
Abstract
The significant morbidity that accompanies stroke makes it one of the world's most devastating neurological disorders. Currently, proven effective therapies have been limited to thrombolysis and thrombectomy. The window for the administration of these therapies is narrow, hampered by the necessity of rapidly imaging patients. A therapy that could extend this window by protecting neurons may improve outcome. Endogenous neuroprotection has been shown to be, in part, due to changes in mTOR signalling pathways and the instigation of productive autophagy. Inducing this effect pharmacologically could improve clinical outcomes. One such therapy already in use in transplant medicine is the mTOR inhibitor rapamycin. Recent evidence suggests that rapamycin is neuroprotective, not only via neuronal autophagy but also through its broader effects on other cells of the neurovascular unit. This review highlights the potential use of rapamycin as a multimodal therapy, acting on the blood–brain barrier, cerebral blood flow and inflammation, as well as directly on neurons. There is significant potential in applying this old drug in new ways to improve functional outcomes for patients after stroke.
Introduction
An increasing prevalence of vascular risk factors in low- and middle-income countries is expected to result in 12 million stroke-related deaths annually by 2030, amounting to a global loss of over 200 million disability-adjusted life years (DALYs). 1 While our understanding of pathophysiology of stroke has grown exponentially during the last few decades, the only available acute therapies target reperfusion of the ischemic tissue 2 – there are currently no clinically proven neuroprotectants that can delay cell death and extend the therapeutic time window to restore blood flow. 3 In ischemic stroke, the occlusion of blood vessels in the brain heralds the beginning of a neurotoxic cascade. Tissue immediately adjacent to the core of the infarct (known as penumbra) has residual cerebral blood flow (CBF), is electrically silent due to impaired cellular energy availability, but is not yet irreversibly damaged and therefore may be salvageable if reperfusion occurs. 4 Any intervention once the cascade has been triggered at best amounts to damage limitation. Translating promising findings from in vitro studies and animal models has proven incredibly challenging. 5 The complexity of the processes implicated in ischemic cell death diminishes the likelihood of achieving benefit from targeting these mechanisms with monotherapies. Our focus should be on multifactorial approaches, countering the multiple pathological processes and ensuring we target the earliest steps in the cascade of pathogenesis.
Despite the neurotoxic cascade occurring during a stroke, some neurons have an innate ability to survive brief episodes of ischemia. In cardiac arrest, or the rodent model of global transient ischemia, the CA3 neurons of the hippocampus are resistant, whereas the CA1 neurons are vulnerable to the ischemic insult.6–13 Pharmacologically, manipulating this inherent capacity to withstand the effects of a stroke may be key to the development of novel therapies. Hamartin expression has been shown to be partly responsible for this endogenous neuroprotection. 14 Hamartin is the protein product of the gene Tsc1. Together with tuberin (Tsc2), it forms the tuberous sclerosis complex (TSC), which inhibits the mammalian target of rapamycin complex 1 (mTORC1) and acts as a tumour suppressor. 15 Pharmacologically, mTORC1 can be inhibited by rapamycin (sirolimus), a macrolide compound widely used as an immunosuppressant. The mTORC1 pathway is remarkably well conserved phylogenetically and expressed in all human cell types, reflecting its role as a central regulator of metabolism (Human Protein Atlas, www.proteinatlas.org). 16 While harnessing this pathway could prove useful, we first need to understand the potential multifaceted effects that rapamycin and mTOR inhibition may have, both centrally and peripherally, following stroke.
Historically, there has been contradictory evidence in the literature surrounding animal models of ischemic stroke as to whether rapamycin is neuroprotective or detrimental to outcomes. A systematic review of these studies has found significant evidence that rapamycin improves not only infarct volume, but also behavioral outcomes after experimental stroke, and that this may be dose-dependent (Beard et al., in press). The inhibition of mTOR by rapamycin results in a plethora of downstream effects including productive autophagy, alteration of CBF, reduction in blood–brain barrier (BBB) permeability and cytoskeletal changes. Instead of taking the traditional view that the multitude of off-target effects of rapamycin precludes its consideration clinically in acute central nervous system (CNS) injury, we take the view that a multimodal approach is exactly what is required to improve outcome following ischemic stroke.
Molecular mechanisms of rapamycin
There are two mTOR complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2), differing in their subunit composition. The regulatory associated protein of mTOR (raptor) is found in mTORC1, whereas the rapamycin-insensitive companion of mTOR (rictor) is in mTORC2.
17
The main function of mTORC1 is to augment protein synthesis and inhibit autophagy (Figure 1A). Both mTORC1 and mTORC2 have been postulated to play roles in cell survival, cell proliferation and regulating the cytoskeleton.18,19 Activators of mTORC2 activity include growth factors and insulin through the PI3K pathway.20,21 Many of the upstream regulatory pathways converge at the TSC, allowing it to play an important role in the control of mTOR activity. The overall effect of impaired TSC activity is mTOR hyperactivation, which results in uncontrolled protein synthesis and inhibition of autophagy.
