
Abstract
Glutamate levels increase dramatically in cerebral ischemia and stroke. This may lead to opening of the blood–brain barrier (BBB) and induce further brain damage. Because endothelial tight junctions are critical elements of the BBB integrity, the aim of this study was to investigate the mechanisms of glutamate-induced alterations of the tight-junction protein occludin in cultured brain microvascular endothelial cells (BMECs). Transient exposure to glutamate resulted in cellular redistribution of occludin, followed by a decrease in the total level of this protein and diminished barrier function of BMECs. Inhibition of the N-methyl-d-aspartate (NMDA) or alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate/kainate (AMPA/KA) receptors attenuated glutamate-induced changes in occludin redistribution but not in the total protein levels. Treatment with glutamate also increased tyrosine phosphorylation and decreased threonine phosphorylation of occludin. Inhibition of the NMDA receptors by MK-801 partially protected against glutamate-induced elevation of occludin tyrosine phosphorylation. In addition, pretreatment with MK-801-attenuated glutamate-mediated disruption of endothelial barrier function. Blocking of the AMPA/KA receptors by 6,7-dinitroquinoxaline-2. 3-dione (DNQX) protected against hypophosphorylation of threonine residues of occludin; however, it did not affect disruption of endothelial integrity. These findings indicate the opposite effects of the NMDA and AMPA/KA receptors on occludin phosphorylation and disruption of the BBB functions.
Introduction
Glutamate is the major excitatory neurotransmitter in the mammalian brain. The brain contains large amounts of glutamate; however, the majority of glutamate content is restricted to intracellular space (Coyle and Puttfarcken, 1993). The concentration gradient of glutamate across the cell membranes is several thousand folds. Glutamate is continuously released from the cells and removed from the extracellular space in a dynamic equilibrium, which can be disturbed by changes in energy/ATP supply. Normal levels of extracellular glutamate are in the range of 3 to 4 μmol/L; similar concentrations are characteristic for other extracellular compartments, such as cerebrospinal fluid (10 μmol/L) and plasma (18 to 25 μmol/L).
During ischemia and stroke, a reduction in blood flow to the brain microvasculature induces a decrease in the delivery of oxygen and nutrients. The resulting ATP depletion contributes to increased leakage of glutamate from the cells. In consequence, extracellular glutamate levels increase early in transient cerebral ischemia (Ritz et al, 2004), intracerebral hemorrhage (Qureshi et al, 2003), and/or cerebral artery occlusion (Umemura et al, 1996) playing a key role in ischemic brain damage (Rothman and Olney, 1986). High levels of extracellular glutamate can lead to disruption of the blood–brain barrier (BBB) (Mayhan and Didion, 1996) and consequently, to vasogenic edema, events that may intensify 24 to 72 h after ischemic stroke (Preston and Webster, 2004).
Cerebral endothelial cells can express several types of glutamate receptors (Krizbai et al, 1998; Sharp et al, 2003). This is important because cellular effects of glutamate are mediated by ionotropic and metabotropic receptors. The ionotropic glutamate receptors function like ligand-gated ion channels and they are divided into N-methyl-d-aspartate (NMDA) receptors, alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate (AMPA), and kainate (KA) receptors (Simeone et al, 2004). The high levels of extracellular glutamate can overstimulate the glutamate receptors, leading to an unregulated increase in intracellular calcium and neuronal death (Dempsey et al, 2000; Lipton and Rosenberg, 1994).
Ionotropic glutamate receptors appear to play a critical role in the pathology of cerebral ischemia and stroke. It was shown that the NMDA and alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate/kainate (AMPA/KA) receptor antagonists can protect against permeability increase and vascular edema formation (Dempsey et al, 2000). Inhibition of the ionotropic glutamate receptors also exerted neuroprotective effects on a model of cerebral artery occlusion in animal studies (Takahashi et al, 2002). In fact, the ionotropic receptor antagonists reduced brain edema after BBB opening by cerebral ischemia/reperfusion (Westergren and Johansson, 1992) and had beneficial effects on intracerebral hemorrhage in rats (Ardizzone et al, 2004).
Cerebral microvascular endothelial cells joined by tight junctions are the main structural components of the BBB. Tight junctions contain several transmembrane proteins, such as occludin, claudin-1, claudin-5, junctional adhesion molecule-1, and tight junction-associated proteins, namely, zonula occludens (ZO)-1 and ZO-2 (Gonzalez-Mariscal et al, 2000; Tsukita et al, 2001). Disruption of tight junctions is critical in many central nervous system pathologies including ischemia and stroke. Tight junction ‘loosening’ may lead to an increase in BBB permeability (Huber et al, 2001) and subsequently to vasogenic edema. Alterations of tight junction proteins were shown in hypoxia/reoxygenation modeling of transient ischemia. Specifically, hypoxia causes occludin, claudin-1, ZO-1, and ZO-2 redistribution; however, reoxygenation results in increased expression of occludin, ZO-1, ZO-2, and claudin-1 protein levels (Brown et al, 2003; Mark and Davis, 2002). Discontinuous tight junctions have also been shown in experimental cerebral infarction (Shibata et al, 1988). Finally, alterations of tight junction morphology have been observed before stroke in spontaneously hypertensive rats. Tight junction particles were slightly shifted from the P (protoplasmic) face to E (extracellular) face in these animals (Lippoldt et al, 2000), suggesting that these early changes may contribute to the pathogenesis of stroke.
