
Abstract
Vascular disruption is the underlying cause of cerebral hemorrhage, including intracerebral, subarachnoid and intraventricular hemorrhage. The disease etiology also involves cerebral hemorrhage-induced blood–brain barrier (BBB) disruption, which contributes an important component to brain injury after the initial cerebral hemorrhage. BBB loss drives vasogenic edema, allows leukocyte extravasation and may lead to the entry of potentially neurotoxic and vasoactive compounds into brain. This review summarizes current information on changes in brain endothelial junction proteins in response to cerebral hemorrhage (and clot-related factors), the mechanisms underlying junction modification and potential therapeutic targets to limit BBB disruption and, potentially, hemorrhage occurrence. It also addresses advances in the tools that are now available for assessing changes in junctions after cerebral hemorrhage and the potential importance of such junction changes. Recent studies suggest post-translational modification, conformational change and intracellular trafficking of junctional proteins may alter barrier properties. Understanding how cerebral hemorrhage alters BBB properties beyond changes in tight junction protein loss may provide important therapeutic insights to prevent BBB dysfunction and restore normal function.
Keywords
Introduction
Cerebral hemorrhage is a particularly devastating form of stroke that accounted for about one-third of the 10.3 million strokes that occurred globally in 2013. 1 There are multiple types of cerebral hemorrhage: intracerebral hemorrhage (ICH), subarachnoid hemorrhage (SAH) and intraventricular hemorrhage (IVH); however, irrespective of type, cerebrovascular integrity is central to disease pathology and neurologic deficits. Not only does a loss of vascular integrity cause the initial hemorrhage, a failure to restore integrity or new sites of hemorrhage can cause hematoma expansion. Further, delayed blood–brain barrier (BBB) disruption contributes to brain edema formation, the infiltration of leukocytes and the entry of potentially neurotoxic and vasoactive compounds from blood into brain2–4 such as prothrombin/thrombin and fibrinogen/fibrin that have marked proinflammatory effects.5–7 Thus, finding methods to promote cerebrovascular integrity represents a promising therapeutic approach to treat intracranial hemorrhage. This review explores some potential methods of enhancing vascular integrity pre- and post-cerebral hemorrhage.
There are several events that may lead to brain endothelial barrier disruption. These include alterations in the junctions (particularly the tight junctions, TJs) that link the endothelial cells, endothelial cell death and increased endothelial cell transcytosis. Endothelial cell death may cause transient barrier hyperpermeability (increased permeability compared to control BBB) as such death will likely trigger the coagulation cascade occluding the damaged vessel. There has been considerable debate about the relative role of increased transcytosis vs. TJ disruption in the BBB hyperpermeability found after cerebral ischemia. 8 For cerebral hemorrhage, most studies have focused on TJ changes and that is the focus of the current review, although it is likely that transcystosis is also altered. 9 This review also highlights advances in the tools that can be used to assess junction changes, the mechanism underlying those changes and their importance in overall hemorrhagic brain injury.
Brain endothelial junctions
Tight junction transmembrane proteins interact with proteins on adjacent endothelial cells to help occlude the paracellular cleft in a mechanism that remains incompletely understood. Many of the transmembrane proteins required to create the BBB have been identified and include claudins (e.g. claudin-5), occludin and junctional adhesion molecule (JAM)-A (Figure 1; Stamatovic et al.
10
). There are also cytoplasmic plaque TJ proteins, such as zonula occludens (ZO)-1, -2, -3, that link the junction to the cytoskeleton and organize the junctional complex.11,12 In addition, polarity proteins such as, partitioning defective (Par)-3 and -6, afadin-6, atypical protein kinase C (PKCζ and λ), contribute to defining the cell polarity and initiating junctional formation.
10
Finally, small GTPases may regulate barrier properties. These include barrier promoting small G-proteins such as Rap1, which contribute to junction assembly, while other small G-proteins such as Rho GTPases contribute to permeability.
13
Junction complexes at the brain endothelial cell. Tight junctions (TJs). These consist of transmembrane and cytoplasmic plaque proteins. The main transmembrane TJ proteins (TJPs) are claudin-5, occludin and JAM-A. These form homophilic interactions with proteins on adjacent cells and help to occlude the paracellular space. Cytoplasmic plaque proteins provide physical support for the transmembrane TJPs (e.g. ZO-1 and -2) or regulate TJ function (e.g. cingulin like-1 (CGNL-1) and protein kinase C ζ and λ). ZO-1 and -2 not only bind to the transmembrane TJPs via PDZ domains, they also interact with the cytoskeleton either directly or indirectly via molecules such as cortactin and vasodilator stimulated phosphoprotein (VASP). Such interactions provide physical support for the TJ complex. Adherens junctions (AdJs). As with TJs, AdJs consist of transmembrane (VE-cadherin) and cytoplasmic plaque proteins (catenins, p120, plakoprotein). The cytoplasmic plaque proteins also form a bridge interacting with ZO-1 and the actin cytoskeleton. Gap junctions (GJs). These are formed by connexins (e.g. connexin 43) on adjacent endothelial cells. They are permeable to small molecular weight signaling molecules including Ca2+ and participate in cell:cell signaling. In addition, connexins may only be expressed on one cell forming a hemi-channel that allows small molecular compounds to move into/out of the cell. Actin cytoskeleton. The actin cytoskeleton is integral for junction function. It provides physical support but can also regulate junction function. Actin dynamics are regulated by many different actin binding proteins that control the assembly and disassembly and organization of actin filaments. Crosstalk. There is evidence that an alteration in one type of junction can alter other types of junction. For example, the entry of Ca2+ via GJs may impact TJ function. For a more detailed description of brain endothelial junction structure see Stamatovic et al.
10
For clarity, the diagram depicts the TJs, AdJs and GJs as separate, whereas these junctions are intimately linked.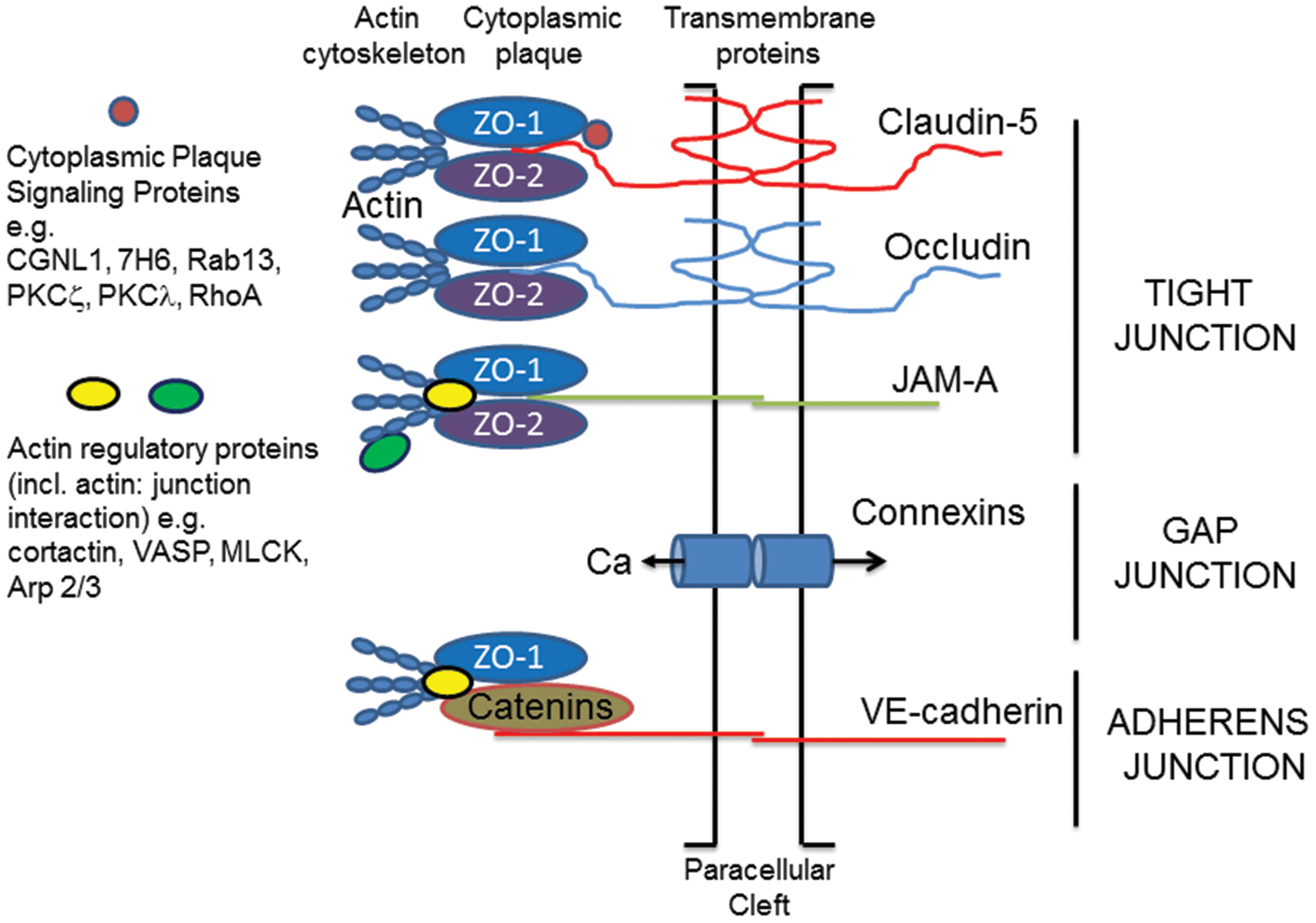
As with the TJs, brain endothelial adherens junctions (AdJs) consist of transmembrane proteins, primarily VE-cadherin, that are responsible for cell:cell adhesion, and cytoplasmic proteins (e.g. the catenins), responsible for physical support and junction regulation. 10 As with the TJs, AdJs are linked to the cytoskeleton (Figure 1).