15
The role of mTOR in neuronal survival following ischemia. (a) Under normal conditions, the adequate provision of nutrients and oxygen activates mTOR to stimulate anabolic processes such as protein synthesis while suppressing catabolic processes such as autophagy. (b) During stroke the reduction in provision of nutrients reduces mTOR activation, limiting protein synthesis and inducing autophagy. (c) Treatment of neurons with rapamycin can further reduce mTOR activation and increase the induction of autophagy. If autophagy induction is optimal this may lead to cell component recycling and cell survival (right-hand box). If autophagy is excessive, this may lead to apoptosis and cell death (left-hand box). mTORC1: mammalian target of rapamycin complex 1; S6K: S6kinase; S6rp: S6 ribosomal protein; 4EBP: eukaryotic translation initiation factor 4E binding protein; eIF4e: eukaryotic initiation factor 4e.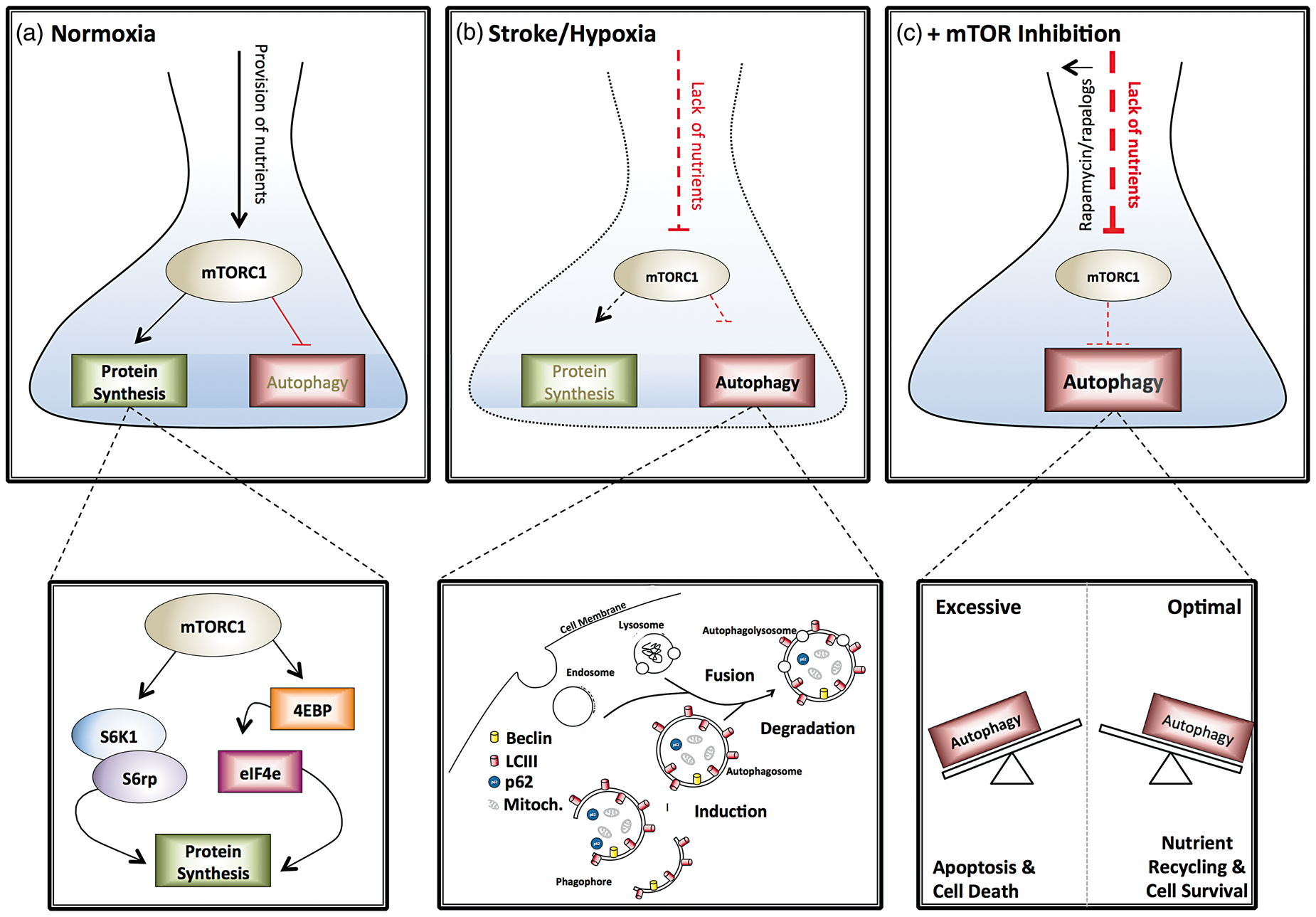
As the name suggests, the major small molecule inhibitor of mTOR is rapamycin. However, the plethora of diseases in which mTOR dysregulation has been implicated has paved the way for the development of novel drugs targeting this pathway. First generation mTOR inhibitors, or rapalogs, aim to directly inhibit the mTOR complex by preventing the binding of raptor and thus the activation of mTOR, rendering it incapable of phosphorylating downstream molecules. These first generation inhibitors include drugs such as sirolimus and temsirolimus. 22
Second generation, more selective, mTOR inhibitors also decrease cell cycle progression and protein production, but selective targeting can push them via either the AKT or the S6K pathways and, in theory, affect either cell death and the induction of autophagy (mTORC1) or cell survival and function (mTORC2). 23 However, since the first generation inhibitors have been used in the majority of preclinical studies and importantly have extensive clinical safety data, this review will focus on the application of these compounds to cerebral ischemia.
Promoting neuronal survival in ischemic conditions
The efficient disposal of superfluous and detrimental proteins is essential for tissues with limited regenerative capacity, such as the CNS. 24 A key mechanism through which this is achieved is autophagy, the engulfment of misfolded, damaged or surplus proteins and organelles in a lipid bilayer, which are subsequently targeted for degradation (Figure 1B).25,26 Dysfunction of basal autophagy has been shown to rapidly lead to formation of protein inclusions and neurodegeneration in rodent models. 27 The relevance of autophagy to stroke is its role as an adaptive response, to provide nutrients and energy from pre-existing sources during episodes of cellular stress, when there is limited nutrient delivery through the vasculature.
Current hypotheses suggest that a short, sharp period of “austerity” might induce cellular tolerance to ischemia.14,28 The up-regulation of hamartin-induced mTOR inhibition with ischemic preconditioning (IPC) could provide the underlying mechanism for this tolerance, 14 and is entirely consistent with the idea that hypothermia 29 and deafferentation 30 might result in increased survival of brain cells following ischemia. Preconditioning with rapamycin (administered 20 h before ischemia) has been shown to induce autophagy, improve survival, longevity, behavioral scores and reduce infarct size following focal cerebral ischemia/reperfusion. 31 Similarly, in a model of permanent focal ischemia, IPC reduced infarct volume, brain edema and motor deficit. 32 These protective effects were abolished when autophagy was inhibited by 3-methyladenine (3-MA). Furthermore, rapamycin treatment on its own mimicked the protective effects of IPC. Together these data show that the mechanisms of neuroprotection by both rapamycin and pre-conditioning appear to both be mediated through the induction of autophagy.
