
Abstract
Located at the interface of the circulation system and the CNS, the basement membrane (BM) is well positioned to regulate blood–brain barrier (BBB) integrity. Given the important roles of BBB in the development and progression of various neurological disorders, the BM has been hypothesized to contribute to the pathogenesis of these diseases. After stroke, a cerebrovascular disease caused by rupture (hemorrhagic) or occlusion (ischemic) of cerebral blood vessels, the BM undergoes constant remodeling to modulate disease progression. Although an association between BM dissolution and stroke is observed, how each individual BM component changes after stroke and how these components contribute to stroke pathogenesis are mostly unclear. In this review, I first briefly introduce the composition of the BM in the brain. Next, the functions of the BM and its major components in BBB maintenance under homeostatic conditions are summarized. Furthermore, the roles of the BM and its major components in the pathogenesis of hemorrhagic and ischemic stroke are discussed. Last, unsolved questions and potential future directions are described. This review aims to provide a comprehensive reference for future studies, stimulate the formation of new ideas, and promote the generation of new genetic tools in the field of BM/stroke research.
Introduction
The basement membrane (BM) is an amorphous structure located at the abluminal side of endothelial cells or basal side of epithelial cells.1–5 It is composed of multiple highly organized extracellular matrix (ECM) proteins and has a thickness of 50–100 nm.6–11 The four major types of ECM proteins found in the BM are collagen IV, laminin, nidogen, and heparan sulfate proteoglycans (HSPGs).1,2,12,13 In addition, some other components, including fibulins, osteonectin and netrin-4, are also found in the BM.3,14–20
In the brain, the BM is predominantly associated with blood vessels. Unlike peripheral organs, the brain has two different BMs: an endothelial BM made mainly by BMECs and a parenchymal BM produced primarily by astrocytes (Figure 1(a) and (b)).21–23 Sandwiched between endothelial cells and astrocytic endfeet, pericytes also contribute to BM formation by synthesizing and depositing ECM proteins to endothelial and/or parenchymal BMs. These two BMs are separated by pericytes and appear as one in regions without pericytes in physiological conditions (Figure 1(a) and (b)).21,22 During neuroinflammation, however, these two BMs can be separated at postcapillary venules by infiltrating leukocytes,1,24 forming perivascular cuffs. Located at the interface of the circulation system and the CNS, the BM is well positioned to regulate blood–brain barrier (BBB) integrity under both homeostatic and pathological conditions.