Cerebral endothelial cells also possess gap junctions (GJs) formed of connexin-37, -40 or -43. 10 These junctions may be between adjacent endothelial cells or between the endothelial cell and other neurovascular unit cells (pericytes and astrocytes). GJs are involved in intercellular communication (e.g. they allow the passage of Ca2+) and they may have important role in regulating other components of the brain endothelial junction complex. 14 It should be noted that connexins can have hemichannel function when only expressed on one cell membrane and such channels may have an important role in endothelial cell injury. 15
The cell cytoskeleton contributes a critical component to barrier formation and function of junction complexes. It is composed of actin microfilaments (F- and G-actin and associated proteins), non-muscle myosin, and the microtubule system. 10 Not only does the cytoskeleton provide a scaffold for junction location, it can produce forces that pull junctions apart (e.g. during stress fiber formation). In addition, the cytoskeleton is essential for vesicular trafficking and endocytosis is an important element of junction remodeling (see below).
Brain endothelial junction regulation
Brain endothelial junctions are dynamic structures and may be altered by multiple pathways. There can be protein modifications including phosphorylation, methylation, palmitoylation and glycosylation.
10
Such modifications can affect protein:protein interactions altering barrier permeability. For example, they may affect transmembrane protein interactions between cells required for paracellular occlusion, or the interaction of cytoplasmic plaque proteins with transmembrane or cytoskeleton proteins affecting junction organization and stability (Figure 2(a)).
Tight junction regulation mechanisms. Tight junction function may be regulated by many mechanisms, although the ultimate effect is a loss of interaction between transmembrane tight junction proteins (TJPs) on adjacent cells. Examples of regulation are: (a) Protein modification. Tight junction stability is a result of protein interactions within the TJ complex (transmembrane and scaffolding proteins) and between the TJ complex proteins and the actin cytoskeleton (cortical actin ring). Protein modifications, such as phosphorylation (P) can have multiple effects. For example, phosphorylation of the transmembrane TJP may lead to loss of interaction with the scaffolding protein ZO-1 causing reduced interaction between transmembrane TJPs such as claudin-5. Alternately, phosphorylation of myosin light chain kinase (MLCK) results in rearrangement of actin and the formation of stress fibers which may exert pressure on TJ complexes. The importance of protein modifications in cerebral hemorrhage has been understudied. However, there is evidence that components of hemorrhage-induced injury (e.g. inflammation and, for SAH, ischemia) cause phosphorylation of TJPs and MLCK. (b) Internalization/recycling. Stimuli can result in the endocytosis of transmembrane TJPs. TJPs within early stage endosomes may be recycled to the plasma membrane if the stimuli is removed or processed further to late endosomes. The effects of cerebral hemorrhage on TJP endocytosis have received little attention although there is evidence that inflammation and ischemia cause such internalization. It may be important in both early changes in TJ function and longer term TJP degradation. (c) Internalization/degradation. After internalization, vesicular trafficking of TJPs may end in the lysosome and degradation. Alternately, TJP proteins may be degraded in the proteasome or while still on the plasma membrane by the action of matrix metalloproteinases (MMPs). There is a lot of evidence that there is TJP degradation after ICH/SAH. The relative role of the different degradation pathways is unknown. (d) Transcription/translation. Brain endothelial TJP expression may also be affected by changes in transcription and translation. Transcription is regulated by multiple transcription factors (TF) but also by epigenetic changes (e.g. DNA methylation, Me), that might be important acutely and long-term. mRNA levels are also regulated by degradation. MicroRNAs (miRs) are one regulator of such degradation. Studies in other tissues show they can also suppress the translation of TJP mRNAs. The role of miRs in regulating brain endothelial TJPs in cerebral hemorrhage is unknown. Translation is also regulated by RNA binding proteins (RBPs). The role of such proteins on BBB changes after cerebral hemorrhage is unknown. A further layer of complexity is the some TJPs (ZO-1, ZO-2) can translate to the cell nucleus where they can alter transcription.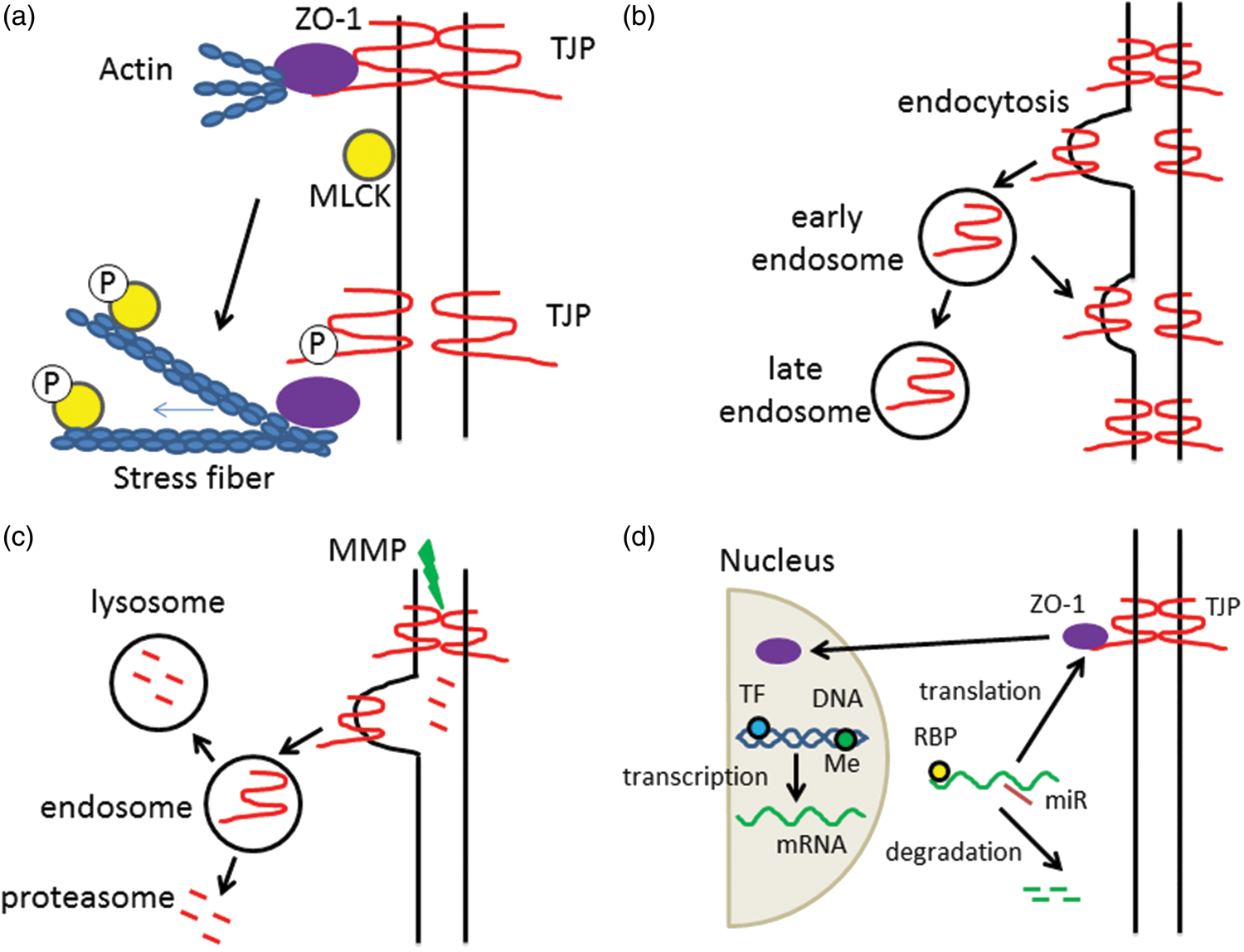
The regulation of endocytosis of transmembrane TJ proteins is now recognized as a major mechanism for regulation of barrier permeability. 16 Following such internalization, TJ proteins may be degraded or they may be recycled to the cell membrane if the stimulus that caused internalization is removed (Figure 2(b) and (c)). For example, exposure of brain endothelial cells to the chemokine CCL2 for 60 min causes caveolae-mediated claudin-5 internalization, but CCL2 removal results in recycling of claudin-5 to the cell membrane. 17 While in the related blood–retinal barrier, vascular endothelial growth factor induces the phosphorylation, ubiquitination and endocytosis of occludin leading to disassembly of the junctional complex. 18
Apart from degradation of TJ proteins after internalization, cleavage of transmembrane TJ proteins may occur while still at the plasma membrane (Figure 2(c)). For example, claudin-5 and occludin are substrates for matrix metalloproteinases (MMPs) and JAM-A is a substrate for A disintegrin and metalloproteinase (ADAM) family of metalloendopetidases. 10
Another important regulator of brain endothelial junctions is the actin cytoskeleton. It normally provides an important scaffold which, via linker proteins, contributes to junction stability. However, actin is present in cells in two forms, a globular monomeric form (G-actin) and a filamentous polymeric form (F-actin) and actin dynamics are regulated by a wide array of actin binding proteins. A variety of conditions result in the production of actin stress fibers19–22 and those fibers may exert forces on junction proteins causing junction disruption and barrier opening (Figure 2(a)).