The mechanisms of glutamate-induced disruption of the BBB integrity are not fully understood. In this study, we propose that glutamate-induced alterations of tight junction protein expression can play a critical role in this process. Our preliminary data revealed that among several tight junction proteins studied (data not shown), glutamate induced the earliest and most dramatic effects on occludin immunoreactivity associated with decreased protein levels. Therefore, the main goal of this study was to evaluate the mechanisms of alterations of occludin expression in response to glutamate treatment. We identified that ionotropic glutamate receptors were responsible, at least in part, for glutamate-induced changes in occludin expression, distribution, and phosphorylation.
Materials and methods
Primary Cultures of Brain Microvascular Endothelial Cells and Astroglial Cells
Primary cultures of brain microvascular endothelial cells (BMEC) were the main experimental model used in this study. Briefly, brain capillary fragments were isolated from the forebrains of 2-week-old Wistar rats and seeded on 35-mm Petri dishes coated with collagen type IV and fibronectin. Brain microvascular endothelial cells migrated from isolated microvessels to reach confluence 4 days after the seeding (Deli MA, 1997; Perriere et al, 2005) (Figures 1A and 1B). The cells were cultured in Dulbeccos-modified Eagles medium with 20% plasma-derived bovine serum (Animal Technologies Inc., Tyler, TX, USA), 40 μg/mL endothelial cell growth supplement (BD Biosciences, Bedford, MA, USA), 100 μg/mL heparin, 2 mmol/L glutamine, 5 μg/mL insulin, 5 μg/mL human transferrin, 5 ng/mL Na-selenite (Insulin-Transferrin-Sodium Selenite Media Supplement; Sigma, St Louis, MO, USA), and 25 μg/mL gentamicin. Brain microvascular endothelial cells can dedifferentiate very rapidly; therefore, only primary cultures were used in the present study. The cultures were positive for the factor VIII-related antigen, took up DiI-acetylated low-density lipoprotein, and were negative for alpha-smooth muscle actin (Figures 1C to 1F).
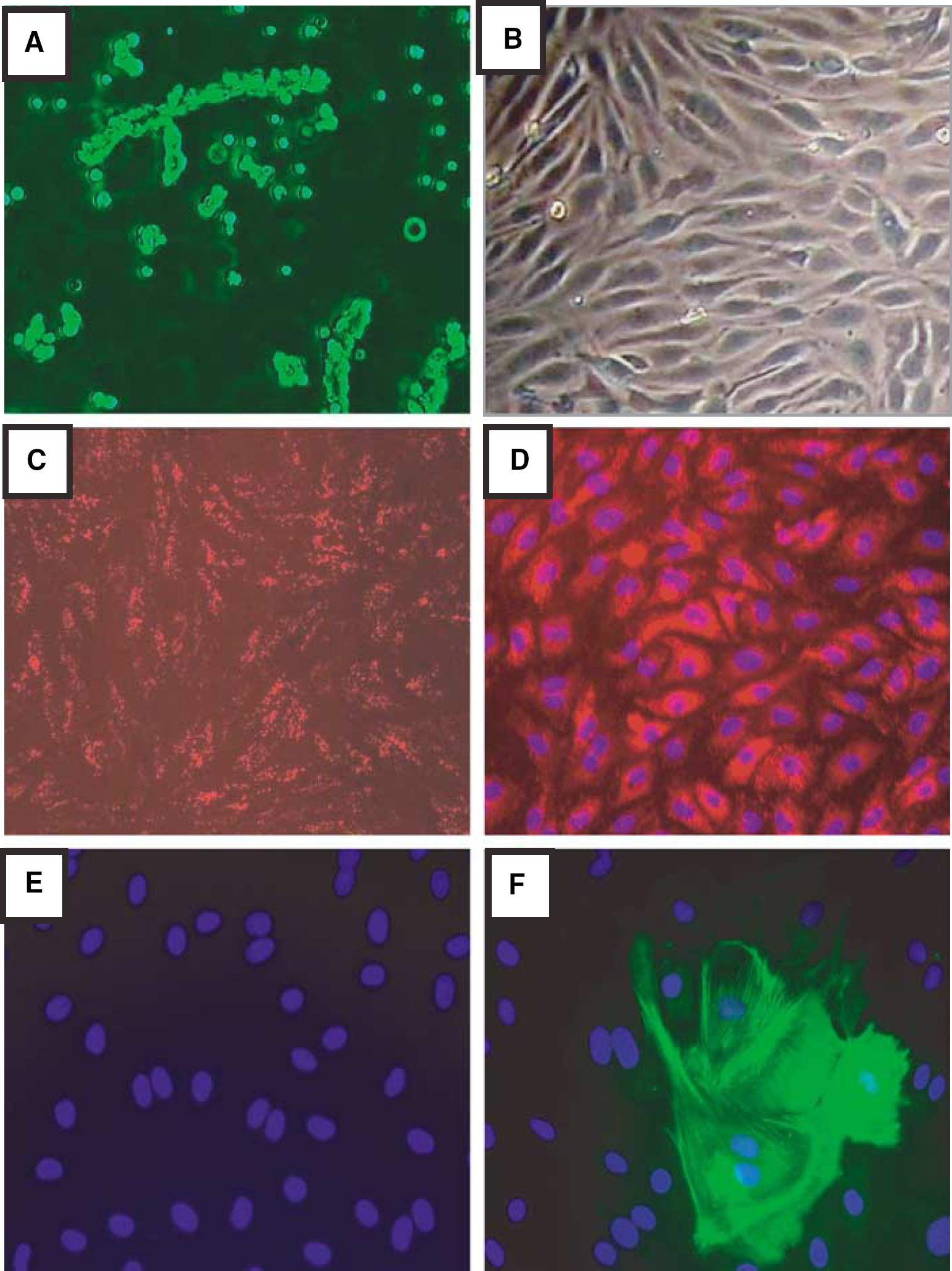
Characterization of primary cultures of BMEC. (
In selected experiments, BMEC were cocultured with rat cerebral glial cells to induce BBB characteristics (Kis et al, 2001). Briefly, primary cultures of glial cells were prepared from newborn Wistar rats. Meninges were removed, and cortical pieces were mechanically dissociated in Dulbeccos-modified Eagles medium containing 5 μg/mL gentamicin and 10% fetal bovine serum and plated in poly-l-lysin-coated 12 well dishes. In confluent glial cultures, 90% of cells were immunopositive for the astroglial cell marker glial fibrillary acidic protein, whereas the remaining 10% cells were immunopositive for CD11b, a marker of microglia.
Treatment Factors and Experimental Design