Anatomical location of the BMs at the BBB. (a) Diagram illustration of the endothelial and parenchymal BMs at the BBB. (b) An electron microscopy image showing endothelial and parenchymal BMs at the BBB. BM: basement membrane; BBB: blood–brain barrier. (c) The cellular source of different laminin isoforms and their distribution in the endothelial and parenchymal BMs. (d) Changes of the four major BM components after hemorrhagic and ischemic stroke.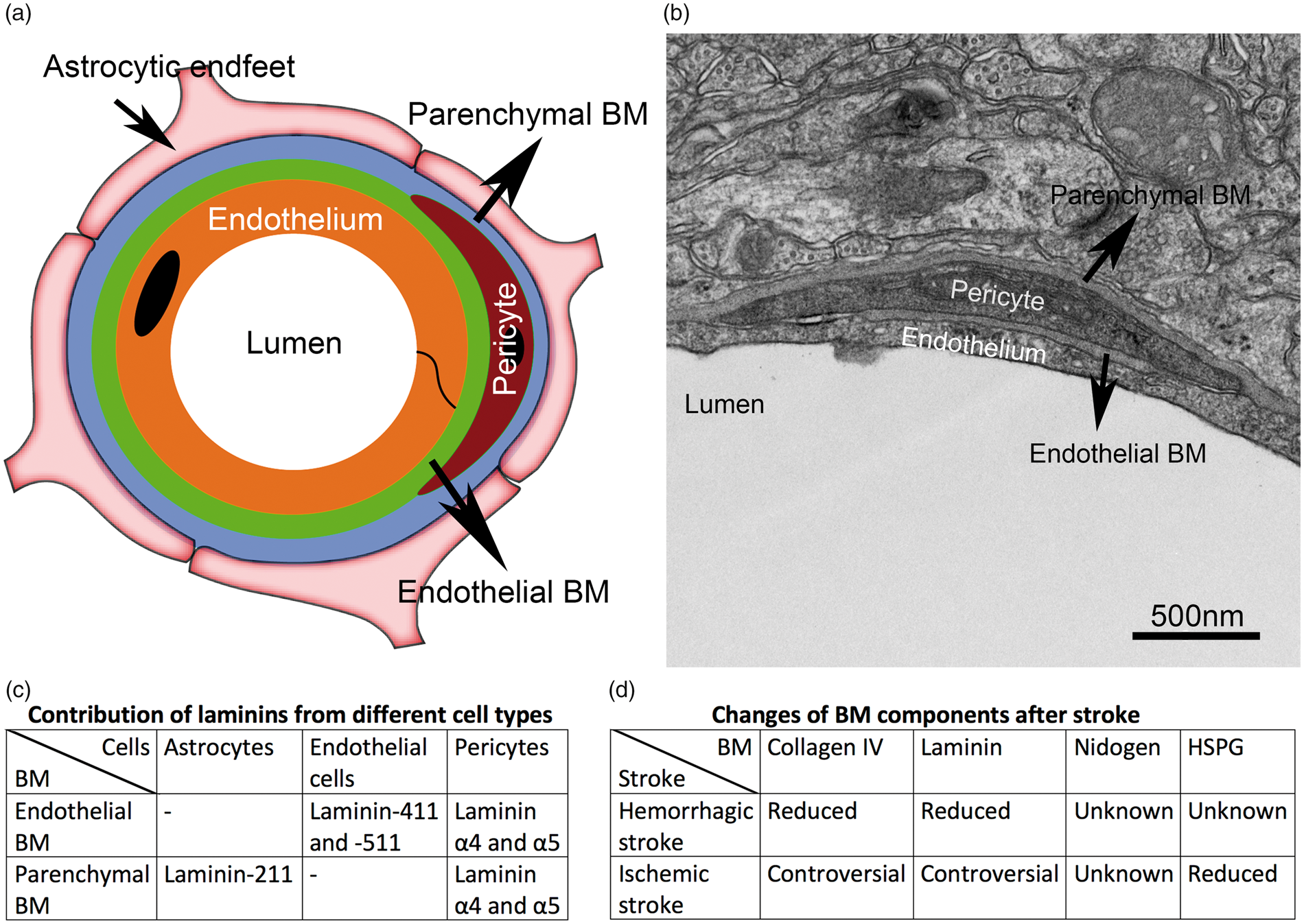
Here in this review, I first briefly introduce the composition of brain BMs. Next, the functions of the BM and its major components in BBB maintenance and stroke pathogenesis are discussed and summarized. Furthermore, unsolved questions, current challenges, and potential future directions are described. This review aims to provide a comprehensive reference for future studies, stimulate the formation of new ideas, and promote the generation of new genetic tools in the field of BM/stroke research.
BM composition
Brain BMs mainly consist of collagen IV, laminin, nidogen, and HSPGs.1,2,12,13 The structure of each component and their expression in cerebral blood vessels are discussed below.
Collagen IV
Collagen IV is the most abundant component of the BMs. It is a trimeric protein containing three α chains. Currently, 6 collagen IV α chains (Col4a1-6) have been identified.25–30 Each α chain contains an N-terminal 7S domain, a triple-helical domain, and a C-terminal globular non-collagenous-1 (NC1) domain (Figure 2(a)). During collagen IV network assembly, three α chains bind to each other forming a trimer, called protomer. These protomers then dimerize via their NC1 domains and tetramerize through their 7S domains, forming sheet-like suprastructures (Figure 2(a)).2,31,32 In the cerebrovasculature, collagen IV is predominantly produced by endothelial cells,33–35 astrocytes,33,36 and pericytes.
37
Structures of major BM components. (a) Structures of collagen IV monomer (α chain), trimer (protomer), and network. (b) Structure of a fully assembled laminin molecule. (c) Structure of nidogen-1/2. (d) Structure of HSPG2. BM: basement membrane; HSPG2: heparan sulfate proteoglycan-2.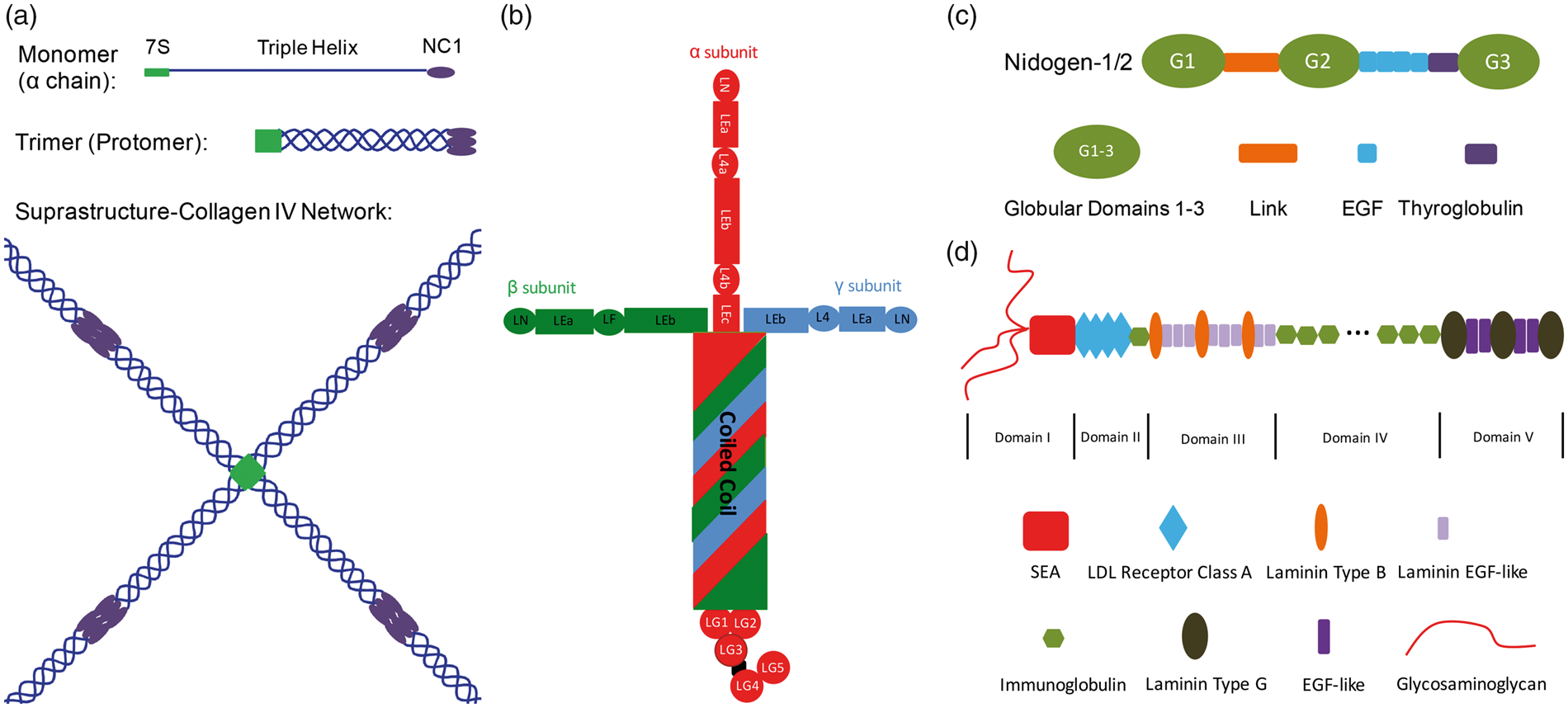
Laminin
Laminin isoforms.

