
Abstract
Plasmin, the principal downstream product of tissue-type plasminogen activator (tPA), is known for its potent fibrin-degrading capacity but is also recognized for many non-fibrinolytic activities. Curiously, plasmin has not been conclusively linked to blood–brain barrier (BBB) disruption during recombinant tPA (rtPA)-induced thrombolysis in ischemic stroke. This is surprising given the substantial involvement of tPA in the modulation of BBB permeability and the co-existence of tPA and plasminogen in both blood and brain throughout the ischemic event. Here, we review the work that argues a role for plasmin together with endogenous tPA or rtPA in BBB alteration, presenting the overall controversy around the topic yet creating a rational case for an involvement of plasmin in this process.
INTRODUCTION
Tissue-type plasminogen activator (tPA) 1 is a protease classically appreciated for its ability to generate the serine-protease plasmin from its inactive precursor plasminogen. Plasmin in turn is recognized primarily for its fibrinolytic role. 2 The concept of tPA-mediated plasminogen activation was harnessed for clinical development over 30 years ago 3 and is still the mainstay for thrombolysis in patients with acute ischemic stroke.4,5 Nevertheless, the clinical use of human recombinant tPA (rtPA) in stroke is restricted owing to the enhanced risk of intracerebral hemorrhage (ICH),4–6 even within the first 90 minutes post-stroke onset, 7 and reduced clinical efficacy at longer time frames (>4.5 hours). 7
To further complicate matters, tPA is also endogenously expressed in the extravascular compartment,8,9 most notably in the CNS8,10–12 where it performs a number of critical non-fibrinolytic functions.13–16 Within this context, while much effort has been devoted to understanding how endogenous tPA and rtPA modulate neuronal function,14,15 another major avenue whereby tPA can influence the CNS is via alteration of the blood–brain barrier (BBB).13,16 Mechanistically, the majority of reports suggested that tPA increases BBB permeability in a plasmin-independent manner; however, the role of plasmin may be underestimated and is accompanied by much contradicting data (summarized in Table 1). It is the purpose of this article to outline the evidence that links plasmin to tPA-dependent alteration of neurovascular permeability, with particular focus on the controversy surrounding the topic. We hypothesize that under certain circumstances, especially when vascular plasminogen and rtPA are simultaneously available around brain tissue, enhanced in situ generation of plasmin may cause substantial damage to the BBB.
Central controversies surrounding plasmin involvement in BBB modulation

BBB, blood–brain barrier; ICH, intraceretbral hemorrhage; PA, plasminogen activator; tPA, tissue-type plasminogen activator.
TISSUE-TYPE PLASMINOGEN ACTIVATOR AND THE BLOOD-BRAIN BARRIER
Over the last decade, much literature accumulated regarding the ability of tPA to interact with the BBB. The following section will summarize this sizable body of work while distinguishing the effects of rtPA from endogenous tPA, the clinical versus experimental data and observations made under naïve or ischemic conditions. The various mechanisms suggested thus far, the different approaches to tackle the issue as well as common limitations accompanying the use of rtPA in animal models will also be discussed.
Recombinant Tissue-Type Plasminogen Activator and the Nonischemic Blood–Brain Barrier
Most thrombolysis-related symptomatic ICH (sICH) in stroke occurs within 24–36 hours post treatment within the infarct core, suggesting that ischemia itself has a role in its etiology (sICH beyond 36 hours is not considered tPA-related).6,17 Interestingly, 20% of rtPA-associated sICH cases take place outside the anatomic infarct site (i.e., in non-affected brain areas)17,18 and cerebral bleeding complications were also observed after rtPA administration for myocardial infarction 19 (we note, however, that an underlying cerebrovascular injury was not thoroughly evaluated in these myocardial infarction cases). Hence, rtPA-associated bleeding in patients may not be fully explained by weakening of cerebral blood vessels due to the stroke.
In line with this notion, recent reports, albeit in rats, have demonstrated that intraarterial (2–4 mg/kg) 20 or intravenous21,22 administration of rtPA or catalytically inactive rtPA 21 (1 mg/kg) into naïve animals mediated rapid opening of the BBB within 1–4 hours (as evidenced by accumulation of Evans blue-labelled albumin in the brain parenchyma), indicating that vascularly originated rtPA may influence the healthy brain endothelium, perhaps independent of its catalytic activity. In contrast, others could not reproduce these observations in uninjured rats (using fluorescent dextran), 23 although this particular study showed that rtPA itself could cross the intact BBB via transcytosis and exacerbate neuronal injury (see section Exposure of the Blood–Brain Barrier to Plasmin During Stroke and Thrombolysis). Moreover, no increase in albumin extravasation 24 or hemorrhage 25 was reported in naive mice receiving intravenously mouse rtPA 24 or rtPA, 25 respectively, even when given at 10 mg/kg (see below). Instead, the BBB of naïve mice was permeated by Evans blue only when exogenous tPA was injected intracerebroventricularly (~0.02 to 0.6 μg),24,26 bypassing the intact BBB and acting preferentially on astrocytes.24,27 Taken together, and despite prominent discrepancies between species and different research groups, these experiments highlight an ability of rtPA to act in vivo, in healthy conditions, on the luminal (endothelial) as well as the abluminal (astrocytic; cerebral) compartments of the neurovascular unit (NVU). It is worth distinguishing, however, that while vascular rtPA may promote extravasation of blood proteins through the uninjured BBB, it has not been shown to trigger ICH in any naïve rodent. Hence, it is tempting to speculate that rtPA has only limited capacity to impact the healthy BBB.
Recombinant Tissue-Type Plasminogen Activator and the Ischemic Blood–Brain Barrier; Observations, Resolutions and Experimental Limitations
A number of clinical studies provided magnetic resonance imaging-based evidence for rtPA-related BBB disruption,28,29 which also correlated with an increased risk of hemorrhagic transformation 28 during thrombolysis in stroke, strongly suggesting a causal relation between rtPA and BBB breakdown in the ischemic human brain. Curiously, however, a recent meta-analysis of the largest clinical stroke trials employing rtPA-based thrombolysis found no support for the hypothesis that the incidence and risk of sICH by rtPA increase with the time from onset to treatment (evaluated up to onset to treatment ≦6 hours), 7 despite the likely deterioration of the BBB under prolonged ischemia. Nevertheless, an apparent lower rate of sICH was observed with onset to treatment ≦90 minutes, but a lack of events in the placebo group prevented evaluation of risk at this early time window. Also apparent was a significant increase in the risk of total rtPA-related hemorrhagic events (symptomatic and non-symptomatic) with onset to treatment > 180 minutes, but the absolute rates of hemorrhage were similar (30% to 35%). 7 Hence, the degree to which rtPA-induced BBB impairment contributes to the higher risk of sICH carried by the treatment (~5-fold 7 to 10-fold4,5) remains controversial.
Evidence for rtPA-mediated BBB disruption emerges also from animal studies, but observations dramatically vary between gyrencephalic species like primates, which possess distinct advantages for human stroke simulation, 30 compared with small animal models of stroke. In addition, the stroke model itself (e.g., thrombotic versus mechanical occlusion) has a bearing on the outcome of thrombolysis. For instance, in a non-human primate model, where middle cerebral artery occlusion (MCAo) is achieved by balloon compression, administration of rtPA (up to 10 mg/kg) within 3.5 hours post-stroke onset did not increase the incidence or severity of ICH (later administration of rtPA was not examined). 31 In contrast, rtPA infusion (3.3 mg/kg) 1 hour post embolic stroke in rabbits was sufficient to increase the ICH rate, 32 indicating a high sensitivity of the rabbit to rtPA-dependent bleeding in these settings.
While limited in their ability to model human disease, numerous studies using MCAo and embolic stroke models in rodents, either relying on endogenous tPA 26 or administering rtPA (up to 10 mg/kg),20,21,24,25,33–41 demonstrated that tPA can compromise the BBB. Indeed, treatment with rtPA, particularly beyond 3 hours of stroke onset (but in some cases earlier 25 ), is often accompanied by increased BBB permeability,20,21,33,34 enhanced hemorrhagic transformation,25,32,34–40 degradation of basement membrane (BM) and tight junction (TJ) proteins,25,34,38,40 greater edema,33,34,36 and aggravated neurologic deficits.21,40
Accordingly, strategies aiming to protect the BBB from rtPA-mediated damage and reduce thrombolysis-associated ICH were put forward, including blockade of matrix metalloproteinases (MMPs),32,41 free-radical scavenging,37,38,42 inhibition of intracellular enzymes like phosphodiesterase-III, 36 Rho-kinase (ROCK)35,43 or poly(ADP-ribose)polymerase, 40 blockade of surface receptors such as LDL receptors (LDLRs)26,39,43 or the platelet-derived growth factor (PDGF) receptor-α 24 and employment of activated protein C, 25 plasminogen-activator inhibitor-1 (PAI-1)-derived peptides21,44 or progesterone. 34 While results of these substantial efforts in experimental models seemed promising, no agent has been clinically implemented thus far in conjunction with rtPA to protect the BBB during thrombolysis.