Intracellular concentrations of glutamate are at the millimolar level (Coyle and Puttfarcken, 1993). Therefore, glutamate release may generate extracellular concentrations at the range of 1 mmol/L, which was used in this study. Such concentrations of glutamate have also been used in several literature reports (Krizbai et al, 1998; Sharp et al, 2003). In our preliminary experiments, glutamate at concentrations lower than 1 mmol/L did not affect cellular occludin levels (data not shown).
In a typical experiment, confluent BMEC cultures were treated with 1 mmol/L glutamate for 30 mins followed by replacing experimental medium with normal growth medium without serum. Such an experimental design mimics pathological conditions of a transient increase in extracellular glutamate. However, a transient exposure to glutamate may induce long-lasting effects. Therefore, experiments were performed immediately after media change or 24 h after glutamate exposure. These treatment conditions did not affect cell viability as determined by 3-[4,5] dimethylthiazol-2,5-diphenyltetrazolium bromide conversion assay (data not shown).
In specific experiments, BMEC were pretreated for 15 mins before glutamate treatment with 10 μmol/L MK-801 (a selective inhibitor of the NMDA receptors) or 5 μmol/L 6,7-dinitroquinoxaline-2.3-dione (DNQX, an inhibitor of the AMPA and KA receptors). Our preliminary studies (data not shown) indicated that these concentrations were most effective in preventing cellular effects of glutamate. In addition, higher doses of these inhibitors showed toxicity and resulted in the cell death. The inhibitors were left in cell culture media for the duration of glutamate treatment.
Antibodies
Mouse monoclonal antioccludin antibody and rabbit polyclonal antiphosphothreonine antibody were obtained from Zymed Laboratories (San Francisco, CA, USA) and rabbit polyclonal antiphosphotyrosine antibody was purchased from Upstate Group (Lake Placid, NY, USA). Rabbit polyclonal anti-GluR1, mouse monoclonal anti-Pan-NMDAR NR-1, and rabbit polyclonal anti-NMDAR NR-2B were from Calbiochem (San Diego, CA, USA). Fluorescein isothiocyanate- and Texas Red-conjugated antimouse and antirabbit, horseradish peroxidase-conjugated antimouse, and antirabbit secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Immunoblotting
Homogenates of cultured BMEC were prepared in lysis buffer (20 mmol/L Tris-HCl, pH 7.4, 150 mmol/L NaCl, 0.5% Triton X-100, 0.1 mg/mL phenylmethylsulfonyl fluoride, 0.5% Nonidet P-40, 1 mmol/L EDTA, 2.5 μg/mL leupeptin, 1 μg/mL pepstatin A, 1 μg/mL aprotinin). After centrifugation at 15,000g for 15 mins, the supernatants were collected and protein concentrations were determined using the Bradford protein assay. Samples (20 μg protein/lane) were separated on sodium dodecyl sulfate-polyacrylamide gel, blotted onto nitrocellulose membrane (Bio-Rad Laboratories, Hercules, CA, USA), and incubated with the respective antibodies. For visualization of detected proteins, immunoblots were analyzed using an enhanced chemiluminescence Western blot detection kit (Amersham Biosciences, Piscataway, NJ, USA).