Two nomenclature systems exist for laminins. The original system names laminins using numbers based on the order of their discovery, such as laminin-1, laminin-2, and so on. 46 The alternative system names laminins using both Greek letters and numbers, which indicate the subunits and genetic variants, respectively. 43 For example, laminin-1 is written as laminin-α1β1γ1, which can be abbreviated as laminin-111. Compared to the original system, the alternative system provides information regarding the trimeric composition of laminins, and thus is more widely used nowadays. A comparison of these two nomenclature systems is illustrated in Table 1.
In the cerebrovasculature, laminins are made by endothelial cells, astrocytes, and pericytes.22,47,48 Interestingly, these cells synthesize different laminin isoforms. Specifically, endothelial cells mainly make laminin-411 and -511,21,49,50 whereas astrocytes predominantly produce laminin-211.21,51 Our unpublished data show that pericytes mainly synthesize γ1- and α4/α5-containing laminins (most likely laminin-421 and -521). This cell-specific expression pattern allows differential distribution of distinct laminin isoforms in the endothelial and parenchymal BMs: the former is rich in laminin-411 and -511, while the latter mainly contains laminin-211 (Figure 1(c)). 22
Nidogen
Nidogen, also known as entactin, is a glycoprotein containing three globular (G1-3) and multiple rod-like (Link, EFG, and Thyroglobulin) domains (Figure 2(c)). In mammals, two genetic variants of nidogen (nidogen-1/2) have been identified. Unlike collagen IV and laminin, nidogens are unable to self-assemble or form sheet-like supramolecules. They can, however, interact with collagen IV and laminin via different domains, suggesting that they may function as a cross-linker to connect/stabilize collagen IV and laminin networks. Nidogens are mainly generated by endothelial cells, 35 astrocytes, 52 and pericytes35,53 in the cerebrovascular system.
HSPG
One important HSPG is heparan sulfate proteoglycan-2 (HSPG2), also called perlecan. It is a large multi-function protein composed of five domains (domain I-V) (Figure 2(d)).54–57 Domain I has three glycosaminoglycans and a sea urchin sperm protein-enterokinase-argin (SEA) motif; Domain II contains four low-density lipoprotein (LDL) receptor class A repeats and an immunoglobulin (Ig) domain; Domain III has multiple laminin B and laminin EGF domains; Domain IV consists of a series of Ig domains; and Domain V, also known as endorepellin, contains many laminin G and EGF domains. Like nidogen, HSPG2 cannot form supramolecular sheet-like structures. 54 Instead, it is able to interact with many molecules,54,57–65 including other BM components (ECM proteins) and heparin-binding growth factors. Similarly, HSPG2 expression is detected in endothelial cells,66–68 astrocytes,69,70 and pericytes 35 in cerebral blood vessels.