Noteworthy caveats often accompany murine experiments examining the effects of rtPA on the BBB during stroke. First, the use of (human) rtPA in the rodent creates a considerable dilemma, as human tPA, whereas conserved (shares 81% homology with its mouse counterpart 45 ), is altered in crucial sequences, including the docking site for PAI-1, 21 its main physiologic inhibitor. 2 In addition, the activation rate of murine plasminogen by human tPA is 5- to 10-fold lower than in an autologous system, 46 resulting in poorer capacity of human tPA to lyse murine cross-linked clots. 47 Therefore, a rtPA dose of 10 mg/kg is commonly used in rodents as a compensatory measure24,25,33,35–41 compared with the clinical dose of 0.9 mg/kg. This exposes the murine system to suprapharmacological plasma concentrations of rtPA (typically in the low nanomolar range during thrombolysis in humans 48 ), potentially exaggerating the amplitude of non-fibrinolytic actions of rtPA (like induced production of MMPs and BBB impairment) and causing overestimation of their pathophysiological relevance. However, efficient tPA-induced opening of the rodent BBB has been demonstrated with lower rtPA dosing regimens (1 to 6 mg/kg20,21,34), reducing the likelihood of a complete artefactual effect.
A final issue relates to the formulation buffer (or ‘vehicle’) of rtPA, containing vast quantities of
The Role of Endogenous Tissue-Type Plasminogen Activator
A recent stroke study has shown that the infarct volume in humans and (female) mice as well as the extent of BBB damage in the latter was inversely correlated with age. 50 Curiously, tPA activity and messenger RNA levels also declined in the mouse brain with aging (but not in the spinal cord and plasma; measurements that could not be performed in brains of healthy humans), raising the hypothesis that endogenous tPA levels may impact the severity of stroke also clinically. 50
Nevertheless, this notion is not supported experimentally in the non-human primate, as tPA activity and antigen levels actually decreased or remained unchanged, respectively, 1 to 2 hours post MCAo in baboons. Instead, upregulation of uPA, uPA receptor, PAI-1, and tPA-PAI-1 complex (measured in the basal ganglia and in plasma)51,52 and loss of microvascular laminin (indicative of BBB damage) 52 were observed.
Unlike its expression profile in the primate brain, a direct contribution of endogenous tPA to BBB damage in murine stroke models was demonstrated either by tPA inhibition with its brain-specific inhibitor, neuroserpin 26 (that also reduces infarct volume 53 ), or in mice deficient of tPA,26,27 both associated with preservation of BBB integrity during 6 hours permanent MCAo. Importantly, mice lacking plasminogen, urokinase (uPA) or matrix metalloproteinase (MMP)-9 were not protected from BBB disruption in this permanent stroke model, suggesting that the effect was unique to endogenous tPA, but not to other naturally occurring plasminogen activators (PAs), and was not solely dependent on plasmin and MMP-9. 26 Temporally, endogenous tPA antigen and activity is often (but not always 54 ) shown to be transiently upregulated in the ischemic rodent brain, appearing early (within 1 to 6 hours post-stroke onset)26,53,55 around the vessel wall26,53 and promptly declining, 53 yet correlating with rapid degradation of the BM. 53 In partial agreement with observations in primates, 51 uPA (but not tPA) levels increase continuously also in the injured rodent brain for days after injury, 53 suggesting that endogenous tPA has only a limited role in the acute phase of the ischemic event. Intriguingly, formation of tPA-PAI-1 complex was recently highlighted in mice as an important factor responsible for BBB breakdown instead of active tPA itself (below). 56 Hence, it can be speculated that PAI-1 levels may in fact determine the capacity of endogenous tPA to affect the BBB. Collectively, despite the elegant work in the transgenic murine models, further studies are clearly required to ascertain how endogenous tPA contributes to BBB breakdown during stroke, especially with relevance to the human condition.
Modes of Action