In certain experimental systems, suppressing autophagy in response to oxidative, hypoxic-ischemic, and excitotoxic injuries confers neuroprotection, linking the over-activation of autophagy to neuronal injury. 24 The suppression of autophagy by exercise pre-treatment and MAPK (p38) inhibition was demonstrated to be neuroprotective in a middle cerebral artery occlusion (MCAO) model of stroke. 33 A number of other studies show that autophagy has a detrimental effect in the context of neuronal ischemia.34–38 In an attempt to reconcile conflicting findings, one study has compared an inducer of autophagy (rapamycin) with an inhibitor (chloroquine). 39 Both compounds reduced infarct size and ameliorated neurological scores, but only the rapamycin-treated mice demonstrated improved survival.39,40 Interestingly, the neuroprotective effects of rapamycin were reversed by the autophagy inhibitor 3-MA. 41 In IPC32,42 and remote ischemic post-conditioning, 43 autophagy was demonstrated to be neuroprotective. However, autophagy inhibition also was found to be neuroprotective in ischemic post-conditioning. 44 In isolated neuronal cultures, excitotoxic glutamate insults were shown to block autophagic flux, and inhibition of autophagy had no effect on neuronal survival. 45 In contrast, the same study found significant neuroprotection with rapamycin and trehalose, two inducers of autophagy that work via mTOR-dependent and mTOR-independent mechanisms, respectively. 45 These studies clearly demonstrate that autophagy has both protective and detrimental effects on neurons, which can be influenced by the administration of rapamycin (Figure 1C).
In an attempt to address the apparent equipoise in the literature and explain the heterogeneous outcomes produced by rapamycin, a systematic review and meta-analysis conducted on rodent models of focal ischemia has demonstrated that in vivo rapamycin is neuroprotective with a reduction in infarct volume and improvement in behavioural outcomes. (Beard et al., in press) The neuroprotective effects of rapamycin were dose-dependent with lower doses associated with improved outcomes, potentially by inducing an optimal level of autophagy to enable viable neurons to survive in an austere environment, without causing excessive neuronal death.
Beyond the neuron: The neurovascular unit
Stroke research is increasingly focussing on the neurovascular unit (NVU). Neurons in vivo do not act in isolation, therefore their interactions with the vasculature and surrounding glia, and the implications these have for neuroprotective therapies, must be considered (Figure 2).46,47 The NVU plays an important role in maintaining a constant cerebral microenvironment, regulating CBF, inducing and subsequently maintaining the BBB.
48
Stroke has been shown to cause damage to, and subsequent dysfunction of, the NVU.49–53
Suggested neurovascular effects of rapamycin. Astrocyte: Stroke/hypoxia causes conversion of astrocytes from cytotropic to cytotoxic driven by mTOR and retraction of astrocytes from the BBB driven by Rho-GTP. Rapamycin treatment stops transition from cytotropic to cytotoxic phenotype in cell culture. Rapamycin can potentially reduce the retraction of astrocytic endfeet and improve BBB permeability by inhibiting RhoA-GTP (hypothetical). Pericyte: Stroke/hypoxia causes pericyte constriction and MMP-9 release (animal and cell culture). Rapamycin may reduce constriction by RhoA-GTP (hypothetical). Rapamycin can reduce MMP-9 expression following stroke (shown in animal models). Endothelial cells: Stroke/hypoxia can cause reduced CBF, endothelial cell death and tight junction breakdown. Excess calcium entry into the endothelial cell leads to cytoskeletal re-arrangements by myosin light chain phosporylation and stress fibre formation leading to BBB opening. Rapamycin can increase eNOS activity and improves CBF in Alzheimer's Disease (AD) mouse models. Rapamycin increases autophagy leading to enhanced cell viability and tight junction expression (based on cell culture and animal studies). Rapamycin can reduce Rho-A mediated cytoskeletal rearrangements by inhibiting mTOR s6K (hypothetical).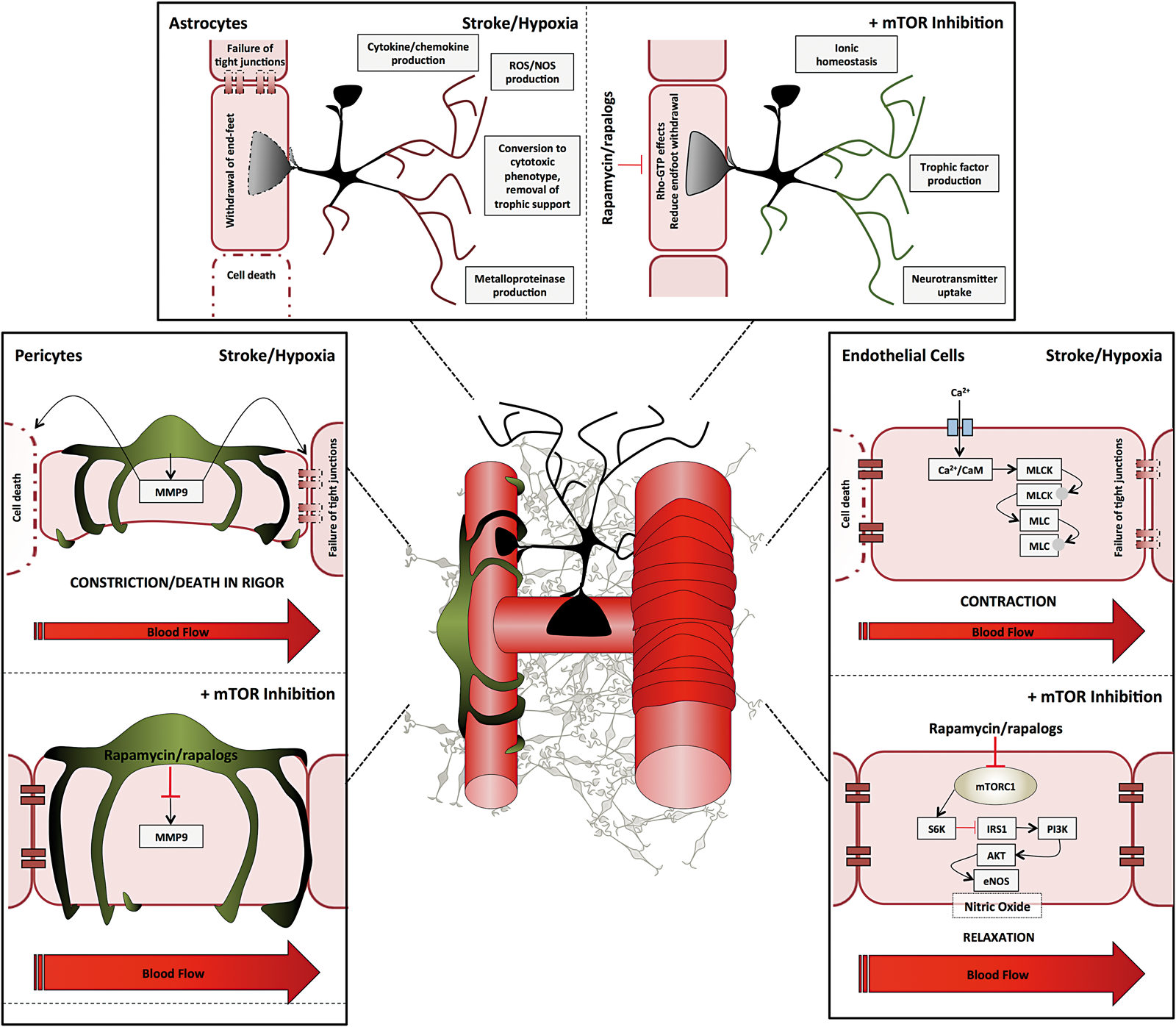