BM and BBB
In physiological conditions, the BMs play a variety of important roles, ranging from providing structural support to modulating molecular signaling.22,71 In addition, accumulating evidence suggests that the BMs also contribute to vascular integrity. For example, leukocytes are more frequently observed between endothelial cells and the underlying BMs than in the process of passing between endothelial cells, in electron microscopy studies.72–74 In addition, it has been reported that: (1) leukocytes take ∼5 min to cross the endothelial layer, but 20–30 min to penetrate the underlying BMs75–78; and (2) T lymphocytes spend 9-10 min crossing the endothelium, but are trapped in the outer surface of blood vessels for ∼30 min. 79 These results strongly indicate that crossing the BMs is the rate-limiting step in leukocyte infiltration, highlighting a critical role of the BMs in the maintenance of vascular integrity.
In the brain, the BMs are located at the interface of the circulation system and the CNS (Figure 1).80,81 This unique anatomical structure/location, together with that the BM is a non-cellular component of the BBB, suggests that the BMs also participate in the regulation of BBB integrity. The BMs may exert this important function directly by serving as a physical barrier at the BBB or indirectly by signaling to endothelial cells, astrocytes, and/or pericytes. In support of the latter possibility, receptors for BM components (ECM proteins), including integrins and dystroglycan, have been identified in these cells.40,47 More importantly, genetic ablation of these receptors and their respective ligands usually results in similar phenotypes,12,40,41,48 strongly suggesting that ECM proteins exert functions by binding to their receptors. Below, we discuss the function of each major BM component in BBB maintenance.
Collagen IV
Col4a1/2, two of the most frequently used α chains, make the major collagen IV isoform α1(IV)2α2(IV). Genetic ablation of Col4a1/2 leads to embryonic lethality at embryonic day (E) 10.5-11.5. 82 Interestingly, BM defects were detected in these mutants at E10.5–11.5 but not before E9.5, 82 suggesting that collagen IV is dispensable for initial BM assembly but essential for BM maintenance and function. To overcome the embryonic lethality, a series of less severe mutations in Col4a1/2 have been generated in mice.83,84 These mutants develop CNS phenotypes, including compromised vascular integrity, stress-induced hemorrhage and adult-onset stroke, to various degrees (see “Collagen IV changes in hemorrhagic stroke” section for details),83–85 suggesting an indispensable role of collagen IV in the maintenance of cerebrovascular integrity.
Laminin
Since different cells synthesize distinct laminin isoforms, laminin’s functions are discussed in a cell-specific manner. Endothelial cells mainly make laminin-411 and -511.21,49,50 It has been shown that ablation of laminin α4 leads to disrupted vascular integrity and hemorrhage at embryonic and perinatal stages, but not in adulthood. 86 This age-dependent phenotype is due to compensatory and ubiquitous expression of laminin α5 in the vascular tree at postnatal stage. 87 Unlike laminin α4, laminin α5, 88 β1,89,90 or γ1,91–92 null mice are embryonic lethal, preventing investigation of their functions in BBB integrity. To overcome this early lethality and enable such investigation, laminin α5 endothelium-specific (Tie2-Cre) conditional knockout (α5-TKO) mice were generated. Using two different laminin α5-floxed lines, our group (unpublished data) and others93,94 independently demonstrate that α5-TKO mice develop normally without BBB defects under homeostatic conditions, suggesting a dispensable role of endothelial laminin α5 in BBB maintenance. Due to potential compensation between laminin-411 and -511, it remains unclear how endothelial laminin regulates BBB integrity under physiological conditions. We are currently generating transgenic mice lacking both laminin isoforms in endothelial cells. These mutants will enable us to answer this important question.
Unlike endothelial cells, astrocytes predominantly produce laminin-211.21,51 To investigate the role of astrocytic laminin in BBB integrity, we generated neural progenitor- and neuron-specific laminin γ1 conditional knockouts by crossing the laminin γ1flox/flox mice 95 with nestin-Cre and CaMK2a-Cre transgenic lines, respectively. The former mutants (termed NKO hereafter) have laminin deficiency in both neurons and glial cells, while the latter mutants (termed CKO hereafter) have laminin deficiency in neurons only. NKO but not CKO mice display BBB disruption 96 and age-dependent hemorrhagic stroke, 97 suggesting that astrocytic rather than neuronal laminin is required for BBB maintenance. Consistent with this finding, laminin α2 null mice develop severe BBB breakdown, 98 again suggesting an indispensable role of astrocytic laminin in BBB maintenance.
Laminin mutants and their phenotypes.