Mechanistically, most major pathways linking tPA to modulation of BBB permeability were shown to be plasmin independent (Table 2). One of the leading hypotheses postulates that in the later hours of ischemic stroke (≥3 hours of onset) endogenous tPA or rtPA given during thrombolysis induce an increase in levels of MMP-9 and MMP-3 (in addition to the induction of MMP-2 and -9 by the ischemia itself). 13 In turn, MMPs degrade TJ proteins and components of the BM and contribute to BBB impairment, vasogenic edema and hemorrhagic transformation13,57,58 (Figure 1). To further support this, numerous stroke studies confirmed that (1) administration of rtPA increases MMP-9 levels in human13,59 and rodent brains,25,38,60,61 (2) that the increase in MMP-9 levels is milder in tPA-knockout mice compared with their wild-type counterparts26,60,62,63 and can be restored by reconstitution of rtPA 60 or mouse tPA 63 into tPA−/– mice and that (3) intracortical injection of neuroserpin into the ischemic area of a wild-type mouse prevents MMP-9 induction. 26 Furthermore, the increase in MMP-9 levels in rats was not reduced by co-treatment of tranexamic acid together with rtPA 60 and was also maintained in plasminogen−/– mice during MCAo, 26 suggestive of a plasmin-independent process. Mechanistically, tPA signaling via LDLR-related protein-1 (LRP-I)25,26,62 was reported to drive the enhanced production of MMP-9, mainly in brain endothelial cells (BECs),25,37,62 but also in microglia 63 and astrocytes (the later shown only in vitro). 64 Almost identical observations, albeit plasmin-related, 41 were reported with regards to MMP-3, which was upregulated in BECs in a LRP-1 dependent process in vitro 39 and was associated with augmented ICH after treatment with rtPA in stroke in vivo. 41
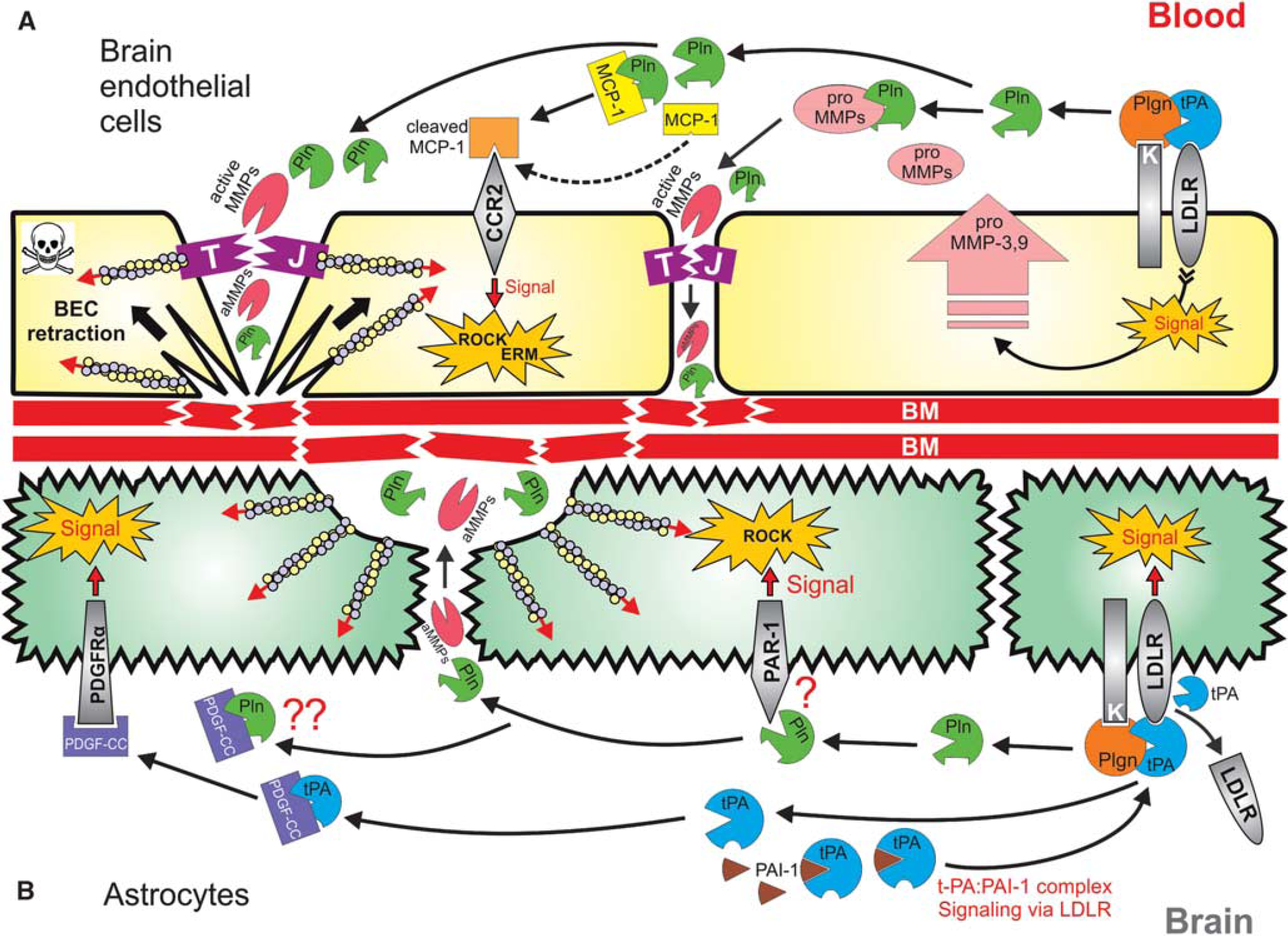
Proposed integrated model for tissue-type plasminogen activator (tPA)- and plasminogen (Plgn)-mediated modulation of blood–brain barrier (BBB) permeability during thrombolysis in ischemic stroke. (
Key molecular mechanisms for tPA- and plasmin-dependent BBB opening during stroke
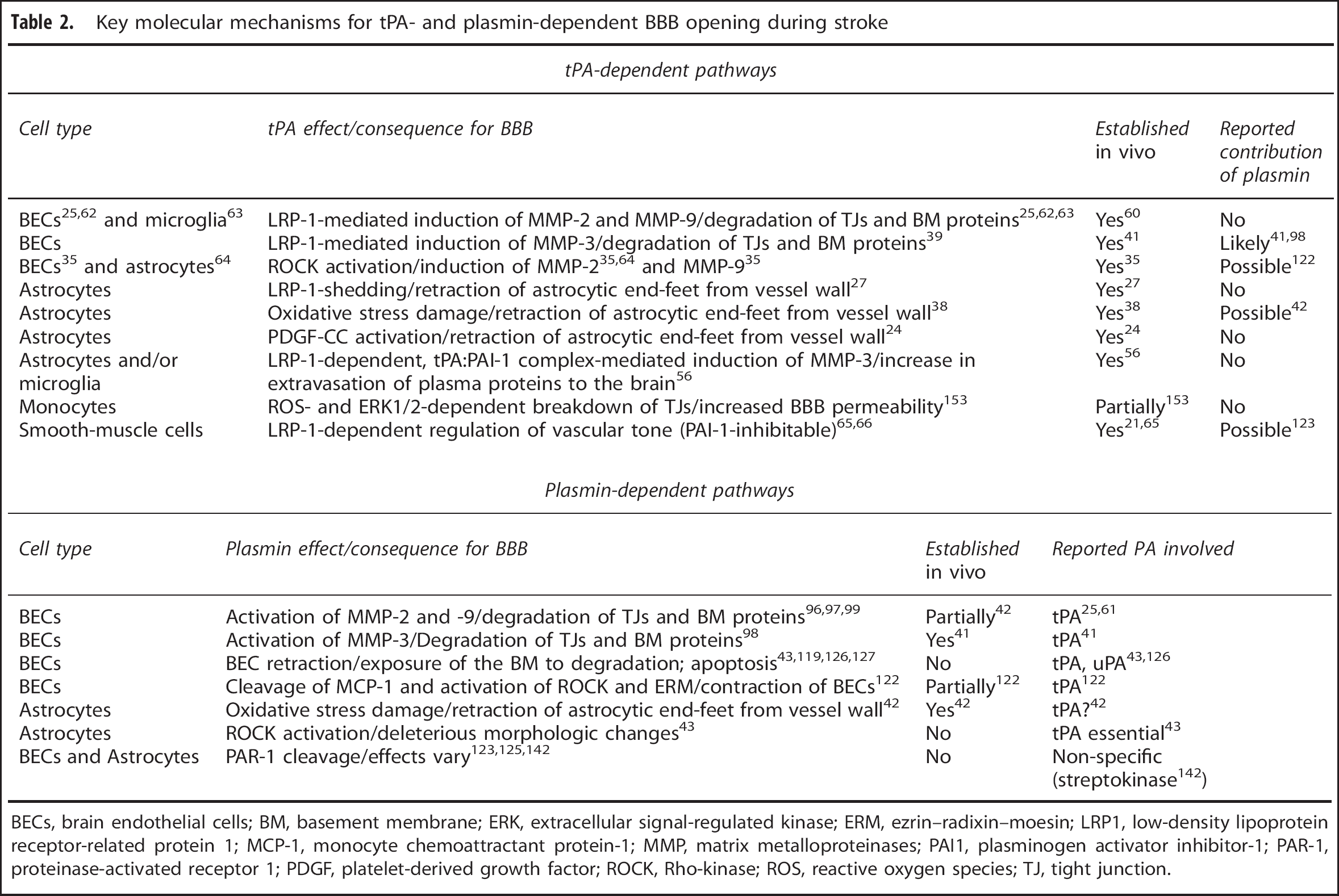
BECs, brain endothelial cells; BM, basement membrane; ERK, extracellular signal-regulated kinase; ERM, ezrin–radixin–moesin; LRP1, low-density lipoprotein receptor-related protein 1; MCP-1, monocyte chemoattractant protein-1; MMP, matrix metalloproteinases; PAI1, plasminogen activator inhibitor-1; PAR-1, proteinase-activated receptor 1; PDGF, platelet-derived growth factor; ROCK, Rho-kinase; ROS, reactive oxygen species; TJ, tight junction.
Other major pathways for BBB disruption by tPA (and rtPA) originate from the abluminal (i.e., brain-facing) compartment of the NVU,24,26 where tPA triggers harmful, plasmin-independent signaling in astrocytes by direct cleavage of LRP-1 at the astrocyte end-feet 27 and by activation of latent PDGF-CC, rendering it a PDGF receptor α-agonist. 24 These events are associated with astrocyte swelling and edema formation, accompanied by detachment of astrocyte end-feet from the BM and further permeation of the BBB24,27,38 (Figure 1) (refer to Simard et al 58 for review of molecular mechanisms underlying brain edema formation). Additional and perhaps less characterized mechanisms, both not requiring plasmin, involve tPA control of vascular tone 65 via activation of smooth-muscle cells 66 and a PAI-1-dependent mechanism described in traumatic brain injury, where signaling of the tPA-PAI-1 complex via LDLRs promotes MMP-3 production in the injured brain. 56
Despite the dominance of the plasmin-independent concept, plasminogen, the ultimate tPA substrate, is essentially present with tPA in both blood and brain under most pathologic brain scenarios, especially together with rtPA during its utilization in stroke. Furthermore, as discussed below, many actions of plasmin mirror or complement the activities of tPA associated with BBB disruption. Therefore, can plasmin truly be excluded from tPA-associated BBB alteration, particularly during thrombolysis?
IS THERE A CASE FOR PLASMIN AT THE BLOOD-BRAIN BARRIER?
The role of plasminogen and plasmin is well cemented in the context of physiologic neurobiology or pathologic neurodegeneration, affecting neuronal sprouting, 67 neuronal plasticity, 68 or extracellular matrix-related neuronal death, 69 respectively. Moreover, plasmin was implicated in microglial activation 70 and received attention for its direct contribution to brain parenchymal damage during excitotoxic injury. 10 Plasmin-associated brain damage was also inferred in models of ICH (where plasminogen potentiates thrombin-induced neurotoxicity) 71 and potentially in stroke.72,73 This evidence strongly links plasmin to brain function and dysfunction.