Junction modifications may also occur via changes in protein transcription and translation (Figure 2(d)). Changes in mRNA can result from alterations in transcription. For example, transmembrane TJP promoter regions have binding sites for multiple transcription factors that can impact transcription in brain endothelial cells.23–26 Changes in histone acetylation/deacetylation can also impact transcription. There is evidence that reducing histone deacetylase (HDAC)-9, an epigenetic regulator, with short hairpin RNA (shRNA) increases brain endothelial cell claudin-5, occludin and ZO-1 levels after ischemia/reperfusion injury in vitro and in vivo. 27 However, transcriptional regulation of brain endothelial TJPs (acute and chronic) is an understudied area. A further layer of complexity is that some stimuli can cause TJPs (e.g. ZO-1) to translocate to the cell nucleus and affect transcription. 28
Changes in mRNA can also result from alterations in mRNA stability. MicroRNAs have been implicated in inducing TJP mRNA degradation in brain endothelial cells and epithelia.29,30 MicroRNAs can also inhibit the translation of TJP mRNAs, while some RNA binding proteins accelerate translation. 31
While the role of some of these types of regulation has been examined in some detail in inflammatory and ischemic conditions, the role of such processes in cereral hemorrhage has not been examined in depth with most studies focusing on TJ protein content (usually ZO-1, claudin-5 and occludin). It is important to note that increases in BBB permeability can occur because of TJ protein modification or relocation without reductions in TJ protein content. More in depth studies of TJ alterations in cerebral hemorrhage are needed and the tools for such studies are available (see below).
Effects of cerebral hemorrhage on BBB permeability, edema and inflammation
Hemorrhagic strokes (ICH, SAH, IVH) are all associated with high mortality and morbidity. All of these forms of cerebral hemorrhage cause increased BBB permeability, brain edema and neuroinflammation. In blood injection models of ICH, there is a delay before BBB hyperpermeability is detected occurring at ∼12 to 24 h, while in the collagenase model, earlier disruption is found at ∼5 h. In humans, BBB hyperpermeability has been detected at 24 h and longer. 3 BBB hyperpermeability can result in vasogenic brain edema and perihematomal edema is very marked after ICH, peaking at about 3 days in animal models and 10 days in humans.2,3 There is an influx of leukocytes (neutrophils, monocytes, T cells) into brain after ICH, as well as activation of resident microglia. As well as being an important component of ICH-induced injury, neuroinflammation is also crucial for hematoma removal and brain repair.32,33
In SAH, there is more diffuse injury than ICH reflecting potential global ischemia early after the hemorrhage and diffusion of blood within the CSF system. BBB disruption has been reported as early as 10 min after ictus and can persist up to seven days (reviewed in Tso and Macdonald 4 ). A biphasic opening has been reported with peaks at 3 and 72 h in the rat endovascular perforation model. 34 In SAH patients, enhanced BBB permeability on MRI imaging predicts the development of delayed cerebral ischemia, 35 a major contributor to poor outcome in patients. Global cerebral edema can result in SAH and this is also a predictor of adverse outcome in patients. 36 SAH induces a marked neuroinflammatory response in both animals and patients, including an influx of leukocytes into brain.37,38
In patients, IVH very rarely occurs alone, it is almost always an extension of an ICH or SAH. In premature infants, bleeding in the germinal matrix adjacent to the lateral ventricles causes IVH. This complicates determining what BBB effects are due to intraventricular blood rather than the initial hemorrhage. Animal models where blood is directly injected into the ventricles may provide insight. In rats with intraventricular injection of blood, periventricular edema and BBB disruption have been reported.39–41 It should be noted, however, that the main effect of intraventricular blood in relation to brain water is post-hemorrhagic hydrocephalus. That is related to altered CSF dynamics (blockage in CSF drainage and/or increased CSF secretion42,43)
Effect of cerebral hemorrhage on endothelial junctions
Changes in blood–brain barrier (BBB) permeability and tight junction (TJ) proteins after different forms of experimental stroke in vivo.
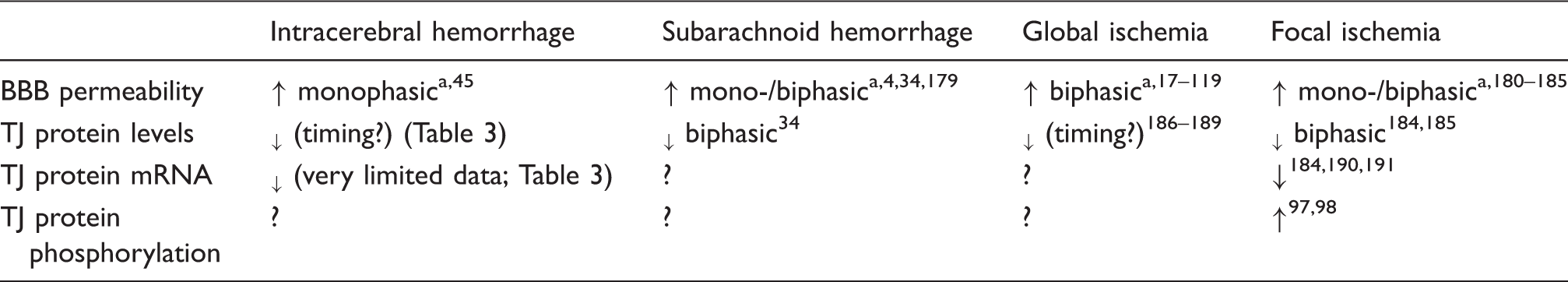
increase, ↓ decrease, ? unknown.
Multiple studies have reported either monophasic or biphasic increases in BBB permeability in experimental models of stroke. For example, in rat endovascular perforation SAH, both a monophasic delayed BBB disruption 179 and a biphasic early and late disruption 34 have been described. Similarly, in focal cerebral ischemia, both monophasic181,182 and biphasic180,183–185 BBB disruption have been described. There have been limited full time courses of tight junction protein changes after experimental stroke, but biphasic decreases have been reported after SAH and focal cerebral ischemia (permanent and transient)34,184,185 Biphasic disruption has been attributed to the temporal sequence of different injury mechanisms.
ICH
In rodent models, reductions in brain ZO-1, claudin-5 and occludin protein levels at one to three days after ICH (collagenase and blood injection) have been reported by multiple groups by Western blot.47–53 In addition, by immunohistochemistry, ICH induced breaks in normal microvascular ZO-1 staining have been reported at days 1 and 3.51,52,54 At the mRNA level, Sun et al. 49 also reported reductions in brain claudin-5 and occludin expression at days 1 and 3 after ICH. While there is consistent evidence of loss of TJ proteins after ICH, the time of onset and duration of that loss is uncertain. Also, there is a lack of information on the spatial distribution of TJ protein changes within the brain and how that evolves with time (e.g. are they limited to perihematomal tissue).