Immunoprecipitation
Immunoprecipitation of occludin was performed using 150 μg of protein extract. Samples were incubated with 1.5 μg of mouse monoclonal antioccludin antibody for 4 h at 4°C. Then, 30 μL of Protein A/G PLUS-Agarose immunoprecipitation reagent (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was added to each sample and immunoprecipitation was performed at 4°C overnight. The beads were recovered by centrifugation at 1,500g for 5 mins at 4°C and washed four times with 1 mL of phosphate-buffered saline (PBS). Bound proteins were eluted by boiling in sodium dodecyl sulfate sample buffer for 5 mins and analyzed on 4% to 15% sodium dodecyl sulfate-polyacrylamide gels. Fractionated proteins were blotted onto nitrocellulose membrane (Bio-Rad Laboratories, Hercules, CA, USA) and incubated with the respective antibodies. For visualization of detected proteins, immunoblots were analyzed using an ECL Western blot detection kit (Amersham Biosciences, Piscataway, NJ, USA).
Immunofluorescence Microscopy
BMEC cultured on 35 mm dishes or Permanox-chambered plastic slides were fixed with ethanol for 30 mins at 4°C. After washing with PBS and blocking with 3% bovine serum albumin (BSA)-PBS for 30 mins, samples were incubated overnight with primary antibody at 4°C. The excess of primary antibody was removed, slides were washed with PBS, and incubated with fluorescein isothiocyanate- or Texas Red-conjugated secondary antibody for 2 h at room temperature. Hoechst nuclear staining was applied for 5 mins at room temperature. After washing with PBS, slides were mounted in ProLong Antifade mounting medium (Molecular Probes Inc., Eugene, OR, USA) and covered with coverslips. Specimens were evaluated at room temperature under the epifluorescence Nikon Eclipse E600 microscope, equipped with the Plan Fluor × 20/0.50 and × 40/0.75 lenses. The images were captured using a Spot CCD camera system and software (Diagnostic Instruments, Sterling Heights, MI, USA). They were processed using brightness/contrast adjustment, color balance adjustment, and gamma adjustment to decrease background autofluorescence.
Measurements of Brain Microvascular Endothelial Cells Integrity
Permeability studies were performed in cocultures of BMEC with glial cells. Brain microvascular endothelial cells cultured on cell culture inserts were placed into multiwells containing astroglia at the bottom of the wells with endothelial culture medium in both compartments. When BMEC became almost confluent, cultures were treated with hydrocortisone (550 nmol/L), 8-(4-chlorophenyl thio)-cyclic AMP (CPT-cAMP; 250 μmol/L), and the cAMP phosphodiesterase-4-specific inhibitor RO 201724 (17.5 μmol/L) for 24 h to tighten the cell junctions and elevate resistance (Deli et al, 2005; Perriere et al, 2005).
The flux of sodium fluorescein (SF) across the endothelial monolayers was determined as previously described (Kis et al, 2001). Treated BMEC on cell culture inserts were transferred to 12 well plates containing 1.5 mL Ringer–Hepes solution (118 mmol/L NaCl, 4.8 mmol/L KCl, 2.5 mmol/L CaCl2, 1.2 mmol/L MgSO4, 5.5 mmol/L d-glucose, 20 mmol/L N-2 hydroxyethylpiperazine-N-2-ethanesulfonic acid, pH 7.4) in the basolateral compartments. In apical chambers, culture medium was replaced by 500 μL Ringer–Hepes solution containing 10 μg/mL SF (MW 376 Da). The inserts were placed after 20, 40, and 60 mins into new wells containing Ringer–Hepes solution. The concentration of SF in the upper and lower compartments was determined using a fluorescent plate reader (emission, 525 nm; excitation, 440 nm). Flux across cell-free inserts was also measured. Clearance (in μL) of SF diffusing from luminal to abluminal compartments was calculated according to the formula (Deli et al, 2005):

The average volume cleared was plotted versus time, and permeability × surface area product value for endothelial monolayer (PSe) was calculated from the following formula:
The endothelial permeability coefficient (Pe; in 10−3 cm/s) was then calculated by dividing the PSe values by the surface area.
Transendothelial electrical resistance, representing the permeability of tight junctions for sodium ions, was measured by an EVOM resistance meter (World Precision Instruments, Sarasota, FL, USA) using the STX-2 electrodes. The values were normalized to the surface of endothelial monolayer (Ω cm2). The TEER values of cell-free inserts (90 to 100Ω cm2) were subtracted from the sample values.
Statistical Analysis
Routine statistical analysis of data was completed using SigmaStat 2.0 (SPSS Inc., Chicago, IL, USA). One-way ANOVA was used to compare responses among treatments. Treatment means were compared using Bonferroni or Newman–Keuls tests. Changes were considered statistically significant at P < 0.05.