Based on the NVU concept (i.e., the tight coupling between neuronal metabolic demands and their supplying capillaries), one might also expect plasmin to serve as a BBB effector. This notion, however, is not always experimentally supported; indeed, early during focal cerebral ischemia (within 2 hours of stroke) in non-human primates, there is evidence of simultaneous neuronal injury and vascular damage (when microvascular integrin loss intensifies toward the ischemic core). 74 Yet, in more functional rodent studies neurotoxicity and BBB alteration have been described as distinguishable processes, particularly during early ischemic75,76 or excitotoxic 77 brain injury. Therefore, the established involvement of plasmin in neuronal pathology might not fully extrapolate to the BBB and the subject requires much finer examination.
Exposure of the Blood–Brain Barrier to Plasmin During Stroke and Thrombolysis
Upon thrombolysis for cerebral ischemic attack, substantial circulating amounts of active rtPA (up to ~50 nmol/L during myocardial infarction with a similar dosing regimen to ischemic stroke48,78), as well as plasminogen (2 μmol/L; the physiologic plasma concentration 2 ) and in situ-generated plasmin (in some cases up to ~300 nmol/L 79 ) perfuse the brain. This ‘cocktail’ immediately comes in contact and interacts with BECs. According to studies using rodent models as well as bovine endothelial cells, vascular rtPA can then cross the intact BBB in a LRP-1-mediated transcytosis 23 and penetrate into the uninjured brain parenchyma. Under experimental ischemia, rtPA transport via the injured BBB increases and becomes LDLR independent 80 (to the best of our knowledge these rtPA properties have not been evaluated in humans and were not reported for plasminogen). Regardless of species, stroke affects the endothelial barrier and therefore provides an additional potential for both vascular rtPA and plasminogen to act upon the BM, pericytes and perivascular astrocytes and finally to provoke microglia and neurons. Notably, much higher local protease levels are expected as the enzymes rapidly accumulate on fibrin,1,2 cells2,81,82 and cell debris83,84 Hence, ischemic stroke and thrombolysis conceptually set the perfect stage for an orchestrated rtPA and plasmin ‘storm’ from the vascular compartment, reaching all cerebral cell types but particularly the NVU.
Importantly, tPA13,30 as well as plasminogen10,85–87 are also endogenously expressed within the brain and, at least in rodents, are further upregulated during ischemic26,55,73,88 and excitotoxic10,89,90 insults (uPA is the predominant PA increased in the ischemic brain of non-human primates 51 ). However, their spatial distributions only partially overlap. Tissue-type plasminogen activator is widely expressed in the brain endothelium as well as in microglia and neurons (in the case of the latter particularly in the hippocampus, hypothalamus, cerebellum, and amygdala),13,30 whereas plasminogen is exclusively localized in rodent brains to neurons of the cerebral cortex, hippocampus, hypothalamus, and the cerebellum.10,85–87 Hence, it should not be obviously assumed that brain-derived plasminogen is activated in situ by endogenous tPA at the BBB during stroke. Blood-derived plasminogen may be more readily available, but vascular-derived plasmin is also tightly controlled by the potent circulating plasmin inhibitor, α2-antiplasmin (α2AP). In fact, zymographies (either in situ casein zymography8,26,54,73 or plasminogen–gelatin zymography 51 ) or amidolytic activity assays 54 of the rodent brain, whether performed using naïve8,54,73 or ischemic tissue,26,51,54,73 cannot demonstrate any tPA or plasmin activity without inclusion of exogenous plasminogen in these assays. This is suggestive of either negligible levels or a very tight control of active endogenous plasmin in the vascular and extravascular compartments of the ischemic brain (without initiation of thrombolysis with rtPA). Questions are therefore raised whether (1) free plasmin is actually formed by endogenous PAs during stroke around brain capillaries and in brain parenchyma and (2) if it has a genuine capacity to operate uncontrollably and inflict BBB damage in the absence of an exogenous thrombolytic to force free plasmin formation.
Conceptual Links Between Plasmin Activity and Blood–Brain Barrier Disruption
Much of the plasmin-independent concept for the actions of tPA on the BBB derives from studies in plasminogen−/– mice. 26 While compelling, not all of these reports are consistent 41 (see section Experiments in Plasminogen Knockout Mice below). Importantly, a plasminogen-deficient approach mainly supports the existence of a plasmin-independent component in the action of tPA, but does not exclude an additional plasmin-dependent element. In fact, if substantial plasmin concentrations indeed build up around blood vessels in the brain, especially when plasmin levels rise in plasma during treatment with rtPA, 79 plasmin should logically contribute to BBB modulation, independently or in conjunction with rtPA. For example, plasmin efficiently cleaves PDGF-CC in vitro 91 and in vivo (in the vitreous of patients with proliferative vitreoretinopathy 92 ), suggesting that akin to tPA, 24 plasmin may be capable of playing a role in astrocyte-related BBB disruption via the PDGF-C pathway. Notably, PDGF-CC activation has not been directly evaluated in plasminogen−/– mice and plasmin involvement was excluded circumstantially 24 based on the lack of plasmin contribution to BBB disruption in an earlier study. 26 Intriguingly, in a recent study that crossed transgenic mice overexpressing human PDGF-C with tPA-deficient mice, the phenotype of the PDGF-C overexpressing mouse was not affected (including high serum levels of cleaved growth factor and significant liver fibrosis), strongly suggesting that proteases other than tPA cleave and activate PDGF-C in vivo. 93
Plasmin can also directly degrade BM components such as collagen IV,42,94 laminin 69 and fibronectin 95 and further activates in vitro96–99 and in vivo41,42,69 MMP-2, -3, and -9, which possess similar BM dismantling capabilities and additionally damage TJs.13,57 Hence, pathologic plasmin activity may contribute to degradation of the microvascular bed and to impairment of vessel integrity, ultimately resulting in hemorrhagic transformation. 100 In solid support of this notion, recent work revealed that genetic ablation of the laminin-γ1 subunit in neurons and astrocytes disrupted astrocytic end-feet BM formation, consequently causing blood vessel disintegration and spontaneous hemorrhage in deep regions of the brain. 101 One could speculate that plasmin degradation of laminin or other components of the BM could inflict a similar effect.
The overlapping capacity of plasmin and MMPs to degrade the extracellular matrix makes it difficult, however, to estimate the actual importance of plasmin in the BM degradation process in vivo. Some cues can be deduced from observations that double-knockout mice for MMP-2 and -9 demonstrated only partial (yet significant) protection against laminin loss and brain hemorrhage and had similar edema compared with wild-type mice after transient MCAo. 102 In line with these observations, a broad MMP inhibitor (BB-94) did not fully block the hemorrhage rate in rabbits 32 and rats 61 undergoing rtPA treatment during embolic stroke. Suzuki et al 41 has further demonstrated that both deficiencies in plasminogen and MMP-3 protected mice from rtPA-induced ICH, suggesting that plasmin may be required for activation of rtPA-regulated MMP-3 in endothelial cells 39 during stroke. Taken together, these observations circumstantially point to another protease(s), potentially plasmin, which operates in concert with MMPs to damage the BM during stroke. Nevertheless, it is reasonable to hypothesize that some other MMP-independent mechanisms may operate in parallel (see section Modes of Action above) and potentially contribute to this residual effect.
Plasmin (and plasminogen) additionally binds a wide array of cell-surface receptors or binding proteins and, utilizing its broad substrate specificity, cleaves a variety of biologic substrates.81,82 Because of this capacity, plasmin has been implied in many cellular responses, including cell migration, wound healing, tissue remodeling, apoptosis, cancer invasion, cancer metastasis, inflammation and immunity (extensively reviewed elsewhere).81,82,103 Importantly, cell stimulation by plasmin often involves signaling via activation of protease-activated receptor (PAR)-I, 103 known to be considerably expressed on astrocytes but also on brain microvascular endothelial cells (see sections Plasmin Actions on Brain Endothelial Cells and Plasmin Activation of Astrocytes below). Therefore, uninhibited plasmin could also affect more precise intracellular signaling processes at the BBB.
Together, these attributes position plasmin as a sound candidate to participate in remodeling of cerebral blood vessels, especially during stroke, when the BBB weakens and blood components gain access to known plasmin-sensitive compartments, such as the BM and astrocytic end-feet.
Can Plasmin Escape Natural Inhibition?