Timely reperfusion is a crucial goal for acute stroke therapies. However, it may also contribute to secondary neuronal damage, hemorrhagic transformation and cytotoxic edema formation. BBB permeability/vasogenic edema is one of the leading causes of patient deterioration following stroke. 54 Early animal studies revealed a triphasic increase in BBB permeability following reperfusion,55,56 with an initial peak upon reperfusion, followed by a limited amount of recovery of BBB integrity with evidence of multifocal BBB permeability to albumin, possibly by enhanced transcytosis as early as 2 h of reperfusion. 57 This is followed by later peaks in permeability at 6 and 72 h after stroke, most likely due to loss of tight junctions leading to vasogenic edema.58–62 Furthermore, vessel recanalization does not always result in microvascular reperfusion, ultimately limiting the effectiveness of thrombolytic treatments.63–73 It is estimated that around 25% of stroke patients that achieve successful recanalization do not achieve reperfusion (termed futile reperfusion or the no-reflow phenomenon). 74 The mechanisms of this phenomenon are difficult to study in humans. No-reflow and post-reperfusion hypoperfusion are well-established concepts in experimental ischemia, where opening of the occluded vessel fails to return adequate capillary perfusion.75,76 Evidence from animal studies suggest a number of mechanisms for this phenomenon including astrocyte end-feet swelling, 77 neutrophil entrapment, 78 and pericyte death in rigor.79,80
Previously in the setting of ischemia, rapamycin has been used to investigate the role of neuronal autophagy. However, we are only just beginning to explore the potential for rapamycin to have an effect on the rest of the NVU. In the following section, the effect of rapamycin on BBB integrity will be discussed, as well the potential for rapamycin to ameliorate other aspects of neurovascular dysfunction after stroke.
Improving CBF
Rapamycin treatment can alter blood flow in neurological disease. mTORC1 inhibition was shown to improve basal CBF, BBB integrity and was associated with amelioration of behavioural outcomes in models of Alzheimer's disease. 81 The restoration of brain vascular integrity and function in the hAPP (J20) mouse model of Alzheimer's disease is attributable to endothelial NO synthase activation through rapamycin's inhibition of mTOR leading to increased activity of eNOS (Figure 2). 82 In a study using a murine low-density lipoprotein receptor model of atherosclerosis and vascular cognitive impairment, rapamycin restored CBF and brain vascular density. 83
Only one in vivo stroke study has measured CBF following rapamycin treatment. 84 Although this study reported a non-significant increase in CBF at 2 h post-reperfusion, there was an increase in infarct volume measured at the same time. This discrepancy may be explained by the chronic (three days) high-dose infusion (20 mg/kg) of rapamycin used in this study. This regimen resulted in inhibition of mTORC2, which is required for adaptation and survival during cellular stress. 19 The authors highlight that optimising their dosage and timing of administration is needed to ensure inhibition of predominantly mTORC1 to obtain the beneficial effects of rapamycin treatment. The results of the meta-analysis of Beard et al. (in press) suggest that lower doses of rapamycin (<1 mg/kg) are associated with improved outcome.
mTOR inhibition with rapamycin may improve neurovascular function by autophagy-dependent and autophagy-independent mechanisms. For example, the mTOR pathway may be implicated in autoregulatory failure following reperfusion. Autoregulatory failure begins as early as 30 min after experimental MCAO in rodents and persists out to 24 h.85–87 Cipolla and Curry 85 show that the degree of autoregulatory failure assessed in MCAs extracted from rats undergoing MCAO is positively associated with the degree of actin cytoskeleton re-arrangement in the smooth muscle cells surrounding the isolated arteries. These observations may explain the persistent loss of autoregulation occurring despite reperfusion. mTOR inhibition with rapamycin has been shown to reduce cytoskeletal reorganisation and cell motility by supressing Rho-A expression in human rhabdomyosarcoma cell lines. 88 Furthermore, rapamycin is currently used in drug eluting coronary stents to prevent smooth muscle hyperplasia by inhibiting mTOR-mediated cytoskeletal re-arrangements and muscle cell proliferation. 89 Rapamycin treatment has the potential to limit actin cytoskeletal re-arrangements in cerebral artery smooth muscle as a means of reducing autoregulatory failure and its harmful downstream effects on the BBB/NVU.
Reducing the no-reflow phenomenon
The no-reflow phenomenon was first described by Ames et al. 75 Since this initial report, a number of studies from different groups in different species have shown that the reduction in capillary perfusion post-recanalisation is associated with the aggregation of neutrophils, platelets and red blood cells within brain capillaries.78,90–99 Recent studies in rodents undergoing MCAO have shown that the degree of blood flow deficit on reperfusion is a stronger predictor of stroke outcome than intra-ischemic blood flow.76,100 To date, the effect of rapamycin on post-recanalization perfusion deficits and cellular infiltration into capillaries after stroke has not been explored.