Interestingly, Krafft et al. 47 reported a very marked increase in phospho-β-catenin one day after ICH. Whether that extends to phosphorylation of other junction proteins or the actin cytoskeleton has not been examined. Given the importance of phosphorylation and other protein modifications in regulating junction function, 10 this should be rectified.
After ICH, there is evidence that the actin cytoskeleton may be a therapeutic target for preventing BBB disruption.55,56 However, there needs to be studies assessing and visualizing the endothelial actin cytoskeleton changes post-ICH (e.g. stress fiber formation; F-actin/G-actin ratios) as has been done for cerebral ischemia. 22
SAH
Historically much research on SAH has focused on delayed vasospasm and delayed cerebral ischemia (starting ∼3 days after ictus, peaking days 5–14 in humans). However, there has been a growing understanding of the importance of early brain injury4,57 and studies on brain endothelial junction proteins have primarily focused on 24 h after SAH. In the rat endovascular perforation model and brain Western blot, multiple studies have shown marked decreases in the TJ proteins ZO-1,58–63 occludin59,60,62,64–68 and claudin-558–61,64,65,68 at 24 h. Reductions in ZO-1, occludin and claudin-5 at 24 and 48 h after SAH have also been reported in the mouse perforation model using brain Western blot.69–73 An alternative to the endovascular perforation model of SAH is direct blood injection into CSF. Reductions in occludin and ZO-1 at 24 and 48 h after SAH have been reported in such models in rat using brain Western blot.74–77 While there is general agreement between these studies, Li et al. 34 examined longitudinal changes (30 min to 1 week) in ZO-1, occludin and claudin-5 in the rat endovascular perforation model. By brain Western blot, they too found a decrease in ZO-1 and occludin after SAH. However, they found no decrease in claudin-5 at any time point and their results suggest that 24 h may not be the optimal time point for studying ZO-1 and occludin. They found a biphasic change in those two proteins. For ZO-1, there was a significant loss of protein at 30 min, a peak loss at 2 h, and a return towards normal levels at 24 h. There was than a second phase of reductions peaking at 72 h but with a significant reduction still present at one week. A similar pattern was found with occludin, although occludin levels returned to normal at one week. Those results were confirmed by immunohistochemistry. 34 The very early changes in TJ proteins (30 min) are important as early BBB impairment has been reported after SAH. 4 Whether the biphasic changes in TJ protein expression reflect different etiologies, e.g. ischemia and inflammation, is uncertain as is whether early and late changes might be associated with protein turnover and transcriptional mechanisms. ZO-1, occludin and claudin-5 mRNA levels have not been studied after SAH.
Immunohistochemistry has rarely been used in SAH to examine changes in TJ proteins. However, loss/breaks in vascular staining for ZO1 have been reported in the perforation model34,58,60 in rats and mice. In addition, an intracellular relocation of ZO-1 has been reported. 78
While most attention has focused on ZO-1, occludin and claudin-5, other junction proteins have been examined. A reduction in brain JAM-A has been reported after endovascular perforation in mice at 24 h 69 and a loss of the endothelial AdJ protein, VE-cadherin, has been reported in both mice and rats at that time point.60,69,72 Assessing the impact of SAH on GJ proteins is difficult because of their high expression in brain parenchyma. Endothelial cells would need to be separated from the parenchymal cells (see below).
Endothelial cytoskeletal changes after SAH have not been a focus of research. This is despite evidence that actin cytoskeletal changes (e.g. stress fiber formation) are a therapeutic target for reducing BBB disruption in cerebral ischemia,22,79 and ischemia is a major component of SAH-induced injury. Cytoskeletal changes have been examined in smooth muscle cells after SAH (e.g. Lefranc et al. 80 ) and it will be important to separate the importance of changes in both cell types.
IVH
IVH occurs as a consequence of a parenchymal hemorrhage expanding into the ventricular system. Thus, in premature infants, a germinal matrix hemorrhage is the predominant underlying cause of IVH81 (although some may result from choroid plexus hemorrhage; see below), while in adults, IVH occurs in ∼50% of patients with ICH. 82 Thus, in patients, changes in endothelial junctions may be a response to a combination of ICH and IVH. In animal models, IVH has been induced by causing parenchymal bleeding with collagenase 83 or by injecting blood directly into the ventricular system. 84 The latter would allow direct assessment of intraventricular blood on parenchymal vessel junctions, but this has, as yet, not been undertaken. However, it is known that intracerebroventricular injection of thrombin, one clot-derived factor that impacts endothelial junctions causes periventricular BBB disruption. 85 In adult, proteins can cross the ependymal lining of the cerebral ventricles; therefore, it is likely that other clot-derived factors can affect periventricular brain endothelial cells and their junctions.
It should be noted that although this review focuses on endothelial junctions, IVH induces marked injury to the ependymal cells that line the cerebral ventricles in humans 86 and animals. 87 In adults, ependymal cells are primarily linked by GJs, with particularly high expression of connexin 43 (Gja1), and the paracellular space is permeable to large molecular weight proteins. 88 In contrast, early in development, a more complex set of junctions, termed strap junctions, link the ependymal cells greatly limiting paracellular permeability. 88 AdJ proteins (N-cadherin and β- and α-catenin) are highly expressed early in development and almost absent in the adult ependyma. 88 It is interesting that in human neonatal IVH, an intracellular accumulation of N-cadherin occurs away from sites of cell:cell contact 86 and alterations in ependymal junctions have been associated with hydrocephalus. 89
Effect of cerebral hemorrhage related stimuli on endothelial junctions
Several clot-derived/induced factors are involved in hemorrhage-induced brain injury.2,90 These include injury due to activation of the coagulation cascade and the generation of thrombin, as well as that due to erythrocyte lysis and the release of factors such as hemoglobin. As well as direct effects on the endothelium, such factors can induce a neuroinflammatory response5–7 that may induce barrier disruption, affect cerebral blood flow and induce ischemia. 90
Thrombin is well known to increase endothelial permeability in the systemic vasculature. There, thrombin via protease-activated receptor (PAR)-1 enhances permeability via effects on the cell cytoskeleton and AdJs, with Ca2+ and RhoA/RhoA kinase (ROCK) playing an important role. 91 In brain endothelial cells, thrombin and PAR-1 activation both increase intracellular Ca2+ 92,93 and thrombin activates RhoA, reduces ZO-1 and occludin protein levels, and causes discontinuities in ZO-1 and occludin expression at the cell border. 94
ICH induces Src kinase activation in the brain,
95
an effect mimicked by intracerebro-ventricular injection of thrombin.
85
Blocking Src reduced thrombin-induced BBB disruption and brain edema.
85
Src kinase phosphorylates tyrosine residues
96
and after transient focal cerebral ischemia, there is Src kinase activation in isolated brain capillaries, occludin tyrosine phosphorylation and decreased occludin content.
97
Inhibiting Src decreased ischemia-induced occludin phosphorylation, increased occludin levels and reduced BBB disruption.
98
Together, these results suggest that thrombin activation of Src kinase may be an important component of ICH-induced BBB disruption by modulating endothelial TJs. Figure 3 shows a schematic of the endothelial pathways thought to be involved in thrombin-induced BBB disruption.
Pathways involved in thrombin-induced junction changes. Thrombin is produced after cerebral hemorrhage to induce hemostasis (particularly via the conversion of fibrinogen to fibrin). However, it also has direct and indirect effects on brain endothelial cells that can lead to BBB disruption. Effects on brain endothelial cells via two protease-activated receptors, PAR-1 (reviewed in Brailoiu et al.
21
and De Bock et al.
193
) and PAR-4,
102
have been described. The pathways outlined in this schematic show major players. More detail (e.g. further intermediaries) is given in the text and Brailoiu et al.
21
and De Bock et al.
193
There is also a great deal of information on the effects of thrombin on systemic endothelial cells (e.g.Tiruppathi et al.