Results
This study is focused on the hypothesis that glutamate, acting through the ionotropic glutamate receptors, can alter expression and distribution of occludin in BMEC and thus cause alterations of the BBB. First, we examined glutamate-induced alterations of occludin expression and distribution. In the next series of experiments, we studied the involvement of the NMDA and AMPA/KA receptors in glutamate-induced changes in occludin phosphorylation. The final experiments were focused on the role of the NMDA and AMPA/KA receptors in alterations of the BBB integrity. Characterization of cultured BMEC used in the present study is reflected in Figure 1.
Expression of Glutamate Receptors in Brain Microvascular Endothelial Cells
To show the presence of ionotropic glutamate receptors in BMEC, we performed Western blot and immunofluorescence microscopy assays for several subunits of ionotropic glutamate receptors. As indicated by Western blots (Figure 2A) in two different samples, BMEC express GluR-1 (an AMPA receptor subunit), and NMDAR-NR-1 and NMDA-2B (NMDA receptor subunits). Other subunits of ionotropic glutamate receptors were not analyzed. Protein extracts of the whole rat brain were used as a positive control. Besides the bands corresponding to the studied receptor subunits, additional lower molecular weight bands were visualized. These bands may reflect unspecific binding of polyclonal antibodies.
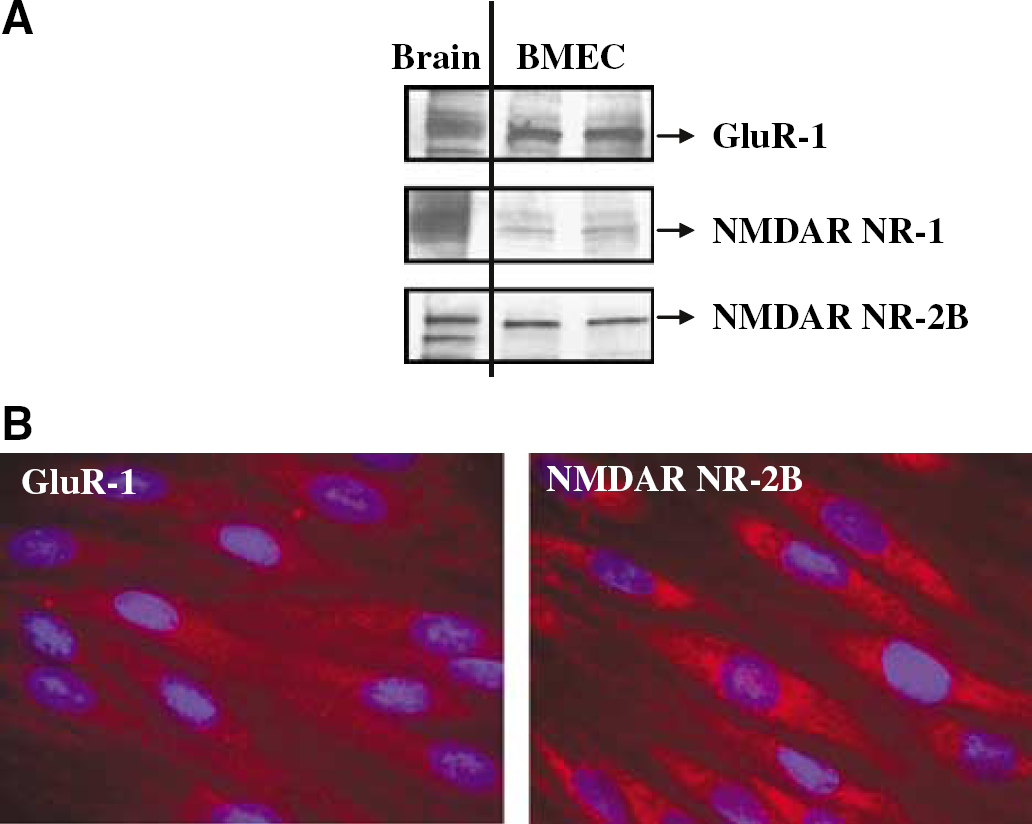
Expression of ionotropic glutamate receptors in BMEC. (
Figure 2B illustrates the cellular localization of GluR-1 and NMDAR-2B. GluR-1 immunoreactivity showed a punctuate immunostaining pattern in the cytoplasm. NMDAR-2B immunoreactivity appears to be distributed in the cytoplasm and around the nuclei.
Transient Glutamate Exposure Alters Occludin Expression
To investigate the effects of glutamate on occludin expression, BMEC were treated with lmmol/L glutamate for 30 mins, followed by replacement of experimental media with normal growth medium without serum. Protein levels of occludin were determined immediately or 24 h after glutamate exposure. As indicated in Figure 3A, a significant decrease in occludin total protein level was observed 24 h after a transient glutamate treatment.