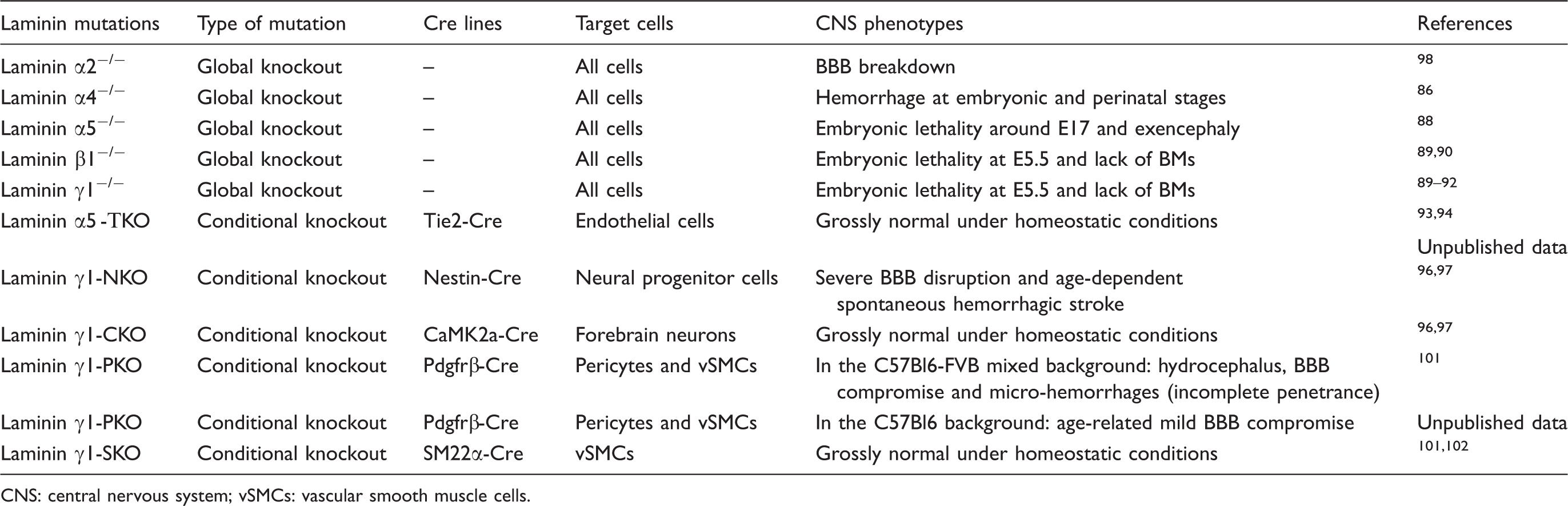
CNS: central nervous system; vSMCs: vascular smooth muscle cells.
Nidogen
Genetic abrogation of either nidogen-1107–110 or nidogen-2 111 results in a grossly normal phenotype, suggesting that they may be able to compensate for each other’s loss in these single mutants. Consistent with this hypothesis, nidogen-1/2 double knockout mice die shortly after birth and show severe BM defects in multiple organs,112,113 suggesting an important role of nidogen-1/2 in BM maintenance. Based on the observed BM defects in the brain, we expect that BBB integrity is compromised in these double mutants. This hypothesis needs further validation/investigation in future studies.
HSPG
Global knockout of HSPG2 leads to severe developmental defects in multiple organs.114,115 The mutants die either around E11 or at the perinatal stage due to defective cephalic, myocardial, and skeletal development.114,115 Although BM formation is not affected, BMs deteriorate in areas with increased mechanical stress in these mutants, 114 indicating an essential role of HSPG2 in BM maintenance rather than assembly. Consistent with BM changes, a hemorrhagic phenotype in the brain, skin and lung is observed in these mutants, 114 suggesting that HSPG2 is required for the maintenance of vascular integrity.
BM and stroke
Stroke is a clinical emergency caused by rupture (hemorrhagic) or occlusion (ischemia) of cerebral blood vessels. It is the fifth leading cause of death and the leading cause of serious long-term disability in the United States. 116 One key pathological change after stroke is inflammatory cell infiltration across the compromised BBB, which profoundly influences disease progression. Due to their important roles in BBB maintenance and vascular integrity, the BMs have been hypothesized to affect inflammatory cell extravasation and stroke pathogenesis. Previous studies have demonstrated that immune cell infiltration predominantly occurs at specific low expression regions characterized by reduced expression of certain BM constituents.87,117–121 For example, neutrophils117–120 and monocytes 117 preferentially penetrate the BM in regions with reduced levels of laminin α5 and collagen IV in peripheral tissues. In addition, in both lymph nodes and brains, T lymphocytes mainly extravasate across blood vessels through laminin α4high and laminin α5low regions.87,121 It is speculated that inflammatory cells use these low expression regions to infiltrate the brain after stroke. However, due to the lack of knowledge on the alterations of specific laminin isoforms after stroke and the existence of controversial results on how collagen IV/laminin change after ischemic stroke (Figure 1(d)), the function of the BM in inflammatory cell extravasation after stroke remains largely unknown and needs further investigation. Here, we discuss BM changes and functions in both hemorrhagic and ischemic stroke.