194
), although there may be differences in the pathways between brain and systemic endothelial cells. DAG: diacyl glycerol; IP3: inositol 1,4,5-triphosphate; MLCK: myosin light chain kinase; PAR: protease activated receptor; PLC: phospholipase C; ROS: reactive oxygen species.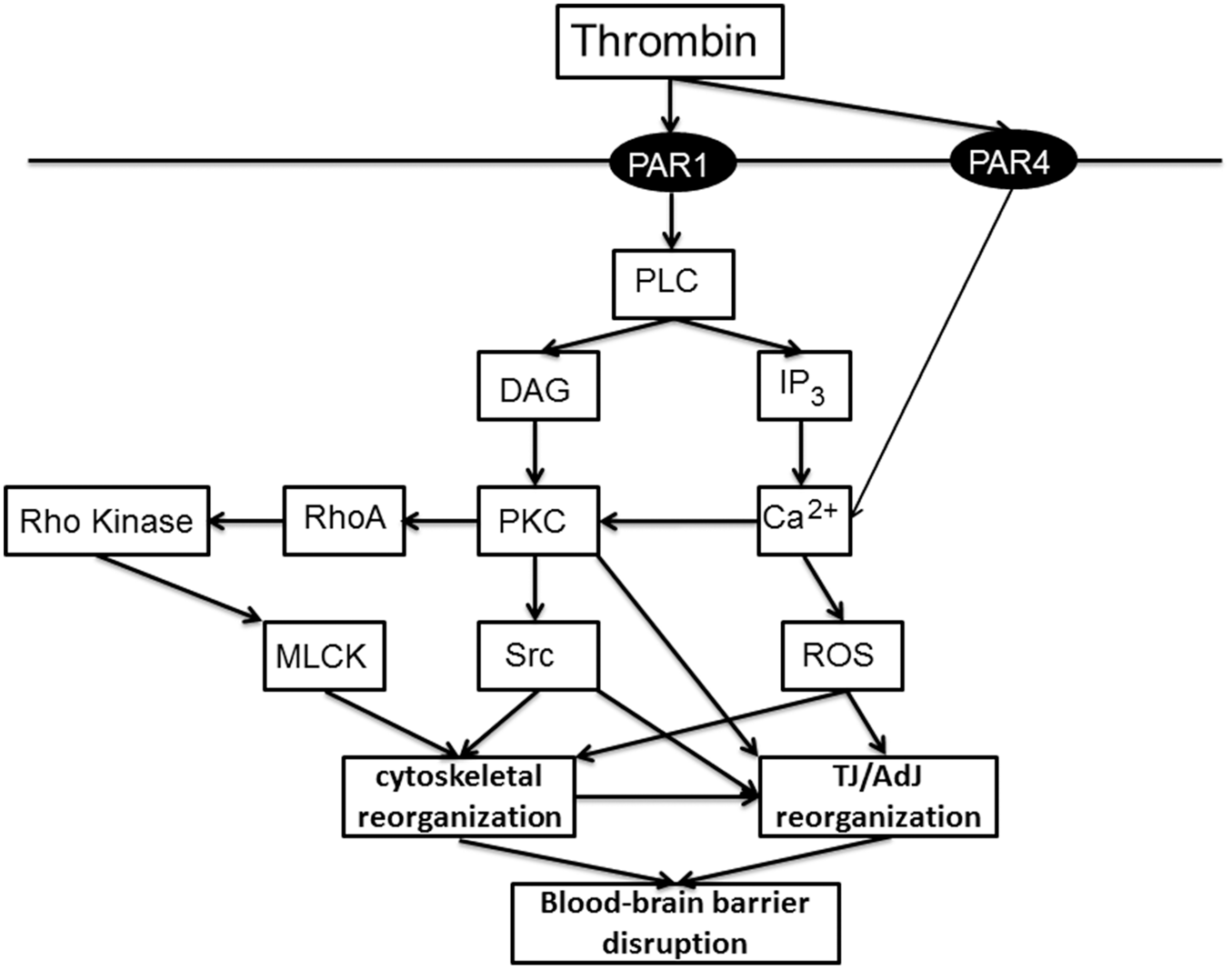
While the effects of thrombin on BBB permeability may be directly on the endothelium,92,93,99 they may also be via effects on other cells of the neurovascular unit. Thus, for example, thrombin can impact BBB function via effects on pericytes.100,101 In addition, it should be noted that PAR-1 is not the only PAR on brain endothelial cells. 99 PAR-4 has been implicated in raising brain endothelial Ca2+ 102 and calcium regulates BBB permeability (Figure 3). Plasmin, which will be generated after an ICH, also regulates PAR-2 signaling in brain endothelial cells. 103
Several studies have examined the effects of intracerebral injections of hemoglobin on BBB TJ proteins. Yang et al. 104 found that hemoglobin-induced BBB disruption was associated with discontinuous microvascular staining for claudin-5, ZO-1 and occludin. They also found reduced claudin-5, ZO-1, occludin and JAM-A mRNA levels in brain after hemoglobin injection, although the occludin reduction was transient with elevated expression at seven days. Fu et al. 105 also found that hemoglobin induced BBB disruption with brain edema, and that this was associated with reduced claudin-5 expression as assessed by Western blot and immunohistochemistry. Some of that decrease may be linked to an increase in MMP-9. However, they also found increases in RhoA, ROCK2 and phosphorylated myosin light chain (p-MLC) and phosphorylated myosin phosphatase target subunit1 (p-MYPT1) suggesting, as with thrombin, that changes in kinase activity and cytoskeletal modifications may be involved in the hemoglobin-induced barrier disruption. Ding et al. 106 extended these findings and found that a peroxynitrite decomposition catalyst suppresses MMP-9 expression and ameliorates hemoglobin-induced neurovascular injury as assessed by ZO-1 changes.
ICH induces neuroinflammation with microglial activation and the production of a wide range of inflammatory mediators such as the cytokines tumor necrosis factor α and interleukin-6 and the chemokine CCL2 and an influx of leukocytes from blood to brain.33,107–109 Elements of the coagulation cascade, thrombin and fibrinogen, are important in neuroinflammation.5–7 In addition, the release of methemoglobin and heme can also cause an inflammatory response via activation of toll-like receptors.110,111 The impact of inflammatory mediators on brain endothelial junctions has been extensively studied.
112
For example, the chemokine CCL2 (also known as monocyte chemoattractant protein-1) causes barrier disruption via TJ complex disassembly with claudin-5, occludin and ZO-1 and -2 phosphorylation, caveolae-mediated internalization of claudin-5 and occludin, and alterations to the actin cytoskeleton with stress fiber formation17,113–115 (Figure 4). RhoA, ROCK and PKCα play critical roles in these changes.113,114 Interestingly, CCL2 also induces internalization of JAM-A, but that transmembrane protein is then translocated to the luminal membrane of the endothelial cell where it can act as a leukocyte adhesion molecule.
116
Pathways involved in the junction changes induced by CCL2 (monocyte chemoattractant protein (MCP)-1). CCL2/MCP-1 is increased in brain after ICH and mechanisms by which CCL2 can induce brain endothelial TJ and cytoskeletal changes leading to barrier disruption are outlined. Mechanisms elucidated in brain endothelial cells
113
are shown in full lines. The dashed lines represent data from endothelial cells other than brain.