A final but critical obstacle remaining in the case for plasmin is the need of this protease to overcome in vivo the dominance of its principal physiologic inhibitor in the circulation, α2AP, as well as other blood and brain inhibitors (i.e., α2-macroglobulin and protease nexin-1, respectively). 104 α2AP is a serine-protease inhibitor (serpin) that circulates at plasma concentration of 1 μmol/L (approximately half the concentration of plasminogen) and possesses unique N- and C-terminal extensions.2,104 The C terminus tail contains conserved lysine residues, mediating the initial docking of α2AP to plasmin via lysine-binding sites in the plasmin kringle domains. Indeed, the association of α2AP with plasmin is one of the fastest known (overall association constant 2 × 10 7 /(mol/L)/second). 104 Despite this efficient interaction, the binding of plasmin to exposed lysine residues on its various targets occupies its lysine-binding sites and reduces the capacity of plasmin to interact with α2AP. Therefore, surface-bound plasmin is considered to be protected from inhibition.105,106 Within the human CNS, plasmin can also be regulated by protease nexin-1, a cell-surface- and extracellular matrix-bound serpin that primarily localizes to capillaries, smooth-muscle cells and astrocyte end-feet. 107 Protease nexin-1 exhibits strong inhibitory activity toward thrombin, but also toward trypsin, uPA, and plasmin (association constant for plasmin 1.3 × 10 5 /(mol/L)/second).104,107 The array of abundant plasmin inhibitors maintains plasmin under tight regulation, as expected when such potent and broad-specificity enzyme is involved (evidently, humans deficient of α2AP suffer severe hemorrhagic predisposition 108 ). Therefore, it stands to reason that the bulk of plasmin generated around brain capillaries in the course of rtPA-mediated thrombolysis (and possibly also endogenous plasmin formed in the brain during stroke by upregulated tPA and plasminogen) may be short lived and incapable of exerting substantial BBB damage. However, once we take into account that surface-bound plasmin is protected from inhibition by α2AP,105,106 that α2AP has decreased activity under acidic conditions 109 (which accompany the ischemic brain 110 ), and that during thrombolysis systemic plasmin generation often consumes α2AP as well as factor VIII and fibrinogen (a process known as plasminemia),111,112 a putative causal role for plasmin becomes hard to ignore.
Collectively, the enhanced generation of active plasmin during thrombolysis, the reduced potential for plasmin inhibition in its surface-bound state and several plasmin activities that can affect the BBB lend a plausible case for plasmin as a bone fide BBB modulator. Despite this argument, the experimental evidence addressing the role of plasmin in BBB alteration, either alone or in conjunction with rtPA, has created much controversy as described below.
EFFECTS OF PLASMINOGEN AND PLASMIN ON THE BBB IN VIVO
Plasmin Administration into the Brain Parenchyma
Direct intracortical injection of concentrated plasmin (0.7 mg/mL (8.2 μmol/L); 10 μL) into Wistar rats, as well as of high concentration of rtPA (5 mg/mL; ~77 μmol/L), resulted in substantial lesion formation 6 hours later, accompanied by oxidative stress damage to lipids, proteins and DNA, degradation of TJ proteins (occludin) and BM (collagen IV) and elevation of MMP-9, without substantial loss of endothelial cells. 42 Similar to previous findings reported by the same group in a MCAo stroke model with rtPA treatment, 38 astrocytes had detached from the vessel wall in both rtPA and plasmin-treated groups, but the effect appeared much stronger in the latter. In fact, no astrocytes could be observed around capillaries by GFAP staining after plasmin treatment. 42 Complementary studies in a rat BBB model confirmed the ability of plasmin (as well as rtPA) to damage TJs and to increase BBB permeability. The free-radical scavenger, edaravone, reduced rtPA damage, but was not evaluated against plasmin-induced injury. The study concluded that plasmin can cause comparable BBB damage as rtPA, potentially inflicting oxidative stress to perivascular astrocytes. Nevertheless, both proteases were used at exaggerated concentrations. 42
A similar intrastriatal injection of plasmin (50 U/mL, 10 μL; specific activity undisclosed) to Sprague-Dawley rats resulted in enhanced brain uptake of intravenously-injected sucrose as early as 4 hours post injury, which was maximal at 24 hours and coincided with necrosis of the blood vessel wall and prominent deposition of fibrin-like material. However, the effect of plasmin was relatively mild compared with that of elastase. 113 While the effect on BBB integrity was not specifically assessed, a very similar experiment in rats confirmed that plasmin and plasminogen administration (67–670 U/mL and 10–100 U/mL, respectively, 5 μL; specific activity undisclosed) into the rat striatum (as well as thrombin (500–5000 U/mL, 5 μL)) inflicted striatal damage, neuronal apoptosis and brain inflammation 48 hours later, seen by increased infiltration of neutrophils and T-killer cells as well enhanced microglial reactivity. Interestingly, the effect of plasmin/plasminogen was much more potent than that of rtPA. 114 Taken together, these experiments (summarized in Table 3) raise the possibility that plasmin is capable of damaging the BBB when applied directly into the brain, primarily affecting the abluminal components of the NVU.
Effects of plasminogen and plasmin in the brain and on the BBB in vivo
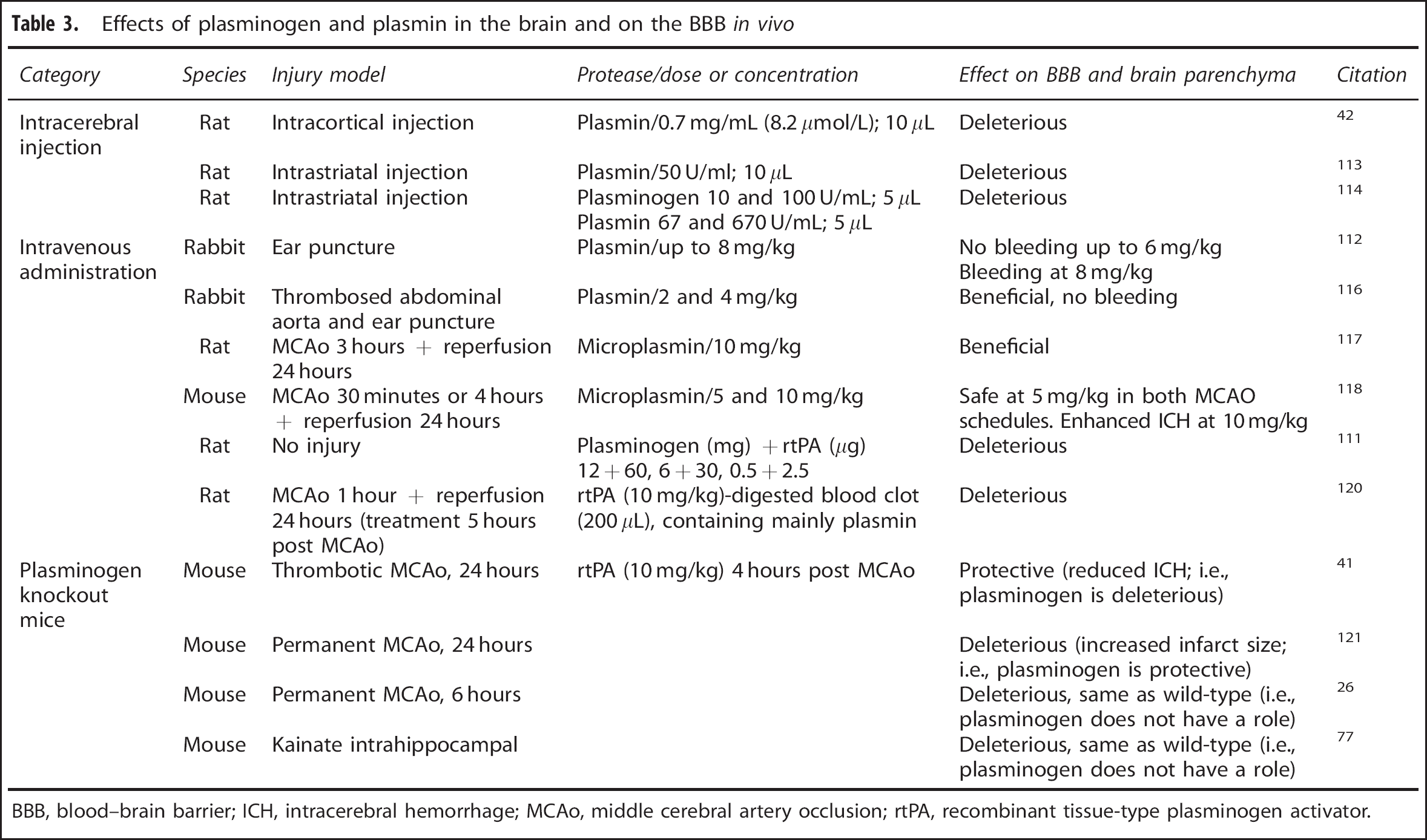
BBB, blood–brain barrier; ICH, intracerebral hemorrhage; MCAo, middle cerebral artery occlusion; rtPA, recombinant tissue-type plasminogen activator.