Recent studies suggest that capillary obstruction in reflow may be caused by pericyte death in rigour.79,80 This may contribute to cells such as red blood cells, neutrophils and platelets becoming trapped in capillaries, as seen in previous studies. Pericyte constriction following ischemia has been shown to be caused by excessive calcium entry into the cytoplasm, 79 which may lead to activation of the Rho GTPase/ROCK/myosin light chain (MLC) pathway resulting in rapid pericyte constriction.101,102 As mentioned previously, mTOR inhibition with rapamycin has been shown to supress RhoA expression. 88 Therefore, rapamycin may lead to a reduction in phosphorylation of MLC, reducing the sensitivity of pericyte MLC to calcium and thus reduce the contractility of pericytes in response to excessive calcium influx during ischemia (Figure 2). The possibility of using rapamycin to reduce pericyte constriction and no-reflow phenomenon after stroke has not yet been directly investigated.
Improving BBB permeability
The BBB is composed of brain endothelial cells and associated tight junctions. 103 Following ischemic stroke, BBB integrity becomes compromised leading to brain edema as a result of increased vascular permeability. Hyperglycaemia and diabetes further perturb BBB function, exacerbating edema.104,105 Our understanding of BBB disruption after stroke is still evolving; however, recent evidence suggests that BBB compromise after stroke is mediated by a combination of ischemic damage to brain endothelial cells, endothelial cell cytoskeleton rearrangements, reactive astrogliosis and release of matrix metalloproteinases from pericytes (Figure 2), this will be discussed in more detail in the following paragraphs.
Rapamycin can reduce endothelial cell death and improve junctional marker expression following ischemia in vitro. mTOR-dependent (rapamycin) and mTOR-independent (lithium carbonate) autophagy inducers were used in brain microvascular endothelial cells (BMVECs) in an oxygen and glucose deprivation (OGD) model and also an MCAO model, demonstrating a beneficial effect on BBB integrity (Figure 2). 106 The potent autophagy inducer, long noncoding RNA MALAT1, has also been shown to protect brain endothelial cells from OGD. 107 Furthermore, autophagy has been shown to be protective in advanced glycation end product-induced early injury of human umbilical vein endothelial cells (HUVECs). 108
Rapamycin has been shown to decrease brain endothelial cell death and maintain the integrity of the BBB in in vivo models.32,106,109 In a rat focal model of cerebral ischemia, rapamycin reduced brain edema, infarct volume and improved behavioral outcomes. 32 In a streptozotocin-induced model of diabetes, rapamycin decreased BBB permeability in control rats only following 1 h of MCAO and 2 h of reperfusion. 110 The evidence for rapamycin improving BBB permeability is limited; however, it does not appear to be as effective in diabetes, which may impact on efficacy in clinical scenarios.
Energy failure within endothelial cells results in increased intracellular calcium, which can lead to MLC kinase activation and F-actin stress fibre formation, ultimately increasing paracellular permeability by changing endothelial cell shape and downregulating tight junctions between cells. 111 Recently, the role of Rho kinase in BBB dysfunction has been explored. Rho kinase helps to determine vascular tone and integrity. Its effects include cell proliferation, inflammation, MLC phosphorylation and nitric oxide bioavailability. In an in vitro experiment using human brain microvascular endothelial cells (HBMECs) and astrocytes, the exacerbation of BBB damage in the context of hyperglycaemia was thought to be mediated by PKC-β through regulation of the RhoA/Rho kinase/MLC2 pathway. 112 Similarly, another study using HBMECs and astrocytes demonstrated that increased RhoA/Rho kinase/MLC pathway activity, in combination with changes in the actin cytoskeleton, explain the endothelial barrier breakdown occurring following OGD (Figure 2). 113 In an animal model of stroke, inhibition of Rho kinase modulated endothelial cell oxidative stress and tight junctions, improving the integrity of the BBB, which was supported in vitro using HBMECs and astrocytes. 114 As mentioned in previous sections of this review, mTOR signalling is critical for activation of the Rho kinase pathway. Therefore, rapamycin treatment may not only improve BBB integrity by improving endothelial cell viability, but also by direct stabilisation of the cytoskeleton and tight junctions.
Astrocytes also play a critical role in maintaining the integrity and function of the BBB, as well as being key players in neuronal function through energy transfer, neurotransmitter recycling and antioxidant activity. 115 In the first few hours after stroke, astrocytes take on a cytotropic phenotype, which is non-proliferative and favours neuronal survival. They mediate this by increasing glutamate uptake to reduce cytotoxic damage to neurons, they provide metabolic support to neurons and also provide antioxidant protection (Figure 2). 116 As these astrocytes are non-proliferative, they maintain their endfoot process contacts with neurons and the vasculature. At around 6 h following ischemia, astrocytes begin to transition to a cytotoxic or reactive phenotype in which they reduce glutamate uptake, 117 undergo cellular hypotrophy, upregulate the cytoskeletal protein glial fibrillary acid protein (GFAP), 118 increase aquaporin 4 (AQP4) expression, 118 increase matrix-metalloproteinase (MMP) release, 119 and retract their endfoot processes from the vasculature, 120 resulting in BBB disruption and impairment of neurovascular coupling (the regulation of blood flow in accordance with neuronal activity) (Figure 2). Therapies that can reduce the transition of astrocytes from cytotropic to cytotoxic reactive phenotype may aid in neuronal survival, reduce BBB disruption and improve post-stroke recovery. 115
Initial acute therapeutic efforts were directed at globally inhibiting the actions of astrocytes to improve outcome following stroke. This ultimately worsened injury, due to the pro-survival role of astrocytes in the acute phase.121,122 mTOR has been shown to be a key mediator of the transition to reactive astrocytes during ischemia. Li et al. 123 showed that rapamycin treatment of astrocytes exposed to OGD reduced reactive gliosis as evidenced by reduced GFAP expression, reduced migration and reduced expression of pro-inflammatory cytokines (Figure 2). Inhibition of reactive gliosis may be another potential mechanism of reduced BBB permeability following rapamycin treatment.