195
CCR2: C-C chemokine receptor type 2; MCPIP: MCP-1 induced protein; PKC: protein kinase C.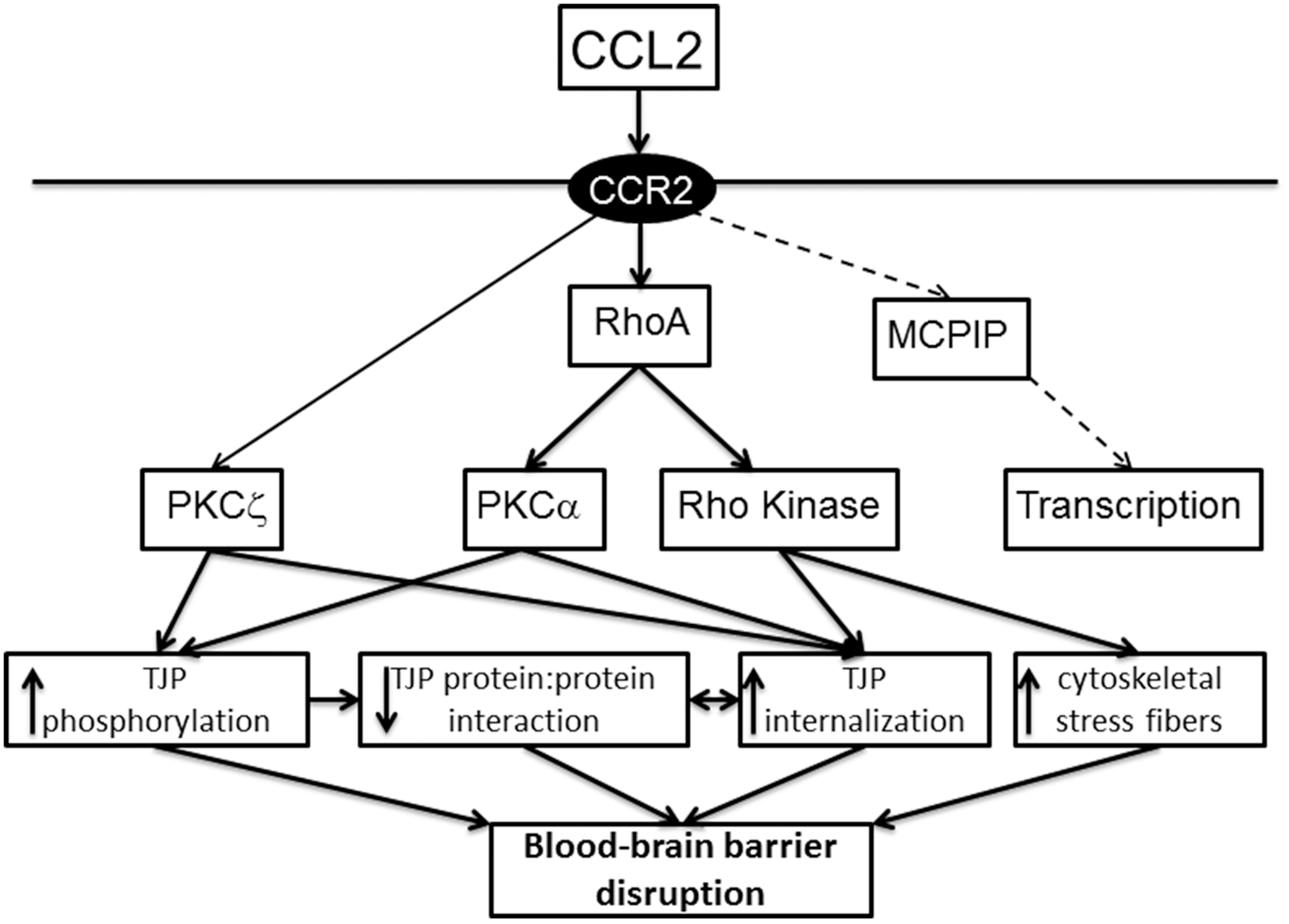
SAH causes early and late reductions in cerebral blood flow, with the early reduction due to increased intracranial pressure, the hemorrhage mass effect, and the later reduction due to macro/microvascular vasospasm. 4 It is notable that the biphasic time course of BBB impairment after rat endovascular perforation SAH34 is very similar to that reported in transient rat global ischemia models.117–119 The role of ischemia in ICH-induced brain injury is still debated and may depend on hematoma size. 2 The effects of ischemia on BBB disruption and cerebral endothelial cell junctions have been studied extensively in vivo and in vitro using models such as oxygen glucose deprivation (OGD) and recently reviewed.8,120 TJ proteins are affected at many levels after ischemia. Thus, reductions in TJ protein and mRNA levels, TJ phosphorylation and loss of TJ protein:protein interactions with TJ protein internalization and degradation have been reported8,120 as well as changes to the actin cytoskeleton.22,79 The relative importance of ischemia-induced TJ changes in the overall changes occurring in SAH remains to be fully elucidated.
As indicated in the above description, there are multiple pathways that can lead to alterations in TJ function and BBB disruption after cerebral hemorrhage. The importance of those pathways may vary with time. For example, thrombin is generated immediately after a hemorrhage, whereas erythrocyte lysis with the release of hemoglobin, heme and iron may be delayed. Similarly, there may be an initial period of global ischemia after SAH followed by delayed ischemia due to macro/microvascular vasospasm. This complex time course of events poses significant issues for treatment unless there are common downstream pathways.
Tools for studying junction changes
Assessing alterations in brain endothelial junction proteins
Although not directly assessing junction protein changes, electron microscopy has been used to examine changes in TJ structure after cerebral hemorrhage (ICH;48,53 SAH34,64,67) and abnormalities have been described such as widening of junction clefts, gaps between endothelial cells. The addition of tracer studies using horse-radish peroxidase would add information on the importance of the ultrastructural changes. Immunogold electron microscopy can be used to trace the subcellular location of particular brain endothelial junction proteins. 121
Most studies that have examined the effects of cerebral hemorrhage on brain endothelial junction proteins have relied on Western blots to examine junction protein loss. Many of those studies have used whole brain samples rather than isolated microvessels/endothelial cells, relying on the high expression of such proteins in the endothelium and not in brain parenchyma. It should be noted that this is not the case for all TJ proteins after injury. Thus, Horng et al. 122 found that reactive astrocytes express JAM-A and claudin-1 and -4 around inflammatory lesions.
Changes to the cerebral vasculature may be both dynamic and highly localized requiring specialized techniques to target regions of alteration. Western blots on isolated cerebral microvessels can be used to limit the potential influence of parenchymal expressed proteins. Such microvessels can be further digested for isolation of brain endothelial cells (particularly removing pericyte contamination) to further increase endothelial cell purity. Alternately, Tie2GFP mice can be used and endothelial cells separated from brain based on green fluorescent protein. 123 The endothelial cells can not only be used to examine changes in total junction protein, they can be used in cell fractionation studies to elucidate protein localization. These studies include determining whether particular junction protein modifications alter their distribution, and immunoprecipitation studies to examine protein:protein interactions.22,113,124 Such studies would provide insight into the changes underlying junction dysfunction in cerebral hemorrhage. Alterations in transmembrane TJ protein:protein interactions represent a major mechanism of altered paracellular permeability.
Immunohistochemistry has been used to examine loss of junction proteins from brain endothelial cells after cerebral hemorrhage (e.g. see literature51,52,60) or exposure of brain endothelial cells relevant stimuli in vitro.113,125 However, the use of fluorescent-tagged junction proteins can give greater detail. Thus, in vivo, Knowland et al. 126 have used fluorescent-tagged claudin-5 with two-photon microscopy to examine the time course of changes in TJs after ischemic stroke in individual animals, including movement of TJ proteins away from sites of cell:cell contact. Fluorescent-tagged tracers can be used to assess barrier permeability in the same experiments. In vitro, fluorescent tagged junction proteins expressed in brain endothelial cells can be used to examine the redistribution of those proteins in response to stimuli in real time.17,116,127 Such tagged proteins can facilitate tracking of the proteins via different intracellular compartments (vesicular trafficking17,116,127). If more than one fluorescent tagged protein is expressed, protein colocalization studies can be performed including the use of fluorescence resonance energy transfer (FRET) to examine protein:protein interactions.14,128 Fluorescence recovery after photobleaching (FRAP) can be used to examine the junction dynamics (e.g. is the protein a stable part of a junction complex or free to diffuse).14,128
At the mRNA level, brain samples have been used to assess TJ protein changes after cerebral hemorrhage. 49 As with protein levels, it is possible to examine brain microvessels or brain endothelial cells directly. In addition, brain endothelial cells can be sampled by laser capture microdissection and mRNA levels assessed. 129 This limits changes in mRNA expression during tissue processing. In vitro, studies of mRNA levels for junction proteins in brain endothelial cells and inhibitors of transcription (e.g. actinomycin D) can help elucidate whether junction regulation occurs at the transcriptional level. 124
As well as examining individual mRNAs by PCR, gene arrays have been used to assess mRNA levels in brain endothelial cells. Those arrays have either been genome wide 123 or focused, including junction arrays. 124 As well as mRNAs, there is a growing interest in assessing microRNA levels in the cerebral endothelium as several microRNAs have been implicated in regulating BBB function and TJ protein expression, including in ICH.130–133
Assessing changes in endothelial barrier function
Techniques for assessing blood–brain barrier permeability a after cerebral hemorrhage in vivo.

Tracer studies do not necessarily indicate route (e.g. para- vs. transcellular vs. cell death).
It should be noted that an accurate in vivo quantification of the BBB permeability (P, cm/min) requires measurement of the plasma profile of a tracer, correction for intravascular tracer if whole tissue is assessed, determination of BBB surface area and measurements should only be made under conditions of unidirectional uptake into brain. For many stroke studies, that information is lacking. For example, many studies do not assess plasma tracer concentrations, whether they change with an experimental condition or with time, or use those values to correct for intravascular tracer. Similarly, almost all studies do not assess changes in vascular surface area or tracer back flux from brain to blood. A decrease in BBB surface area due to lack of perfusion after a stroke or any tracer back flux, may lead to an underestimate of permeability increases.
