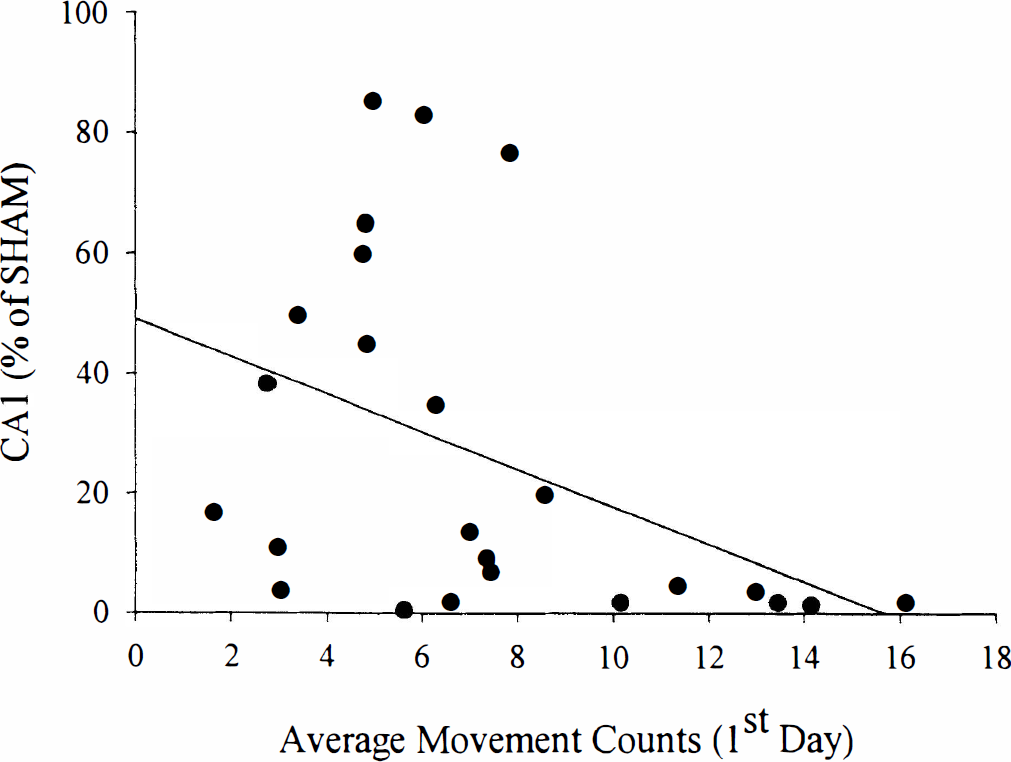
DISCUSSION
Six-hour delayed but protracted postischemic hypothermia, by itself, conveyed lasting and robust CA1 sector neuroprotection in rats subjected to a severe forebrain ischemic insult. This is contrary to other findings in the rat in which brief hypothermia (e.g., 3 hours, O-hour delay) has been found to only delay but not chronically (e.g., 2 months) reduce CA1 neuronal injury (Dietrich et al., 1993, 1995). However, the results are like those found in the gerbil bilateral carotid artery model in which CA1 zone injury has been persistently (e.g., 6 months) reduced with long durations of delayed hypothermia (Colbourne and Corbett, 1994, 1995; Corbett et al., 1997). The discrepancy between rat and gerbil studies of postischemic hypothermia might have been thought to be related to gerbils being more easily protected than rats (i.e., an important species difference). However, our findings of lasting CA1 sector protection with protracted cooling (>48 hours) in the 4-VO model indicates that the previous failures to find chronic protection in rat models of global ischemia (Dietrich et al., 1993, 1995) are because of an insufficient duration of hypothermia. Indeed, findings in the gerbil support this because a 24-hour duration of hypothermia was 6 times more efficacious in salvaging CA1 neurons than 12 hours of cooling, whereas briefer bouts (e.g., 3 to 6 hours) were ineffective (Colbourne and Corbett, 1994). Although it is known that the severity of ischemia affects the efficacy of postischemic hypothermia (Chopp et al., 1991; Colbourne and Corbett, 1994), the considerable protection afforded by delayed hypothermia in the present study was not because of the use of a milder insult. If anything, the total CA1 loss in the ISCH-REG group indicates that this insult was at least as severe as that used in studies in which protection was only transient (Dietrich et al., 1993, 1995). Furthermore, an even better outcome might be expected in this study if hypothermia was induced earlier because intervention after longer delays are less efficacious (Carroll and Beek, 1992; Coimbra and Wieloch, 1994; Colbourne and Corbett, 1995).