The effect of rapamycin on reactive astrogliosis, and how this relates BBB permeability in vivo, is just starting to be explored. In a rat model of transient MCAO and reperfusion, rapamycin decreased infarct volume and maintained the integrity of the BBB, potentially via inhibiting the expression of matrix metalloproteinase 9 (MMP-9) and AQP4. 124 AQP4 expression in the brain is astrocyte end-foot specific; however, the source of MMP-9 was not determined. Cultured astrocytes have been shown to enhance MMP-2 and MMP-9 expression and activity following in vitro ischemia. 119 Recent evidence suggests that pericyte somata are a major source of MMP-9 in ischemia, which is associated with localised BBB disruption, primarily at pericyte locations along the capillary. 125 Cultured pericytes have been shown to release MMP-9 but not MMP-2, by p38 MAPK-dependent mechanisms. 126 Furthermore, ischemia has been shown to induce p38 MAPK activation 127 and rapamycin has previously been shown to reduce p38 MAPK activation by inducing the expression of MAPK phosphatase activity. 128 Although this is a plausible mechanism for the reduced MMP-9 observed in the above studies, the effect of rapamycin on astrocyte or pericyte MMP release is yet to been investigated directly. Together, these studies suggest that while the integrity and function of the BBB in stroke are severely compromised, rapamycin has significant potential to alter many of the molecular mechanisms underlying these changes.
Inflammation
Regulation of an effective inflammatory response post-stroke is critical for a good outcome.129,130 The immune system, largely comprised of microglia, is often described as ‘Janus faced’, with positive as well as negative effects in the CNS. 131 There is a need for the phagocytic clearance of dead tissue, and yet the paradoxical pro-inflammatory response this engenders results in the death of bystander cells by chemical ‘friendly fire’. In addition to the CNS immune system, invading leukocytes—largely neutrophils and monocytes—result in exacerbation of damage. The mechanisms for activation of both the local and systemic immune systems remain under scrutiny.
CNS inflammation
Microglial phagocytosis of apoptotic cells is a largely anti-inflammatory process, 132 resulting in the release of substances such as TGFβ. However, the exact mechanisms of cell death within the infarct core in stroke remain contentious. Release of factors such as high mobility group box (HMGB) during necrosis may induce a more pro-inflammatory phenotype in the local immune cell population. 132 This double-edged sword has resulted in an increased interest in drugs with the capacity to polarise microglia. While now considered much more of a spectrum, inducing an M1 or an M2 phenotype in the resident microglia remains an important goal. 133 By reducing the pro-inflammatory effects of microglia potentially activated by necrotic cells, we may reduce the risk of both local damage, as well as the recruitment of peripheral immune cells.
The literature surrounding mTOR pathway alterations, microglia and inflammation is contentious. For example, studies of glial cells in vitro have demonstrated that rapamycin prevents transition to an M2 phenotype, maintaining the M1 cytotoxicity and potentially enhancing microglial anti-tumour capacity. 134 However, in a rodent model of stroke, mTOR pathway inhibition promoted an M2 phenotype, upregulating genes such as Arg1 and YM-1. 135 The latter showed little significant difference between rapamycin and the more potent mTORC1 inhibitor, everolimus, suggesting this is a general pathway mechanism, rather than an Akt-specific effect. In a mouse permanent MCAO model, autophagy in microglia was shown to increase infarct size and edema, and was detrimental to neurological scores, with rapamycin promoting the inflammatory response. 136 The discrepancy between these studies could be due to the in vitro vs. in vivo effects. Indeed, isolated cell culture experiments often report that rapamycin enhances the pro-inflammatory activity of microglia, 137 whereas in whole animals this appears not to be the case. Xie et al. suggest this is likely to be due to cell–cell interactions after stroke. Their study demonstrated that the interaction between T-reg cells and macrophages/microglia in ischemia is an important regulator of inflammation, and that by enhancing the anti-inflammatory activities of T-regs, rapamycin indirectly reduces microglial pro-inflammatory abilities. 138
Systemic inflammation
While rapamycin is known to cross the BBB, 139 systemic administration is likely to be used—should this drug become therapeutically viable in stroke—meaning that the immune effects of mTOR inhibition will be widespread. One of the first uses of rapamycin was as an immunosuppressant in transplant therapy, with rapamycin having broad effects on dendritic cells, 140 macrophages, 141 monocytes, 142 and T cells. 143 This wide scale immunosuppressive activity may be beneficial in the post-stroke pro-inflammatory environment, but given the importance of the inflammatory process in the repair process after stroke, this immunosuppression may be detrimental.
The primary function of the mTOR pathway in post-stroke inflammation is via metabolic reprogramming to drive pro-inflammatory processes. 144 For example, myeloid lineage cells are likely to need increased energy stores for replication and migration after an injury, but again, these processes may be affected by the polarisation of the cells. 145 These changes in energy regulation are also likely to be context dependent. The nutrient-deficient environment in ischemia will prompt a metabolic shift in the cells and a polarisation towards a particular phenotype, potentially via mTORC1 activation and metabolic reprogramming. 146 Conversely, the nutrient- and oxygen-rich environment of the liver will result in systemic macrophages activated by damage-associated proteins, which are likely to polarise to the opposing phenotype. When the latter migrate to the site of injury, it is little wonder that there are conflicting findings in myeloid lineage experiments post-stroke.147,148 Application of rapamycin to this system could prove problematic. While low dose and short-term rapamycin treatment has been shown to preferentially inhibit mTORC1, prolonged exposure or high doses are likely to inhibit mTORC2. 149 As such, dosing regimens should be considered carefully.
Migrating cells found within the CNS acutely post-stroke include significant numbers of neutrophils. Pro-inflammatory stimuli have been shown to exacerbate perfusion deficits following vessel recanalization, which are associated with increased platelet and neutrophil aggregation in capillaries. 150 Specific to the no-reflow phenomenon, rapamycin has anti-neutrophil151,152 and anti-platelet activity.153,154 This may be, in part, due to mechanisms involving RhoA and the cytoskeletal changes required for chemotaxis. Knockdown of this pathway in isolated neutrophils results in decreased chemotactic ability 155 and therefore could be beneficial post-stroke. However, mTOR inhibition has also been shown to dysregulate the formation of neutrophil extracellular traps (NETs).156,157 Dysregulation of NET formation may lead to complications such as sepsis, 158 which would be highly undesirable in an already compromised patient. However, there is a paucity of data on the role of the mTOR pathway in neutrophils post-stroke.