BM and hemorrhagic stroke
Multiple studies have shown that the BM negatively correlates with hemorrhagic stroke. For example, BM degradation has been reported in rats at 24–72 h after subarachnoid hemorrhage 122 and in mice with age-dependent spontaneous intracerebral hemorrhage (ICH). 97 In addition, loss of BM components and/or their mutations usually lead to BM defects and hemorrhagic stroke (see below for details). BM loss during hemorrhagic stroke may be caused by diminished ECM protein synthesis or increased ECM protein degradation. We lean towards the second possibility. This is because: (1) ECM proteins usually have a long turnover rate, 123 and (2) various ECM-degrading proteases, including matrix metalloproteinases (MMPs) and thrombin, are dramatically induced/activated after hemorrhagic stroke.124–132 Changes of major BM components after hemorrhagic stroke are discussed below and summarized in Figure 1(d).
Based on these findings, it is logical to hypothesize that inhibiting BM-degrading enzymes and/or supplementing BM components may have a therapeutic potential in hemorrhagic stroke. Consistent with this speculation, various MMP inhibitors, including minocycline, GM6001 and BB-1101, have been found beneficial in hemorrhagic stroke.124,133,134 In contrast to these results, BB-94, another broad-spectrum MMP inhibitor, has been reported to increase hematoma size and apoptosis after hemorrhagic stroke. 135 In addition, minocycline failed to affect clinical and radiological outcomes in patients with hemorrhagic stroke in a pilot study. 136 This inconsistency may be due to different experimental models and/or low specificity of these MMP inhibitors. We are currently investigating the therapeutic potential of exogenous BM components in hemorrhagic stroke.
Collagen IV changes in hemorrhagic stroke
Collagen IV mutants and their phenotypes.
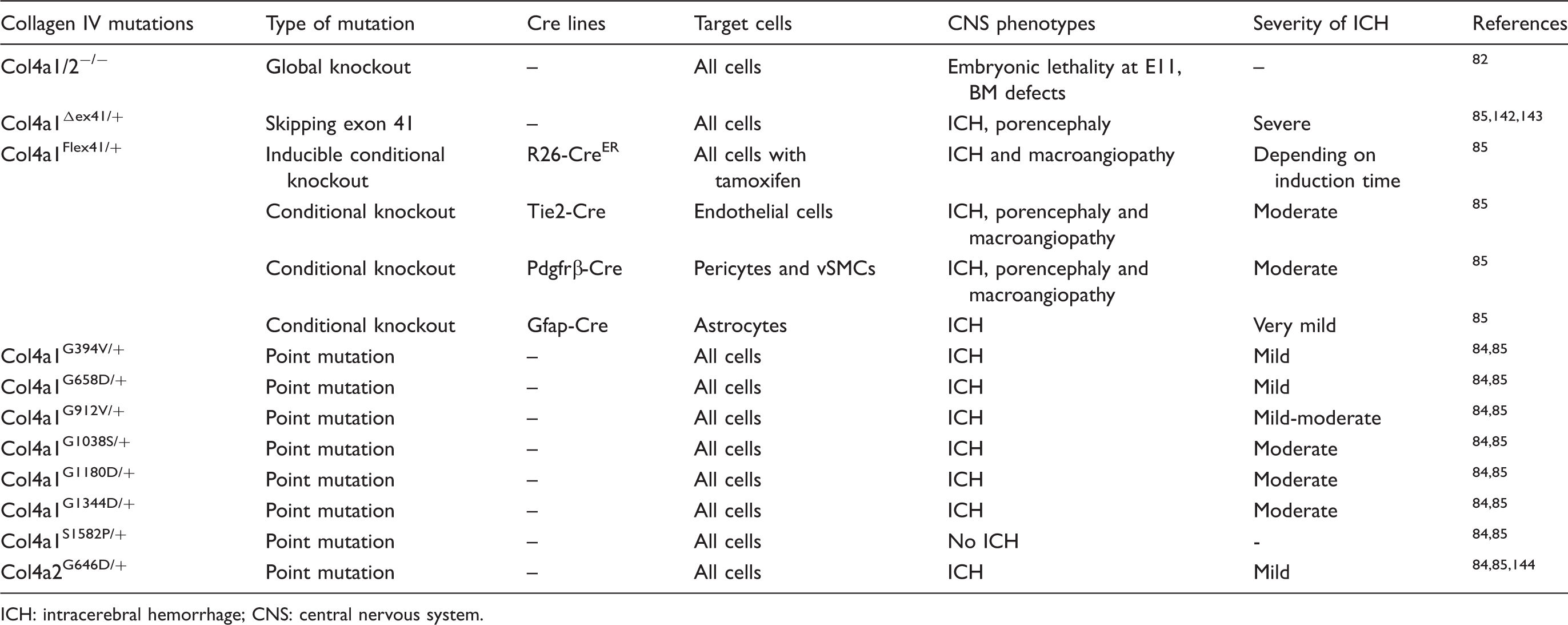
ICH: intracerebral hemorrhage; CNS: central nervous system.