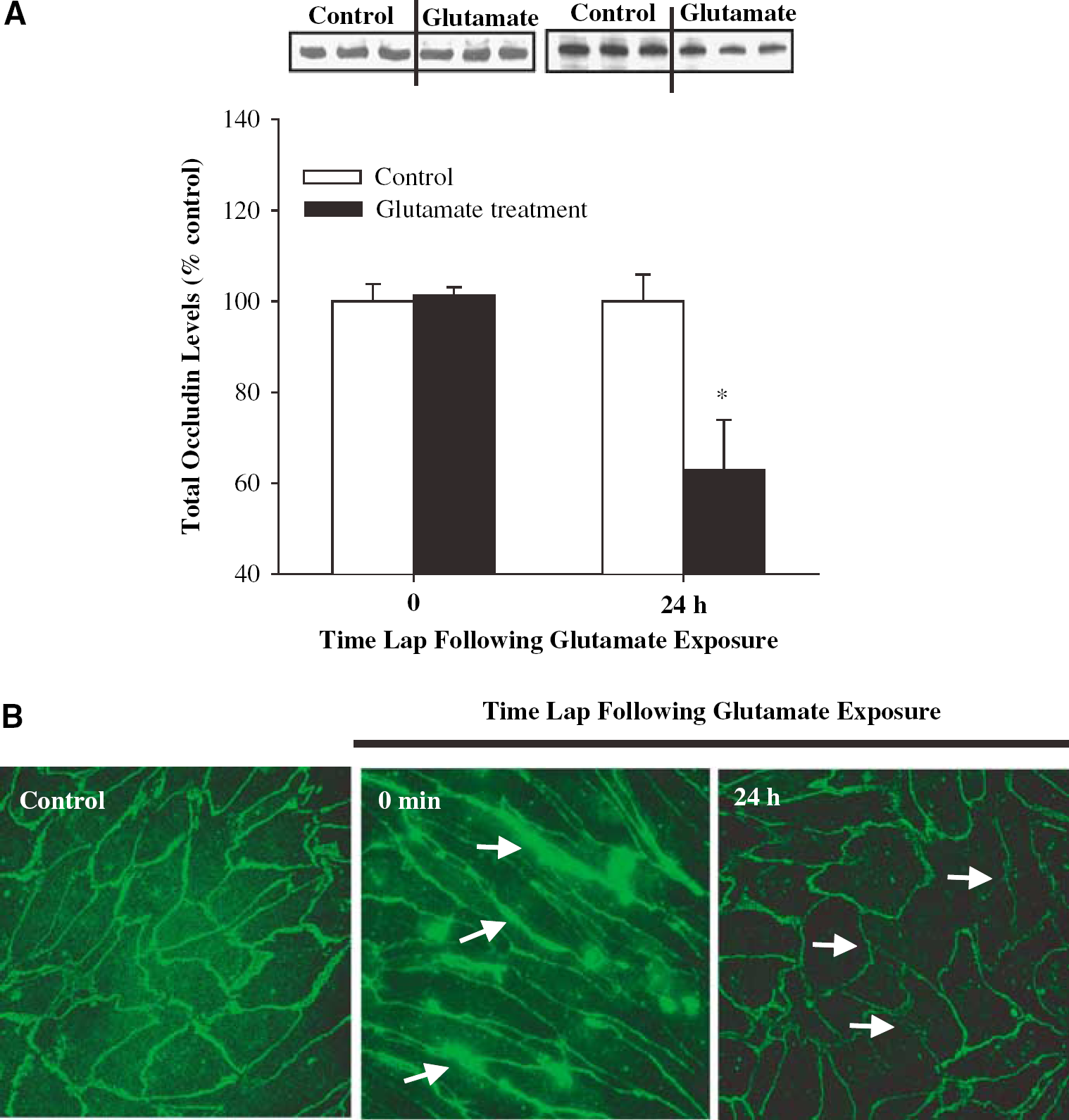
Exposure to glutamate alters occludin expression in BMEC. Confluent cultures were exposed to 1 mmol/L glutamate for 30 mins and occludin total protein levels or immunoreactivity were determined immediately or 24 h after this exposure. (
Occludin distribution after glutamate treatment was also estimated by immunofluorescence microscopy (Figure 3B). A transient exposure to glutamate resulted in immediate redistribution of occludin immunoreactivity. Indeed, immediately after a 30 mins treatment, occludin-positive staining became stronger at cell–cell border segments as indicated by arrows in Figure 3B (middle panel). Moreover, 24 h after glutamate treatment, occludin linear immunoreactivity was markedly weakened and interrupted at cell–cell border segments (Figure 3B, right panel, arrows).