In vitro, the effects of alterations in junctions on brain endothelial barrier function are widely examined by measuring changes in transendothelial electrical resistance (TEER), a measure of ion permeability, or measuring the permeability of different sized tracers. The tracers are usually either fluorescent- or radioisotope-tagged. Such measurements can be made with brain endothelial monocultures on transwells, or in cocultures with astrocytes and/or pericytes, 136 or with monocultures treated with astrocyte-conditioned media. The monocultures have an advantage of direct assessment of endothelial effects. The co-cultures generally have higher baseline TEERs and lower tracer permeabilities (i.e. are tighter). Indeed, there have been major concerns over how well in vitro models reflect in vivo function particularly relating to barrier tightness and TJ protein expression and location. 136 Recently, progress has been made in this area. 136 For example, induced pluripotent stem cell (iPSC) models with co-cultures of endothelial cells, astrocytes and pericytes have produced TEERs that are close to in vivo. Such models have been produced from human iPSCs and even individual patients, increasing translational relevance.137–139
TEER and tracer permeability studies are influenced by TJ function. Another assessment of barrier function, leukocyte transmigration, 140 is influenced by multiple factors including leukocyte:endothelial adhesion, crawling as well as diapedesis across the paracellular cleft that require altered TJ interactions. 112 In terms of cerebral hemorrhage, it is an important measure of barrier function as neuroinflammation with leukocyte infiltration into brain is a major component of brain injury.4,33
GJ function in vitro can be assessed by examining intercellular communication. Thus, for example, cell membrane impermeable tracers, such as Lucifer yellow can be injected into an endothelial cell within a monolayer and tracer spread to adjacent cells monitored. 14 That the spread is via a particular connexin can be examined with inhibitors. 141
Manipulating brain endothelial junctions
Multiple methods have been developed that can be used to experimentally manipulate brain endothelial junctions or their regulation. At the genetic level, there have been studies on global knockout or transgenic mice such as claudin-5 knockout. 142 Off target effects can be limited by using ‘endothelial’ specific or restricted promoters such as the Tek promoter, previously called the Tie2 promoter143,144 or the inducible platelet-derived growth factor B promoter, 145 which is more restricted to vascular endothelium than the Tie2 promoter that leads to myeloid cell expression, or a ‘brain endothelial restricted promoter’, such as that for claudin-5, but these have not yet been fully characterized. There have also been studies with inducible transgenic mice (e.g. Shi et al.22,79) and acute manipulations in vitro and in vivo using siRNA or shRNA (e.g. see literature124,146,147) to limit potential compensatory changes in other proteins. The impact of such manipulations on BBB and brain injury after cerebral hemorrhage or hemorrhage-like conditions in vitro should be very informative. Recently, the impact of reducing brain endothelial claudin-5 with an adeno associated virus expressing an inducible shRNA was examined and found to induce ‘depression-like’ symptoms in mice. 147
An advance for studying the role of occludin is the development of a mutant where occludin S490 is changed to an alanine to block phosphorylation at that site. 148 At the blood–retinal barrier, S490 is a site of vascular endothelial growth factor-induced phosphorylation that results in occludin ubiquitination and TJ trafficking. 148 Blocking that phosphorylation prevents vascular endothelial growth factor-induced hyperpermeability and neovascularization. 148 Phosphorylation of occludin S490 has a similar role in ischemia/reperfusion injury. 149
There have also been developments in targeting TJ proteins to enhance BBB permeability for drug delivery. Monoclonal antibodies and peptidomimetics targeting claudin-5 have been produced that increase BBB permeability in vitro and in vivo.150,151 Such agents may be useful for assessing the impact of alterations in claudin-5 function in neurological conditions, such as cerebral hemorrhage. Peptides are also available that inhibit the GJ and hemichannel functions of specific connexins to examine their role at the brain endothelium and in cerebral hemorrhage.14,141
An alternate approach has been to overexpress proteins within the cerebral endothelium. Shi et al. 79 attached heat shock protein 27 (HSP27) to a cell-penetrating transduction domain (TAT) peptide. HSP27 is a potent inhibitor for actin polymerization and it suppressed stress fiber formation and tight junction relocation in brain endothelial cells exposed to OGD. Intravenous HSP27-TAT increased brain endothelial HSP27 levels and reduced both BBB disruption and neurological deficits after ischemic stroke. 79
The impact of cell signaling on brain endothelial junctions has been examined with ‘specific’ inhibitors in vivo and in vitro.62,113,114 In vitro, dominant negative mutants have also been used.113,114 There can be, however, multiple concerns over interpreting in vivo data. For example, similar cell signaling cascades may occur in several cell types, not just endothelial cells, raising the possibility of indirect effects on endothelial junctions.
Potential therapeutic approaches to limit BBB changes post-hemorrhage
Changes in brain endothelial tight and adherens junction proteins after ICH.

: decrease, ↑: increase.
BI: blood injection; Coll: collagenase; WB: Western blot; PCR: polymerase chain reaction; IHC: immunohistochemistry; siRNA: small interfering RNA.
Changes in brain endothelial tight and adherens junction proteins after SAH.
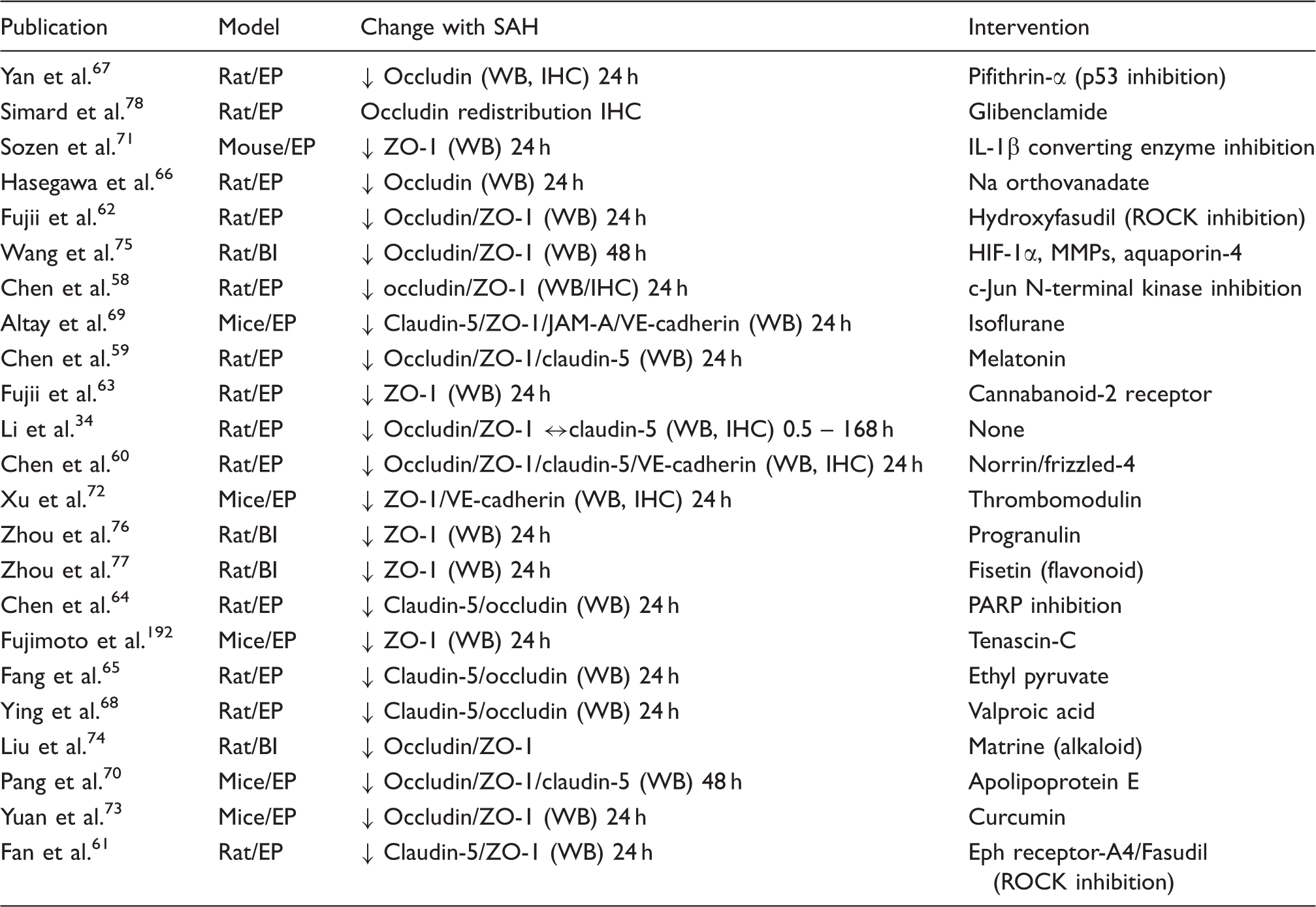
: decrease, ↔: no change.