An alternative explanation for the failure of brief hypothermia in the rat two-vessel occlusion (2-VO) studies is the occurrence of very delayed fever that has been documented by Coimbra and colleagues (1996a, 1996b). When Coimbra et al. (1996a) used a 7-hour period of mild hypothermia (2-hour intervention delay) combined with prolonged antipyretic treatment or cooling to normothermia, they found moderate CA 1 protection at a 7- and 60-day survival time in rats subjected to 2-VO ischemia. Although the efficacy of brief hypothermia plus prolonged antipyretic treatment has never been directly compared with prolonged cooling alone, the latter should be superior. First, studies (Colbourne and Corbett, 1995) have shown that the depth of postischemic hypothermia is important because 32°C hypothermia provided greater protection than 34°C hypothermia (24-hour duration in gerbils), whereas even milder reductions in postischemic temperature are less effective (Nurse and Corbett, 1996). Second, the greater number of remaining CA 1 neurons in the ISCH+HYPO group than in the ISCH-UNREG group, which was only mildly hypothermic, clearly shows that the larger reduction in postischemic temperature was more efficacious. Finally, the amount of CA1 protection (85% salvaged) presently seen with prolonged hypothermia was greater and more consistent than that observed (approximately 50% of CA1 neurons salvaged) in the 2-VO study by Coimbra et al. (1996a), even though the normothermic 4-VO insult was more severe (approximately 15% more CA1 loss) and the hypothermia delayed by an additional 4 hours in the present study. Nonetheless, although the present experiment found that greater than 2 days of mild hypothermia was markedly protective, it is unknown whether this duration is optimal or even necessary for maximal CA 1 protection. Regardless, it is clear that even very brief cooling periods can also be significantly and persistently protective, especially after milder ischemic insults (Colbourne and Corbett, 1994; Coimbra et al., 1996a). Given the multitude of factors that affect neuroprotection (e.g., severity of ischemia), it is evident that the optimal hypothermic parameters that minimize side effects will have to be worked out in the clinic.
The mechanisms by which postischemic cooling salvages CA1 neurons remains elusive, and largely unstudied (see Colbourne et al., 1997, for a recent review). Furthermore, the beneficial effects of intraischemic hypothermia (e.g., reduced glutamate release) should not be assumed to apply to postischemic hypothermia. The use of prolonged postischemic hypothermia provides an excellent opportunity to further our understanding of ischemic injury and neuroprotection. For example, this study indicates that the threshold of irreversible injury occurs after 6 hours, and thus any deleterious events occurring before this time are not insurmountable. A better understanding of the detrimental effects of hypothermia might lead to improved efficacy and greater safety. For example, in this and many other studies of protracted postischemic hypothermia (Colbourne and Corbett, 1994, 1995; Yanamoto et al., 1996; Corbett et al., 1997; Colbourne et al., 1998 a), cooling was produced in the unanesthetized rodent. Although this is safer than the prolonged use of an anesthetic (Colbourne et al., 1996), it does potentially diminish efficacy because cooling awake rodents causes shivering (at least temporarily), which elevates metabolism (Dill and Forbes, 1941; Penrod, 1949; Bigelow et al., 1950; Spurr et al., 1954; Stone et al., 1956). Because reduced metabolism is thought to be one of the mechanisms by which hypothermia conveys its protection (Colbourne et al., 1997), the side effect of shivering, that is, increased metabolism, might lessen protection and is nonetheless contraindicated in humans.