Inhibition of the N-Methyl-d-Aspartate and Alpha-Amino-3-Hydroxy-5-Methylisoxazole-4-Propionate/Kainate Receptors Attenuates Glutamate-Induced Alterations of Occludin Immunoreactivity
To investigate a possible involvement of the NMDA receptors in the glutamate-induced changes in occludin expression, confluent BMEC cultures were pretreated with MK-801 (10 μmol/L, the NMDA receptor antagonist) for 15 mins followed by a 30 mins coexposure to 1 mmol/L glutamate. Western blot and immunofluorescence staining was performed 24 h after this exposure. As illustrated in Figure 4A, MK-801 did not significantly affect glutamate-induced downregulation of total occludin levels. However, pretreatment with MK-801 partially restored glutamate-induced disturbances in occludin immunoreactivity. Figure 4B shows that occludin staining at the cell–cell borders appears to be stronger and more uniform in cultures pretreated with MK-801 as compared with cells treated with glutamate alone.
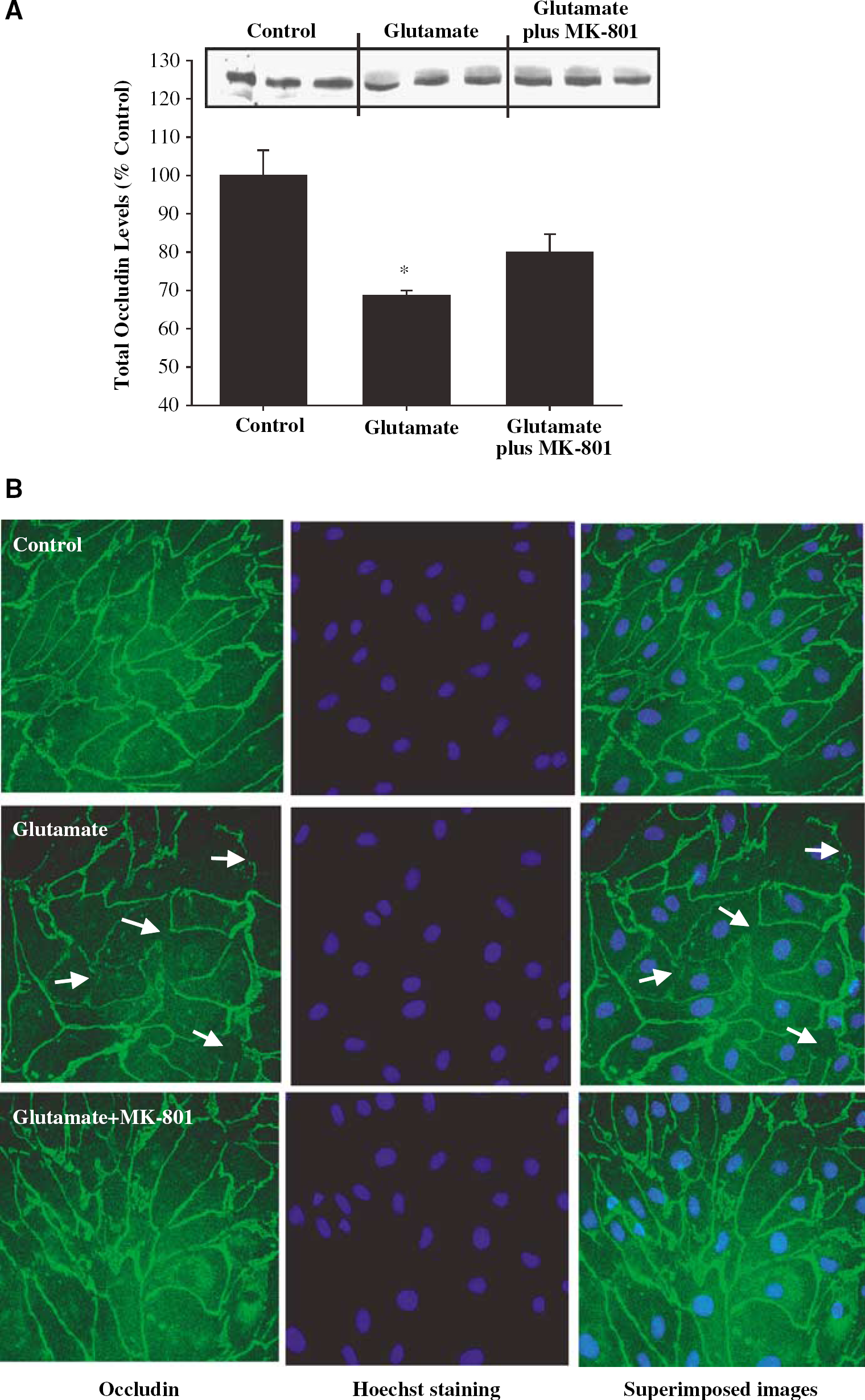
Inhibition of the NMDA receptors attenuates glutamate-induced alterations of occludin immunoreactivity in BMEC. Confluent cultures were treated with 1 mmol/L glutamate for 30 mins and occludin total protein levels (Western blot) or immunoreactivity were determined 24h after this exposure. In addition, selected cultures were pretreated with 10 μmol/L MK-801 for 15 mins, followed by a 30 mins exposure to 1 mmol/L glutamate. (
Glutamate-induced alterations of occludin immunoreactivity may also be mediated by the AMPA/KA receptors. To evaluate this possibility, BMEC were pretreated with DNQX (5 μmol/L, the inhibitor of the AMPA/KA receptors) for 15 mins followed by a 30 mins exposure to 1 mmol/L glutamate. The effects exerted by DNQX were similar to those mediated by MK-801. 6,7-Dinitroquinoxaline-2.3-dione had no statistically significant effect on glutamate-induced downregulation of total occludin levels (Figure 5A). However, pretreatment with DNQX partially restored glutamate-induced alteration of occludin immunoreactivity at the cell–cell borders (Figure 5B, arrows).