EP: endovascular perforation; BI: blood injection; WB: Western blot; IHC: immunohistochemistry; HIF-1α: hypoxia inducible factor-1α; MMPs: matrix metalloproteinases; PARP: Poly (ADP-ribose) polymerase; ROCK: RhoA kinase.
There has been considerable interest in the role of MMPs in the loss of TJ proteins and BBB disruption found after stroke. 8 Non-specific MMP inhibitors reduced ICH-induced brain edema 152 and hemoglobin-induced BBB disruption, 153 and a MMP-9 inhibitor, SB-3CT, also reduced SAH-induced brain edema. 154 However, some of these effects may also be via inhibiting neuroinflammation and there may also be other pathways leading to TJ protein loss. Endocytosis, with lysosomal and proteasomal degradation, may contribute. Similarly, there are results indicating reduced TJ protein mRNA levels after ICH and hemoglobin exposure49,104 that may also result in reduced TJ protein levels.
As described above, several kinases (e.g. RhoA, ROCK, Src, JNK) may be involved in junction changes and BBB disruption after cerebral hemorrhage. There is evidence that inhibiting Src can have beneficial effects, although that can depend on the timing after ICH as Src may also be involved in barrier repair.85,95 As described above, components of ICH- and SAH-induced injury (thrombin, hemoglobin, inflammation and ischemia) activate RhoA and ROCK. Fasudil, a ROCK inhibitor, has been extensively tested in SAH, but with a focus on vasospasm. 155 It has been used in Japan since 1995 for SAH patients.156,157 In addition, there is evidence that ROCK inhibition reduces BBB damage and TJ changes in animal models of cerebral ischemia. 158 Therefore, it is surprising that RhoA and ROCK inhibitors have not been more of a focus of testing in ICH and SAH models for preventing BBB disruption. Two ROCK inhibitors, hydroxyfasudil and Y27632, reduced SAH-induced early brain injury and edema formation and hydroxyfasudil reduced SAH-induced BBB disruption and loss of occludin and ZO-1. 62 More such studies are required. Fan et al. 61 recently tested fasudil in a rat SAH, endovascular perforation model, but only in combination with another protective agent.
It should be noted that there are some potential concerns in targeting kinases. Multiple kinases may be involved in ICH- and SAH-induced BBB disruption. There may be multiple target proteins phosphorylated with different functions. Even with a single junction protein, there may be multiple phosphorylation sites, which may be phosphorylated by different kinases and have different functional consequences.10,96
While there has been much interest in the role of protein kinases on BBB disruption, there has been much less attention focused on proteins that dephosphorylate junction proteins and the potential role of such phosphatases in BBB regulation. Tissue non-specific alkaline phosphatase (TNAP) is highly expressed in brain capillary endothelial cells but not systemic capillary endothelial cells. Deracinois et al. 159 found that inhibiting TNAP with levamisole increased the permeability of a bovine capillary endothelial cell model. There is also evidence that another endothelial phosphatase, vascular endothelial cell-specific phosphotyrosine phosphatase (VE-PTP) regulates endothelial permeability via effects of VE-cadherin, with down regulation of VE-PTP increasing permeability in systemic endothelial cells.160,161 The ability of angiopoietin-1 to repair TJs may be via non-receptor protein tyrosine phosphatase N-2 (PTPN-2). Depletion of PTPN-2 blocks the ability of angiopoietin-1 to dephosphorylate tyrosine on occludin and causes brain endothelial hyperpermeability. 162
Another potential target is preventing the translocation of TJ proteins from the plasma membrane. This might be achieved by inhibiting endocytosis/vesicular trafficking although such pathways are crucial for multiple cell types in the body. An alternative approach may be to prevent cytoskeletal reorganization and stress fiber formation. There is evidence in cerebral ischemia that targeting the cytoskeleton by endothelium-targeted expression of actin depolymerizing factor or heat shock protein 27 can prevent BBB disruption and reduce brain injury.22,79 Recently, in a mouse ICH model, Manaenko et al. 55 found that targeting platelet derived growth factor-β signaling could reduce stress fibers, limit BBB disruption, ameliorate brain edema and improve neurological outcomes. Wang et al. 56 also found that ICH causes a loss of annexin A1 from the cerebrovascular endothelium and that administration of exogenous annexin A1, which helps maintain cytoskeleton integrity, reduces ICH-induced BBB disruption.
Potential therapeutic approaches to limit bleeding pre-hemorrhage
Brain endothelial cells and their junctions are dynamic structures. This raises the question of whether vascular integrity may be altered to limit the risk of hemorrhage? This has been a particular focus of IVH research where ‘immature’ vessels of the germinal matrix in premature infants are prone to rupture. 163 Antenatal glucocorticoids can reduce the frequency and severity of IVH in premature infants 164 although they have multiple off target effects to consider. Glucocorticoids have pleiotropic effects including affecting brain endothelial cell TJs, AdJs and cytoskeleton. 165 However, while most studies suggest a beneficial effect on junction proteins and barrier properties (e.g. Blecharz et al. 166 and Na et al. 167 ), others have found detrimental effects 168 and more precise methods of manipulating barrier properties would be beneficial. There is growing understanding of the pathways involved in regulating angiogenesis and barrier-genesis (e.g. the role of angiopoietin, platelet-derived growth factor B, sonic hedgehog and Wnt signaling). 169 Manipulating such pathways may provide a method of preventing germinal matrix hemorrhage/IVH in premature infants.
Interest in ‘strengthening’ the cerebral blood vessels to prevent hemorrhage is not limited to neonatal IVH. Yan et al. 170 examined the effects of administering minocycline for two months in a model of cerebral amyloid angiopathy (Tg2576 mice). They found that the treatment reduced the occurrence of hemorrhages and that this was associated with increased capillary ZO-1 expression and reduced MMP-9 and -2 levels. Some dietary components, such as sulforaphane found in cruciferous vegetables, can reduce endothelial TJ changes after brain injury. 171 Whether such agents might prevent vascular injury, such as hemorrhage, merits investigation.
Recently, treatment with the ROCK inhibitor, fasudil, has been tested in two mouse models of two different forms of cerebral cavernous malformation (CCM-1 and -2). In the model of CCM-1, chronic fasudil administration for four months, reduced lesion burden and iron deposition, a measure of hemorrhage, and improved animal survival. 172 There was no benefit with fasudil in the CCM-2 model.
Choroid plexus bleeding and response to cerebral hemorrhage
Bleeding into the CSF (IVH) is usually a result of disruption of parenchymal blood vessels (e.g. in the germinal matrix of premature infants) or the extension of a SAH. However, bleeding may sometimes occur at the choroid plexus.173,174 The choroid plexuses are present in the lateral, third and fourth cerebral ventricles and have a fenestrated blood vessel and stroma core surrounded by an epithelial layer. The choroid plexus epithelial cells are linked by TJs, forming a blood-CSF barrier, and are the primary site of CSF secretion. 175
As with the BBB at the cerebral endothelium, the paracellular cleft of choroid plexus epithelial cells is occluded by transmembrane TJ proteins. However, while both barriers express occludin and JAM-A, the claudins highly expressed at the choroid plexus epithelium (−1, −2, −3 and −11) are different from that at the cerebral endothelium (where claudin-5 is highly expressed) 176 and the choroid plexus epithelial barrier is not as tight as the BBB. The cadherin at the choroid plexus epithelium AdJs, E-cadherin, also differs from that at the cerebral endothelium (VE-cadherin) 176 as does the types of connexins expressed at the GJs. 177
Apart from being a potential site of bleeding, choroid plexus function may be affected by cerebral hemorrhage and particularly IVH. This has been reviewed elsewhere. 178 Surprisingly, there is a lack of studies examining changes in choroid plexus epithelium junction structure in IVH where the blood may bathe the choroid plexus. Overall, this is an understudied area of research deserving of more investigation.
Conclusion
The impact of cerebral hemorrhage on cerebral endothelial junctions is an understudied area with important clinical implications for reducing edema, neuroinflammation and the entry of potentially neurotoxic and vasoactive compounds from blood. Junction function can now be assessed at much greater depth than just examining TJ protein loss. Such research may not only provide important insights for the treatment of primary ICH, SAH and IVH but also the hemorrhages that can occur after traumatic brain injury or brain tumors. It may also provide information on how to prevent hemorrhage or limit hemorrhage size by ‘strengthening’ the barrier.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health grants NS073595, NS079157, NS090925, NS091545, NS093399, NS096917, NS98066, NS098211, AG057928 and NIH EY012021. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. It was also supported by the Joyce & Don Massey Family Foundation and Research to Prevent Blindness (RPB).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.