Laminin changes in hemorrhagic stroke
Due to the substantial up-regulation and activation of various BM-degrading proteases after hemorrhagic stroke, laminin levels are expected to decrease in hemorrhagic brains. Consistent with this hypothesis, significantly lower levels of laminin were found in hemorrhagic areas compared to non-hemorrhagic areas in ischemic brain. 159 Interestingly, loss of laminin is associated with hemorrhagic stroke in an isoform-specific manner. First, a previous study from our laboratory showed that NKO but not CKO mice developed age-dependent spontaneous ICH, 97 suggesting a causative role of loss of astrocytic laminin (laminin-211) in hemorrhagic stroke. Next, although micro-hemorrhages and hydrocephalus occur hand-in-hand in PKO mice in the C57Bl6-FVB mixed background, micro-hemorrhages are absent in these mutants in the C57Bl6 background when the incidence of hydrocephalus is reduced to a negligible level. These findings suggest that micro-hemorrhages in PKO mice in the mixed background are probably secondary to hydrocephalus and that loss of pericytic laminin does not cause hemorrhagic stroke. Additionally, due to potential compensation between laminin-411 and -511, whether loss of endothelial laminin results in hemorrhagic stroke remains unknown. Mice with endothelium-specific ablation of both laminin-411 and -511 will enable us to answer this important question.
Nidogen changes in hemorrhagic stroke
How nidogen levels change in hemorrhagic stroke remains largely unknown. One in vitro study showed that nidogen fragmentations were found in astrocyte medium after exposure to plasma kallikrein, 160 suggesting that nidogen may be degraded after hemorrhagic stroke. This result needs to be validated in vivo using hemorrhagic models. Due to the grossly normal phenotype of nidogen-1/2 single knockouts and the perinatal lethality of the double knockouts, the role of nidogen in the pathogenesis of hemorrhagic stroke remains unclear. Mice with simultaneous deletion of nidogen-1/2 in different cell types (conditional nidogen-1/2 double knockouts) will provide insights into the function of nidogen in hemorrhagic stroke.
HSPG changes in hemorrhagic stroke
Like nidogen, how hemorrhagic stroke affects HSPG2 level is unclear. In addition, due to early embryonic lethality, the function of HSPG2 in the development and progression of hemorrhagic stroke is unknown. Similarly, HSPG2 conditional and/or inducible knockout mice will enable us to answer these important questions.
BM and ischemic stroke
Similar to hemorrhagic stroke, BM dissolution is associated with ischemic stroke. For example, loss of BM is found soon after the onset of ischemia. 161 In addition, it has been reported that BM degradation occurs as early as 10 min after reperfusion in the middle cerebral artery occlusion (MCAO) model, 162 and that loss of BM can be detected as early as 1–3 h after ischemia.161,162 Ultrastructurally, the well-defined electron-dense BMs become diffused and faint after ischemia.163,164 Biochemically, many BM components are degraded and reduced after ischemic stroke.161,165 Like in hemorrhagic stroke, BM loss in ischemic stroke is mainly due to enhanced protein degradation rather than reduced protein synthesis. This is supported by the following observations. First, MMP expression and their activities are markedly up-regulated after ischemic stroke.166–171 Second, the expression levels and enzymatic activities of cathepsin B and L, which primarily degrade HSPG2 and other cellular components, 172 are dramatically enhanced after ischemia.172,173 The changes of major BM components after ischemic stroke are discussed below and summarized in Figure 1(d).
Recombinant tissue-type plasminogen activator (tPA) is the only FDA-approved therapy for ischemic stroke. tPA exerts its protective effects by inducing plasmin-dependent fibrinolysis inside blood vessels. 174 However, with the progression of disease, tPA leaks out of blood vessels through compromised BBB. Outside blood vessels, tPA substantially enhances MMP-9 levels and exaggerates MMP-mediated BM degradation.175–177 This deleterious effect outside blood vessels may explain the short (4.5 h) therapeutic window of exogenous tPA in ischemic stroke. Based on these results, we hypothesize that inhibiting MMPs and/or supplementing BM components may be beneficial in ischemic stroke. In agreement with this hypothesis, many broad-spectrum MMP inhibitors, including minocycline, doxycycline, GM6001 and BB-94, displayed protective effects in ischemic stroke,178–181 although one study showed that intravenous injection of minocycline was safe but not effective. 182 SB-3CT, an MMP-2/9 specific inhibitor, restored laminin degradation, reduced infarct volume, and improved neurological function in a mouse model of ischemic stroke.183,184 In addition, many studies showed that MMP inhibitor-tPA combined therapies significantly reduced hemorrhagic complications of tPA.178,185–189 We are currently investigating the therapeutic effects of BM components in ischemic stroke.