Systemic Plasmin and Plasminogen Administration in Normal and Ischemic Conditions
While the above experiments imply that plasmin can affect the BBB from the brain side, can it also initiate similar actions working from the vascular compartment?
Noticeably, systemic administration of plasmin or its truncated derivatives (i.e., microplasmin, consisting only the serine-protease domain, or mini-plasmin, harboring kringle 5 and the protease domain 115 ) has a striking hemostatic safety advantage over PAs 115 with regards to general bleeding susceptibility112,116 and particularly with relation to ICH in experimental models of stroke.117,118 Plasmin could be given up to six times the therapeutic dose of rtPA (i.e., up to 6 mg/kg) before bleeding re-occurred in a rabbit ear puncture model, despite the observed plasminemia characterized by consumption of α2AP, factor VIII and fibrinogen post intravenous plasmin application. In contrast, rtPA caused uncontrolled bleeding already at 0.25 mg/kg, a quarter of its therapeutic concentration. 112 In rodent stroke models, microplasmin preserved the BM, reduced brain hemorrhage and decreased infarct volume compared with saline, rtPA, and TNK-tPA.117,118 These beneficial plasmin activities, attributed to the rapid containment of plasmin by circulating α2AP,112,115 suggest that systemic administration of active plasmin (given in comparable therapeutic doses to rtPA) may not induce substantial BBB damage and hemorrhage. An ex vivo experiment in an organ perfusion model (where rat brains were first cleared of blood followed by protease infusion) supports this idea; indeed, perfusion of plasmin or mini-plasmin, but not of inactive plasmin, caused extravasation of circulating horseradish peroxidase into the brain, indicative of BBB opening. However, if the blood was only partially washed out of the microcirculation, plasmin perfusion did not affect the BBB. 119 These observations suggest that the action of systemic plasmin on the BBB is efficiently contained in vivo by circulating plasmin inhibitors.
While the discussion above argues against a role for plasmin, it must be remembered that physiologic plasmin generation occurs locally and is driven by a PA-mediated cleavage of the precursor plasminogen on the surface of fibrin or cells, serving as a template. It is therefore possible that plasmin will contribute to BBB disruption only when generated by tPA in situ, directed to specific NVU components (rather than solution-applied) and when it is protected from α2AP inhibition.105,106 In support of this idea, intravenous administration of rtPA together with plasminogen in three dose-decreasing schedules into naïve, stroke-prone spontaneously hypertensive rats promoted intracranial IgG extravasation. This breakdown of the BBB was attributed to plasminemia, which greatly increased the hemorrhagic potential. 111
Plasmin, generated in situ by tPA, also potentiated BBB breakdown during experimental stroke. In an interesting experiment by Gautier and Colleagues, 120 MCAo was induced for 1 hour in spontaneously hypertensive rats, followed by intravenous administration 5 hours later of saline, rtPA or rtPA-dissolved autologous blood clot (rtPA + TLP (‘thrombolysis products’)), containing mainly in situ-made plasmin. It was observed that rtPA + TLP increased the cerebral hemorrhage severity at 24 hours, evident by augmented petechiae, which was associated with enhanced mortality in the rtPA + TLP group. Ex vivo, MCA vasodilation in response to acetylcholine was diminished by ischemia and further reduced by rtPA + TLP treatment, but not by rtPA alone, suggestive of a plasmin-mediated impairment of endothelium-dependent vascular response in the brain.
In summary, it can be speculated that generation of plasmin in situ by rtPA, but not free, active plasmin, has a greater potential to alter BBB integrity when administered intravenously, in line with our original hypothesis. These studies are further collated in Table 3.
Experiments in Plasminogen Knockout Mice
The involvement of plasmin/plasminogen in BBB breakdown can also be inferred from experiments in plasminogen-defkient mice, but inconsistencies are present in the literature (see Table 3). Suzuki et al 41 confirmed the involvement of plasmin/plasminogen in ICH formation as plasminogen−/– mice undergoing thrombotic MCAo did not show elevated intracranial bleeding 20 hours post delayed treatment with rtPA (administered 4 hours post stroke), in contrast to their plasminogen+/+ littermate controls. 41 This observation indicates that during thrombolysis plasmin contributes to severe destabilization of vessel structure and passage of whole blood into the brain, potentially by degradation of the BM (considered a principal event underlying hemorrhagic conversion58,100). However, others demonstrated by the Evans blue technique a robust extravasation of circulating albumin 6 hours post permanent MCAo in plasminogen−/– mice. 26 This observation suggests that basal processes responsible for TJ damage (without exogenous application of rtPA), which enable paracellular passage of ions, plasma proteins and fluid into the brain and formation of vasogenic edema, 58 can proceed independent of plasmin activity. Similarly, Chen et al 77 showed BBB opening by detection of cerebral immunoglobulin G after kainate delivery into the hippocampus of plasminogen−/– mice (as well as in wild-type and tPA−/– mice), indicating that acute excitotoxic injury impairs the BBB without the requirement for tPA/plasmin (in contrast to critical roles of these proteases in kainate-mediated neuronal degeneration). Noticeably, intrahippocampal, but not intravenous administration of plasminogen, re-sensitized plasminogen−/– mice to kainate-triggered hippocampal neurodegeneration, suggesting that circulating plasminogen cannot fulfill neurotoxic roles in the brain parenchyma. 77 One might postulate that plasma-derived plasminogen ‘leaks’ into the brain together with α2AP, which effectively restricts its action. In an extensive study of murine gene deletion constructs by Nagai et al, 121 permanent MCA ligation (24 hours) resulted in an increase or a reduction of infarct size in plasminogen−/– or α2AP−/– mice compared with wild-type controls, respectively, 121 implying a neuroprotective role for plasmin during stroke. However, in both these genotypes fibrin deposition within the infarct zone (indicative of altered blood vessel permeability) remained intravascular, compared with extravascular fibrin reactivity in PAI-1−/– mice. 121 These findings indicate the involvement of endogenous plasmin in neuronal death, but not in BBB alteration during stroke (consistent with the notion that BBB disruption and neurotoxicity are distinguishable events in the rodent75–77), in contrast to endogenous tPA, which participates in both.
Overall, a rather complex picture emerges in plasminogen knockout mice that could be partially derived from the various stroke models used, from inclusion or exclusion of rtPA and from the different readouts tested (i.e., hemorrhage versus plasma extravasation). As it currently stands, the data suggest that endogenous plasminogen present in the brain during stroke not only avoids playing a role in ischemia-mediated TJ damage and formation of vasogenic edema, 58 but also possesses neuroprotective capabilities. However, initiation of thrombolysis by rtPA may tilt excess plasmin into a more detrimental direction, resulting in proteolytic destabilization of vessel structure and development of ICH.58,100 Additional experimentation in plasminogen−/– mice is required to further evaluate this concept.
MECHANISMS FOR BLOOD-BRAIN BARRIER MODULATION BY PLASMIN
In a more controlled in vitro environment the effect of plasmin on various cell types is prominent. The actions of plasmin often include remarkably similar signaling events, such as changes in intracellular calcium (Δ[Ca2+]i) and activation of pathways like the phosphati-dylinositide 3-kinase (PI3K)/Akt, mitogen-activated protein kinases, Rho/ROCK and more (reviewed by Syrovets et al). 103 Of particular relevance, plasmin affects the principal constituents of the NVU (BECs, astrocytes, neurons, and the BM) and increases permeability in human 43 and rodent42,122 BBB models. The mechanisms underlying these effects will be overviewed below and are further listed and illustrated in Table 2 and Figure 1, respectively.