Further up the immunological tree lie the adaptive immune cells. Antigen-presenting dendritic cells, or even antigen-presenting microglia activate T cells post-stroke. Antigen presentation is a key process in adaptive immunity and is driven, in part, by autophagic pathways. There is a suggestion that basal autophagy is decreased in activated immune cells in order to prevent the excessive presentation of auto-antigens, and that interference with this pathway may increase this process. In fact, the treatment of transplant patients with rapamycin increases the levels of circulating autoreactive T cells, suggesting that the use of mTOR inhibitors in inflammatory cells may be dependent on activation status. 159 When taken with data showing that rapamycin significantly impairs remyelination, 160 there is a chance that poor dosing strategy may result in an autoreactive phenotype with poor remyelination, reminiscent of other major CNS diseases.
Multi-target versus mono-target therapies
Rapamycin may have a distinct advantage over previous and current neuroprotectants as it acts at a pivotal point governing a plethora of neurovascular protective effects (Figure 2), rather than targeting a single pathway within the multi-faceted ischemic cascade. In this section, we will discuss how the molecular targets of other neuroprotectants relate to those of rapamycin.
As discussed previously, the main clinical use for rapamycin is in the context of immunosuppression following transplant. 161 This remains a potential mechanism by which rapamycin is neuroprotective following stroke.135,162 The role of immune modulation is increasingly being investigated in stroke. 163 Fingolimod, a T-cell modifying therapy used in multiple sclerosis, has been trialled in ischemic stroke patients in conjunction with alteplase. 164 Neuroprotection is thought to be due to inhibiting the migration of lymphocytes, BBB stabilization, 165 inhibiting the recruitment of leukocytes in cerebral microvessels and inhibition of endothelial activation. 166 Small clinical trials have shown that fingolimod is well tolerated; however, due to small patient numbers and heterogeneity of the trials, no definitive answer on the efficacy of this therapy can be made at this time. 167
Subcutaneous interleukin-1 receptor antagonist (Anakinra) is another immunomodulatory therapy currently being trialled as an adjunct to thrombolysis in ischemic stroke. 168 This intervention is backed up by many years of animal model evidence, systematic review and meta-analysis 169 and a multicentre randomised pre-clinical trial confirming its efficacy in improving stroke outcome. 170 A recently published small phase-II randomised trial has confirmed that subcutaneous Anakinra was well tolerated, reduced circulating IL-6 levels but did not improve patient outcomes, potentially due to an interaction between Anakinra and alteplase. Larger studies are needed to investigate the efficacy of this therapy and the interaction with thrombolysis. 168
Rapamycin can protect the BBB leading to reduced brain edema after focal cerebral ischemia reperfusion in rodent models via multiple mechanisms including: reduced endothelial cell death via induction of autophagy, 106 improved tight junction expression, 106 inhibition of matrix metalloproteinase 9 (MMP9) and reduced aquaporin 4 (AQP4) expression. 171 Similarly, the antibiotic minocycline has shown promising results in animal stroke models through reducing MMPs and hence the permeability of the BBB. 172 However, the efficacy of improving stroke outcome in large human stroke trials has yet to be obtained. 173
Rapamycin can also reduce the detrimental effects of oxidative stress in neurons. 174 This may be particularly important in the reperfusion phase when oxygen is re-introduced to defective or damaged mitochondria leading to reactive oxygen species production. 175 Rapamycin has been shown to be neuroprotective in an animal model of stroke by inducing mitophagy (removal of damage mitochondria by autophagy), 176 which has been shown to reduce reactive oxygen species production and ultimately cell death. 177 Numerous anti-oxidants have been trialled for stroke. The most notable being NXY-059, which showed promising results in numerous animal models and provided the impetus for two randomised trials. The first smaller SAINT-I trial showed a protective effect, while the follow-up larger SAINT-II trial was neutral for improving patient outcome. 178 Post hoc analysis of the animal studies that informed the decision to proceed to clinical trial showed that the poor quality of these studies may have led to an overestimation of the intervention's efficacy. 179 Furthermore, it has recently been shown that NXY-059 does not have biological efficacy in embryonic stem cell-derived human neurons exposed to both ischemia and oxidative stress models of cell death. 180 Therefore, alternative anti-oxidant therapies that are based upon sound preclinical data and have been trialled in human neural tissue may still be a viable therapy for stroke.
Translational considerations
Overall rapamycin has the potential to suppress the deleterious effects of CNS immune activation on stroke outcome; however, optimal dosing must be considered in order to keep detrimental systemic immunosuppression to a minimum. Different dosing strategies for rapamycin have been used in studies of the anti-ageing effects of rapamycin in rodents. These studies have shown that the beneficial anti-ageing effects of rapamycin are mediated through mTORC1 inhibition, while the detrimental side effects of long-term rapamycin administration such as immunosuppression and glucose intolerance are mediated by mTORC2 inhibition. 181 Studies by Arriola Apelo et al. have shown that an alternative dosing strategy where rapamycin is administered once every five days (as compared to once a day) inhibits mTORC1 without inhibiting mTORC2 and increases longevity while reducing the negative effects on the immune system and glucose homeostasis.182,183 Specifically, they found that a single 1 mg/kg dose of rapamycin was enough to suppress mTORC1 activation for one to three days post-administration. 182 This would be an ideal approach for treating stroke patients. A single low-dose administration within 6 h post-stroke would be expected to provide the beneficial neuroprotective effects via mTORC1 inhibition for the initial phases of stroke. Rapamycin would then be cleared from the brain by three days, thus allowing the critical neural and angiogenic proliferation necessary for stroke recovery. 184
One must also consider the route of administration. Rapamycin is a large hydrophobic macrocyclic lactone (Figure 3A), which makes it advantageous for crossing cell membranes and the BBB. However, this makes it difficult to get into solution for ease of intravenous administration. Therefore, rapamycin has traditionally been given by mouth in tablet or liquid formulation (with ethanol to improve solubility).
185
This may be problematic for acute administration to stroke patients given that 65% of these patients have difficulty swallowing and protecting their airways.