Collagen IV changes in ischemic stroke
Unlike in hemorrhagic stroke, controversial results exist on how collagen IV level changes in ischemic stroke. Most studies support that collagen IV is reduced after ischemic stroke. For instance, reduced collagen IV levels were found in rats with thromboembolic 190 or suture132,191 model of MCAO. In addition, substantial loss of collagen IV was also detected in non-human primate baboons (Papio anubis/cynocephalus) after ischemia-reperfusion injury.172,192 Furthermore, an association between ischemic stroke and Col4a1 mutations was found in humans,150,193,194 suggesting that collagen IV may be involved in the pathogenesis of ischemic stroke. In contrast to these results, one study found increased collagen IV expression in the spinal cord 24 h after ischemia–reperfusion injury. 195 In addition, it was also reported that MCAO alone without systemic inflammation failed to affect collagen IV level in the brain. 196 This discrepancy may be due to different organs examined (brain vs. spinal cord). Whether ischemic stroke affects collagen IV level in an organ-specific manner needs further investigation.
Laminin changes in ischemic stroke
Like collagen IV, laminin changes after ischemic stroke have been controversial. On one hand, there is evidence demonstrating that laminin is reduced after ischemic stroke. It has been shown that ischemic stroke increases the activity of MMP-9, which degrades laminin in mouse brain. 184 In addition, laminin expression is substantially decreased in Mongolian gerbils (Meriones unguiculatus) up to 72 h of reperfusion after ischemia. 197 Similarly, ischemia-reperfusion injury induces a gradual and continuous reduction of both laminin expression and laminin-positive vessel number in baboons.172,192 Furthermore, diminished laminin level is also found in patients with ischemic stroke. 198 Interestingly, loss of laminin correlates well with the increase/activation of various proteases,172,192,197 again suggesting that proteolytic degradation is responsible for the loss of laminin in ischemic stroke.
On the other hand, there is also evidence showing that laminin expression is unaffected or even enhanced after ischemic stroke. For example, laminin level was found unchanged in the brain after MCAO without systemic inflammation. 196 Laminin was also reported to be up-regulated in the ischemic core within 24 h after injury.199,200 Additionally, astrocytes have been shown to express high levels of laminin two to three days after ischemia, which forms a barrier and separates the ischemic regions from healthy tissue.199,200 Furthermore, it has been demonstrated that ischemia induces a transient up-regulation of laminin in the brain in a COX-2 dependent manner. 201
These paradoxical results may be explained by different animal models used and time points examined. In addition, distinct fixation methods may also contribute to this discrepancy. It has been shown that heavy formaldehyde fixation masks laminin antigen in the BM, while mild-moderate formaldehyde fixation reveals it. 202 Furthermore, it is worth mentioning that most of the above-mentioned studies used a pan-laminin antibody to examine laminin expression. It remains unclear whether different laminin isoforms have distinct susceptibility to ischemia and how these laminin isoforms change after ischemia. Future research should address these important questions.
Nidogen changes in ischemic stroke
Like in hemorrhagic stroke, nidogen changes in ischemic stroke are largely unknown. One in vitro study showed that oxidative stress, one pathological alteration found in ischemic stroke, enhanced nidogen expression in human BMECs, 203 suggesting that nidogen may be up-regulated after ischemic stroke. Whether ischemic stroke increases nidogen expression in vivo and how nidogen affects the pathogenesis of ischemic stroke need further investigation.
HSPG changes in ischemic stroke
There is evidence showing that ischemic stroke decreases HSPG2 levels. One study reported a 43–63% reduction of HSPG2 within a few hours after ischemic injury in baboons, 172 suggesting that HSPG2 is one of the most sensitive ECM proteins to proteolysis in ischemic brain. 172 Unlike other ECM proteins, which get degraded, HSPG2 is cleaved into small fragments after stroke. For instance, high levels of HSPG2 domain V (endorepellin)204,205 and its C-terminal fragment 206 are detected in rodents in multiple ischemic stroke models. Consist with these reports, a similar result was found in humans with ischemic stroke. 207 Further studies demonstrate that these HSPG2 fragments are bioactive and exert a beneficial role in ischemic stroke. It has been reported that these HSPG2 fragments are able to diminish ischemic volume, reduce neuronal death, modulate astrogliosis, enhance angiogenesis, and improve motor function.204,206,208
Future directions
Due to its intrinsic complexity and technical difficulties in its research, the BM has been understudied. With advances in biochemistry and genetics, many valuable research tools (e.g. transgenic mouse lines) have been generated and substantial progress has been made in this field. Many important questions, however, remain to be answered. These questions include: when and where is each BM component expressed? What is the relative contribution of these BM components from each cell type? Is there functional compensation among different isoforms of ECM proteins? How do distinct laminin isoforms change after stroke? How do nidogen and HSPG2 levels change in hemorrhagic and ischemic stroke? How does each BM component affect the pathogenesis of hemorrhagic and ischemic stroke? Answers to these questions rely on the generation of cell-specific conditional knockouts, compound mutants (e.g. double knockouts), and/or inducible knockouts. Future studies should focus on generating these mutants and answering the above-mentioned questions.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was partially supported by AHA Scientist Development Grant (16SDG29320001).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.