Plasmin Actions on Brain Endothelial Cells
Cell surface-generated plasmin activates many pathways in endothelial cells, 103 some could potentially contribute to the modulation of BBB permeability. For example, plasmin initiates in vitro a PAR-1 -independent increase in cytosolic Ca2+ and nitric oxide production in human and porcine coronary ECs, leading to vasorelaxation and implying plasmin, like tPA,65,66 as a modulator of vascular tone. 123 In the brain, PAR-1, a substrate for plasmin (see Plasmin Activation of Astrocytes below), could not be detected in rat BECs utilizing in situ hybridization 124 but was later found together with PAR-3 (and PAR-2) on these cells in vitro by RT-PCR. 125 Whether PAR-1 on BECs is expressed in a polar distribution across the apical and basolateral membranes, an important determinant for possible activation by brain-derived proteases, has not been determined 125 (the susceptibility of BECs to blood-born proteases, such as thrombin or plasmin, suggests at least an apical expression). Nevertheless, brain endothelial PAR-1 could functionally induce Δ[Ca2+]i in response to thrombin stimulation. Plasmin could not trigger BEC Δ[Ca2+]i on its own but abolished BEC responsiveness to thrombin. 125 This observation suggests that plasmin in fact inactivates PAR-1 in BECs (a conclusion reached by others in coronary ECs 123 ), providing a path for plasmin interference of BEC activation.
Although this may imply a protective effect of plasmin on the BBB, other data link plasmin to BECs damage and BBB disruption (Figure 1A). Plasmin or mini-plasmin was shown in a number of studies to rapidly bind monolayers of human BECs in a reversible fashion.119,126 Plasmin subsequently induced a ‘spiderlike’ contraction of BECs, seen as early as 30 seconds post application. While other hemostatic factors, such as thrombin, uPA, elastase and fibrin, induced a similar BEC contraction, plasmin was the fastest to act.119,127 Similarly, our group reported marked morphologic changes in human BECs post application of rtPA and plasminogen, which were blocked by aprotinin and tranexamic acid and fully correlated with permeability changes induced by rtPA and plasminogen in BEC monolayers. 43 Plasmin-induced retraction is not limited to brain ECs and can be seen, for example, in human umbilical vein endothelial cells treated with uPA/rtPA and plasminogen. 128 Recent identification of rtPA-mediated ROCK activation in BECs, 35 potentially driven by plasmin-cleaved monocyte chemoattractant protein-1 (MCP-1) 122 (see section Rho-Kinase- and Monocyte Chemoattractant Protein-1-Dependent Pathways below), can provide in principle the driving force for the BEC retraction phenomenon.
During the initial stages of plasmin exposure, BECs experience reversible changes that include damage to cell-cell boundaries, disruption of monolayer integrity, 128 membrane blebbing, vesiculation and formation of micro-particles (~300 nm in diameter). 126 Of note, micro-particles carry both plasminogen-activator and plasmin/plasminogen, and thus can transmit the proteolytic activity of plasmin and its associated signaling beyond the original site of plasmin activation. 126 If plasmin stimulation persists beyond several hours, matrix proteins are severely degraded, triggering loss of cell adhesion and subsequent cell death by apoptosis.126,128 Importantly, as surface-bound plasmin is protected from inhibition,105,106 the process is less susceptible for attenuation by α2AP. 128
Surface-bound plasmin therefore induces morphologic and functional changes in a range of cultured ECs, including brain ECs, which can affect their ‘tight’ BBB phenotype (Figure 1A).
Plasmin-mediated retraction of BECs may be also an important first step in the extravasation of blood cells through the capillary wall, as it uncovers the underlying BM, exposing matrix proteins for cellular digestion. 128 Notably, rtPA-generated plasmin also participates directly in BM degradation by infiltrating cells. For example, the passage of C6 glioma cells through artificial laminin or collagen I barriers (without cells) was dramatically enhanced by addition of rtPA and plasminogen. Annexin A2 was pointed out as the primary gliomal receptor responsible for efficient cell-surface plasmin generation and glioma invasion. In vivo, knockdown of Annexin A2 in glioma cells reduced their metastatic, angiogenic and growth capabilities, highlighting the therapeutic potential of the Annexin A2-tPA-plasmin axes in the treatment of brain cancers. 129
In summary, plasmin-induced retraction of BECs may contribute to BBB disruption, formation of edema and ICH and infiltration of cells into the brain. This mechanism is especially relevant during thrombolytic therapy; first, substantial levels of in situ-generated plasmin could outweigh the inhibitory capacity of the various blood and brain plasmin inhibitors.119,127 Second, the ischemic insult damages TJs and permits easier access of blood-derived plasmin to the BM, accelerating BM degradation and creating a positive feedback loop for BEC retraction. Interestingly (as indicated in section Recombinant Tissue-Type Plasminogen Activator and the Non-lschemic Blood–Brain Barrier above), it was reported that intracerebroventricular,24,26 but not intravenous 24 administration of mouse tPA into uninjured mice promoted breakdown of the BBB, suggesting that a preceding brain insult is required for development of tPA damage from the vascular compartment, at least in the mouse (recall that systemic delivery of tPA opened the BBB in naïve rats20–22). By inference, circulating plasmin may be able to initiate its deleterious cascade in BECs only once TJs have been compromised.
Beyond these conceptual considerations, in vivo evidence for BEC retraction (or damage) post rtPA treatment or after exposure to plasmin is scant. In fact, few studies have shown that BBB injury post thrombolysis does not involve the brain endothelium but is associated instead with morphologic changes in astrocytes (see section Plasmin Activation of Astrocytes below).26,28,38,42 For example, rtPA 38 and plasmin 42 induced no obvious changes in endothelial cell markers despite significant reduction in BM proteins and a partial decrease in TJ proteins, suggestive of a brain-to-blood effect on brain capillaries.38,42 Yet, scanning electron micrographs of brain capillaries by Ishiguro et al 36 clearly showed substantial endothelium disruption and disappearance of BEC nuclei post treatment with rtPA in stroke. Others reported functional impairment of brain endothelium post treatment with plasmin (see section Systemic Plasmin and Plasminogen Administration in Normal and Ischemic Conditions above) 12 or rtPA 130 ex vivo. Of note, BEC retraction and interendothelial gap formation is also induced by many inflammatory mediators, including thrombin, and were suggested to participate in thrombin-mediated development of vasogenic edema during brain ischemia or cerebral hematoma. 58 Nevertheless, more detailed work is warranted to determine whether plasmin-dependent BEC retraction is relevant for BBB disruption in vivo.
Plasmin Activation of Astrocytes
An intricate and reciprocal relationship exists between astrocytes and plasmin, as astroglia both modulate BEC (and BBB) sensitivity to plasmin and become affected by its action. For example, in keeping with their well-described role as the major inducers of BBB properties in BECs, 131 astrocytes increase LDLR expression in the latter, 132 possibly transforming BECs into a more tPA/plasmin-responsive phenotype. Conversely, tPA 64 and plasminogen (potentially microglia-derived) 133 can stimulate intracellular signaling in astrocytes (e.g., the Rho/ROCK pathway and p38 + c-Jun N-terminal MAPK (JNK), respectively), which mediate glial release of BBB-permeating factors, such as MMPs 64 and MCP-1 134 (see sections Modes of Action and Rho-Kinase- and Monocyte Chemoattractant Protein-1-Dependent Pathways, respectively). As an added complexity, plasminogen-induced p38 and JNK signaling also induces PAI-1 secretion in cultured rat astrocytes, implying that plasmin also initiates a negative feedback loop in glia that may protect the BBB from excessive fibrinolytic activity. 133 Hence, the net effect of plasmin on the BBB in a given scenario will be influenced by a complex cross-talk between the various cell types comprising this multilayered structure.
When describing the effect of plasmin on astrocytes, an emphasis should be placed on PAR-1, a G-protein coupled receptor most potently activated by thrombin, which can also be cleaved by plasmin.135,136 However, the action of plasmin on PAR-1 is complex as plasmin can activate103,136,137 or inactivate123,125,135,137 the receptor, cleaving the N-terminal exodomain of PAR-1 at the thrombin cleavage site (Arg41-Ser42), 137 but also doing so inappropriately in other positions, 135 creating a number of different tethered ligands.
In the human brain, PAR-1 is moderately expressed in neurons but is predominantly present on the astrocyte soma and end-feet ensheathing the capillaries.138,139 Both neurons and astrocytes are similarly PAR-1-positive in the rat brain.124,140 PAR-1 is also upregulated in the brain during cerebral ischemia. 141 Collectively, it seems plausible that plasmin may participate in some of the vast array of PAR-1-dependent brain processes (reviewed by Adams et al), 136 with potential consequences for the BBB, particularly during stroke. Despite this argument, although PAR-1−/– mice show reduced infarct volume 24 hours after 30 minutes MCAo, they do not display any detectable differences compared with wild-type mice in the anatomy of healthy cerebral microvessels or in the level of BBB breakdown 4 hours after similar occlusion. 139 This result suggests that the net effect of PAR-1 activation by thrombin/plasmin during stroke contributes to neurotoxicity in the brain parenchyma rather than to astrocyte-dependent BBB modulation. Yet, permeability assessment at a later time frame (24 hours) was critically required to fully confirm this conclusion.