186
To allow acute administration of rapamycin to these patients, a naso-gastric tube would allow safe administration of a rapamycin solution. This may pose some problems in terms of time needed to place the tube, the added time needed for the drug to reach the bloodstream and the drug being subject to extensive first-pass metabolism by CYP3A enzymes in the intestine and the liver.
187
If rapamycin is to persist as a potential therapy, these routes of administration should also be considered in pre-clinical research.
Rapamycin (Sirolimus) and rapalogues (a). Chemical structure of Sirlolimus. (b) Chemical structure of temsirolimus. Substitution of the C40 hydroxyl group is shown in blue.
192
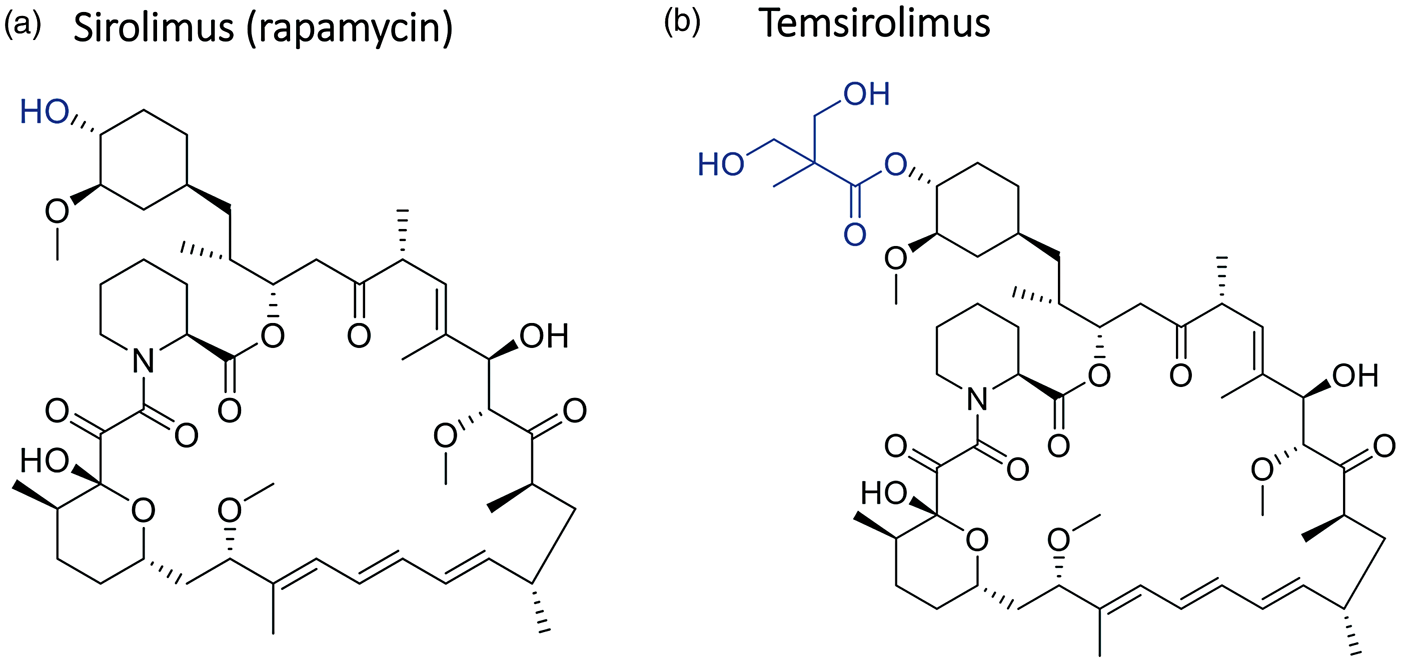
An alternate strategy would be to give the rapalogue temsirolimus, which comes as an intravenous formulation. 185 Temsirolimus is the pro-drug of sirolimus that once administered intravenously is rapidly converted to sirolimus (Figure 3B). 188 Temsirolimus is currently approved for the treatment of renal cell carcinoma. 189 Although temsirolimus is a pro-drug of sirolimus, this analogue appears to have very similar potency and tolerability when compared to sirolimus. 190 Therefore, it is anticipated that temsirolimus would exert the same neuroprotective effects in stroke as regular sirolimus. However, this has yet to be tested experimentally.
Conclusion
Neurons do not work in isolation. It is increasingly recognized that cerebrovascular and immunological factors influence neuronal survival following stroke. Investigations into drugs that target certain hubs of the complex cell death cascade are ongoing. Rapamycin not only targets multiple facets of this complex biological cascade, but it also has immunological properties and effects on blood flow. Coupled with the fact that rapamycin already has known biological efficacy and dose tolerability in human patients for transplant and cardiovascular medicine, means that it may rapidly and relatively cheaply be repurposed for use as an adjunct stroke therapy to the gold standards of thrombolysis and thrombectomy – protecting neurons until perfusion can be restored. 191
Exploring the role of mTOR in neurovascular and neuroimmune regulation is in its infancy. An increased understanding of how this pathway contributes to pathology will be key for developing novel therapies. Further investigations are needed into the precise molecular mechanisms of rapamycin, the most efficacious dosing, and optimal timing in relation to ischemic stroke. By targeting multiple processes within the NVU post-stroke, we may extend the time window for effective restoration of blood flow and improve functional outcomes in stroke patients.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: AMB, BAS, DJB and YC were funded by the Medical Research Council (MR/M022757/1). GH is supported by the Oxford University Clinical Academic Graduate School, Oxford. BAS is also funded by the National Health and Medical Research Council (APP1137776), Rebecca J. Cooper Foundation, J J Mason and H S Williams Memorial Foundation, Brain Foundation and Royal Hobart Hospital Research Foundation.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article:AMB is a senior medical science advisor and co-founder of Brainomix, a company that develops electronic ASPECTS (e-ASPECTS), an automated method to evaluate ASPECTS in stroke patients. All other authors declare no conflict of interest.
Authors' contributions
The manuscript was written by GH, DJB, YC, AAN and BAA. Figures were constructed by DJB and YC. The ideas upon which the manuscript is based were conceived by GCD, BAS and AMB. All authors edited the manuscript.