A comparison of prolonged postischemic hypothermia with some other current drug strategies shows that hypothermia is the superior neuroprotectant. For example, N-methyl-
Much of the histologic variability in the 4-VO model is caused by spontaneous cooling of the brain during ischemia, and this is often complicated by a dissociation between rectal and brain temperatures (Busto et al., 1987; Minamisawa et al., 1990). Prevention of this mild hypothermia during and after ischemia resulted in a more severe and consistent degree of CA1 zone injury as previously found when brain temperature was preserved during 4-VO ischemia (Busto et al., 1987; Miyazawa et al., 1993). However, Miyazawa et al. (1993) have argued that the maintenance of normothermia “introduces an aggravating pathological element that may interfere in an unpredictable way with the manifestation or treatment of ischemic injury.” Our data do not support this because very mild intraischemic or postischemic hypothermia predictably but variably reduced CA1 zone injury, whereas ischemia under normothermic conditions resulted in a consistent and greater degree of CA1 injury. Thus, brain temperature should be precisely controlled during and for a prolonged period after ischemia (i.e., 24 to 48 hours) to minimize variability and the number of rodents needed. Such precise temperature control is only practical with an automated system (Colbourne et al., 1996). Because prevention of spontaneous hypothermia will aggravate injury and possibly extend it to other structures, which undoubtedly can lessen the potential for neuroprotection, we suggest that the ischemia duration be shortened when assessing weak neuroprotectants instead of allowing for variable temperature drops. In this way, a precise and less variable insult can be delivered.
The cauterization surgery resulted in a brief period of hypothermia that did not affect subsequent ischemic neuronal injury. This is in keeping with findings that a mild period of hypothermia does not induce tolerance (Wada et al., 1997). Nonetheless, it would seem prudent to maintain temperature during this procedure because more-prolonged or severe hypothermia during the cauterization surgery might induce tolerance. This remains to be determined.
The novel finding that many of the rats undergoing unregulated 4-VO experienced a brief to prolonged period of spontaneous mild hypothermia after ischemia is contrary to findings in rats subjected to 2-VO ischemia (Coimbra et al., 1996a, 1996b) and gerbils subjected to transient forebrain ischemia (Kuroiwa et al., 1990; Neill et al., 1990; Warner et al., 1991; Colbourne et al., 1993; Colbourne and Corbett, 1994), in which spontaneous postischemic hyperthermia typically develops. The absence of this aggravating factor in the 4-VO model has likely contributed to the need for longer ischemia durations (e.g., 15 to 30 minutes) to achieve consistent injury in 4-VO studies compared with the 2-VO (e.g., 10 minutes) and gerbil models (e.g., 5 minutes). Indeed, Coimbra et al. (1996a) have shown that prevention of delayed hyperthermia in 2-VO rats slows CA1 injury. Gerbils experience more postischemic hyperthermia than 4-VO rats perhaps in part owing to the greater and longer bouts of hyperactivity that follow forebrain ischemia in gerbil (Colbourne et al., 1998). As in gerbils (Chandler et al., 1985; Gerhardt and Boast, 1988; Wang and Corbett, 1990; Mileson and Schwartz, 1991; Colbourne et al., 1998), motor hyperactivity in rats predicted ensuing CA1 neuronal loss.
In summary, a prolonged period of hypothermia induced starting at 6 hours after ischemia markedly and chronically reduced hippocampal CA1 neuronal injury. Ten minutes of unregulated 4-VO ischemia resulted in spontaneous hypothermia that endured well after ischemia. Preservation of brain temperature aggravated CA1 zone loss, but resulted in a more reproducible insult. The fact that hypothermia was efficacious when induced 6 hours after forebrain ischemia makes this therapy suitable for further mechanistic studies and perhaps for clinical intervention. Furthermore, the fact that such delayed hypothermia can markedly affect CA1 neuronal survival makes it necessary to always and persistently control for the potential confounding effects of delayed hypothermia as this has already plagued other putative neuroprotectants, e.g., MK-801 (Buchan and Pulsinelli, 1990; Corbett et al., 1990) and NBQX (Nurse and Corbett, 1996).