Plasmin had been shown, however, to exert a number of PAR-1-dependent effects in cultured astrocytes,103,142 which can be linked to both BBB protection and harm. For example, exposure of astrocytes to plasmin or plasminogen prompted astrocytic TGF-/β3 production in a PAR-1 and PI3K/Akt-dependent mechanism. 143 Astrocyte-derived TGF-β in turn downregulated tPA (and thrombomodulin) expression in cerebral endothelial cells. 144 Thus, plasmin-stimulated astrocytes potentially suppresses the fibrinolytic capacity at the BBB, consistent with the plasminogen-dependent, astrocytic PAI-1 release mentioned above. 133
In contrast, recent in vitro findings from our group 43 and a follow-up work by others 142 showed that rodent and human astrocytes, like endothelial cells (section Plasmin Actions on Brain Endothelial Cells), undergo significant, PAR-1-dependent 142 morphology changes in response to plasmin or rtPA 43 /streptokinase 142 -activated plasminogen (Figure 1B). These transformations underlined ROCK-mediated opening of the BBB (see also section Rho-Kinase- and Monocyte Chemoattractant Protein-1-Dependent Pathways below), 43 indicative of their possible functional significance. If exposure to plasmin was prolonged, astrocytes detached and went through apoptotic cell death. 142 Hence, excessive, PAR-1-mediated plasmin activity may also harbor deleterious consequences to astrocytes and to the BBB (Figure 1B).
Finally, plasmin, via PAR-1, initiated hydrolysis of phosphoinositides, Δ[Ca2+], and ERK1/2 signaling in astrocytes, which potentiated neuronal NMDAR function. 145 The possible contribution of this pathway to NVU function, however, remains unclear.
In summary, plasmin exerts in vitro diverse effects in astrocytes, with either beneficial or deleterious potential to the function of the BBB. The net contribution of these events to plasmin-dependent BBB alteration in vivo remains to be elucidated.
Rho-Kinase- and Monocyte Chemoattractant Protein-1-Dependent Pathways
The most recently-discovered plasmin-dependent mechanisms for BBB opening originate from either BECs122,146 or astrocytes 43 and involve various extracellular initiators and cell-surface receptors. Nevertheless, a common component underling these pathways is the ultimate activation of ROCK and modulation of the actin cytoskeleton (Figure 1).
The MCP-1 pathway occurs in BECs (Figure 1A) and relies on the ability of plasmin to cleave the chemokine MCP-1. 122 Full-length MCP-1, via interaction with its receptor C-C chemokine receptor type 2 (CCR2), was shown to impair BBB integrity under normal conditions 146 and during OGD/reoxygenation 147 in a process involving Rho/ROCK activation 148 and downstream engagement of the α-isoform of protein kinase C alpha. 149 This in turn caused modulation of the actin cytoskeleton and disruption of endothelial TJs. It was then discovered that plasmin can cleave MCP-1 at lysine residue 104 150 and that the truncated MCP-1 is more potent at activating microglia than the full-length MCP-1. Consequently, microglial recruitment to the site of injury and progression of excitotoxicity were enhanced.150,151 Recently, cleaved MCP-1 was also demonstrated to harbor a greater capacity to induce stress-fiber formation in BECs (indicative of enhanced activation of the Rho/ROCK pathway and generation of contractile forces) and to trigger phosphorylation of ezrin–radixin–moesin proteins, which serve as linkers between the actin cytoskeleton and the plasma membrane. Together, these signaling pathways were proposed to underlie an enhanced capacity of plasmin-cleaved MCP-1 to redistribute TJ proteins and to increase BBB permeability in vitro and in vivo. 22 One limitation worth noting in this study is that the full-length MCP-1 was never directly compared with or without plasmin (for example in plasminogen−/– mice) to fully prove the concept; instead, a recombinantly truncated MCP-1 or full-length MCP-1 together with the plasmin inhibitor α2AP were used. 122 Importantly, the MCP-1 pathway does not involve astrocytes, as only BECs deficient of CCR2, but not CCR2−/– astrocytes, could offer protection against MCP-1 treatment in a mouse syngeneic in vitro BBB model. 146 A non-related study showed that the ROCK inhibitor fasudil (HA1077) attenuated rtPA-dependent MMP-9 production and cytotoxicity in human BECs, suggesting that rtPA is also capable of activating ROCK in human BECs. However, exaggerated rtPA concentrations were required and the role of plasmin and MCP-1 was not assessed. 35
We recently highlighted plasmin-mediated activation of ROCK in astrocytes as a key event underling a unique capacity of rtPA to modulate a human BBB model 152 by receptor-mediated mechanism 43 (Figure 1B). Our studies suggested that in situ activation of plasminogen on the surface of astrocytes, specifically mediated by rtPA (but not by other plasminogen activators), has a superior capacity compared with rtPA or plasmin alone to increase BBB permeability. These findings thus represent a novel concept where unique synergism between rtPA and plasminogen/plasmin is pivotal for alteration of BBB permeability. Furthermore, while BECs and astrocytes both responded by morphologic changes to the action of rtPA and plasmin, ROCK activation (which was accompanied by enlargement of focal adhesion, enhanced myosin contractility and stress-fiber formation) was exclusively observed in astrocytes in a process which involved LDLRs (possibly serving as a cell-surface tPA receptor) and other yet-to-be identified plasminogen receptor/s 43 (Figure 1B). Numerous animal studies reported astrocytic retraction from the vessel wall post rtPA or plasmin administration,24,27,38,42 an observation that may represent the in vivo correlate of our findings in vitro.
In summary, ROCK activation in BECs and astrocytes emerges as a key element allowing plasmin, alone and in conjunction with tPA (or rtPA), to modulate BBB permeability. Pharmacological targeting of ROCK and/or relevant cell-surface receptors (CCR2, LDLRs) should be incorporated into novel strategies aiming to increase the safety of rtPA treatment. Indeed, recent in vivo work, which utilized fasudil 35 or the LDLR inhibitor receptor-associated protein, 39 successfully attenuated the hemorrhagic side effects of rtPA in mouse models of stroke.
CONCLUDING REMARKS
Despite fundamental variations between observations made in rodent and in primate species, the idea that rtPA and with less certainty endogenous tPA cause BBB alteration in humans during thrombolysis in stroke has gained solid experimental support. A role for plasmin or tPA-activated plasminogen in this process is not fully clarified, but cannot be ignored. Plasmin can evidently influence the isolated NVU components and observations/mechanisms showing plasmin-induced BBB opening certainly exist. Yet, other evidence is provided to the contrary or implies that the tight regulation of plasmin by its potent inhibitors (α2AP and other brain inhibitors) will result in a net lack of potency. Hence, the subject is still controversial.
One must remember, however, that tPA-dependent plasminogen activation principally occurs in a local, cofactor-dependent manner (i.e., on the surface of fibrin, cells, or cell debris), where plasmin is less susceptible to inhibition; such a shielding process is absolutely essential for the progression of clot degradation. As clot dissolution is ultimately achieved and free plasmin activity is also detected in plasma during thrombolysis, uninhibited in situ-generated plasmin is certainly available during rtPA treatment for interaction with brain capillaries in infarcted as well as in unaffected brain tissue. Hence the unique conditions evolving during thrombolysis, when excess rtPA interacts with circulating plasminogen, increase the likelihood of a plasmin-dependent BBB alteration, in keeping with our original hypothesis.
Taken together, although additional work is needed to distinguish the effects of tPA and plasmin on the BBB, current state-of-the-art suggests that both agents are indeed capable of damaging the cerebral vasculature; the remaining question is to what extent. Future efforts to improve the safety of rtPA treatment in stroke may need to address the non-fibrinolytic effects of plasmin on the BBB without compromising its fibrinolytic capacity.
Akin to tPA, such approaches could include blockade of plasmin-triggered intracellular signaling. Simultaneous interference with both tPA and plasmin activities may offer a more efficacious therapeutic option.
Footnotes
The authors declare no conflict of interest.