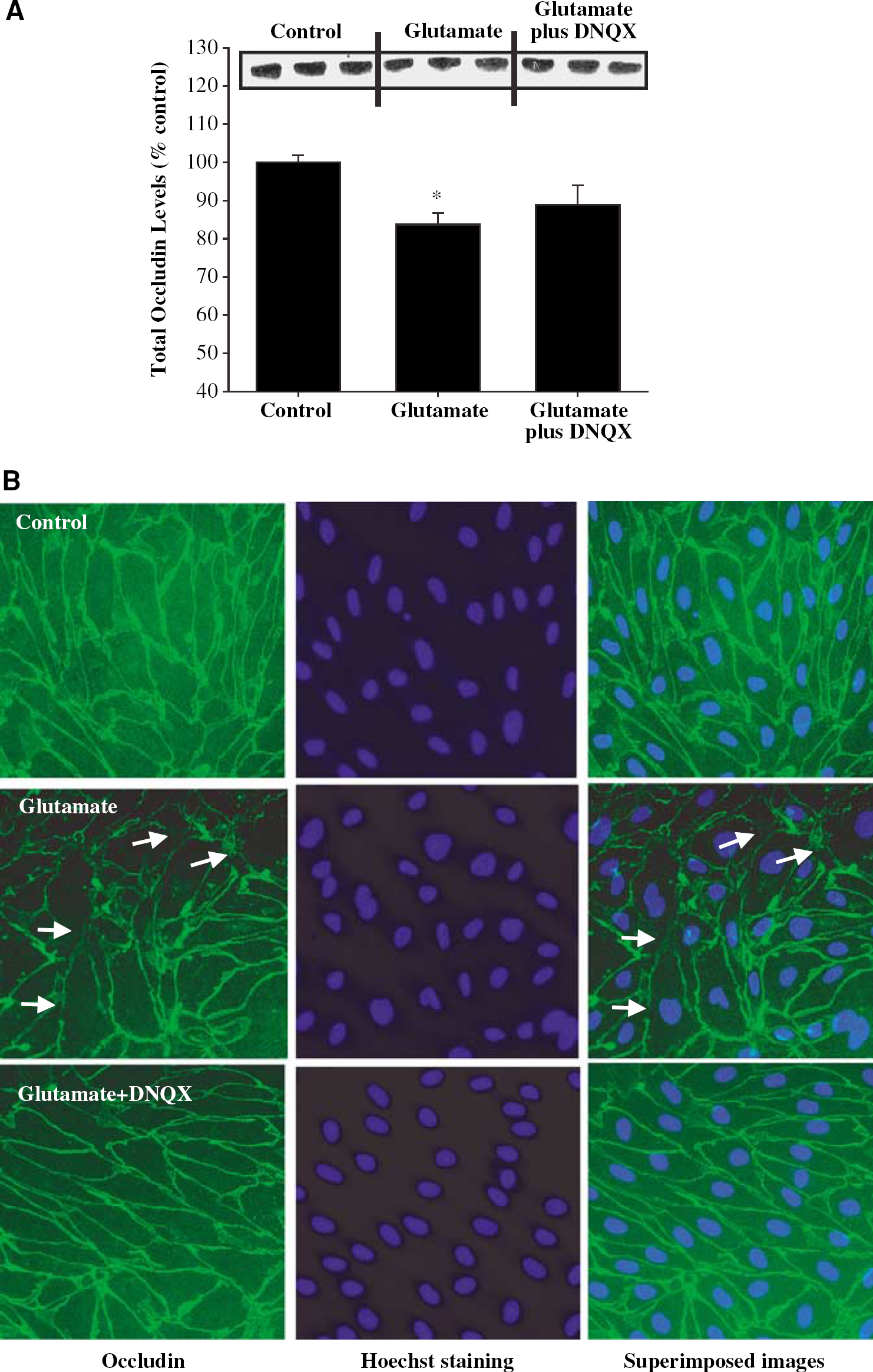
Inhibition of the AMPA/KA receptors attenuates glutamate-induced alterations of occludin immunoreactivity in BMEC. Confluent cultures were treated with 1 mmol/L glutamate for 30 mins and occludin total protein levels (Western blot) or immunoreactivity were determined 24h after this exposure. Selected cultures were pretreated with 5 μmol/L DNQX for 15 mins, followed by a 30 mins coexposure to 1 mmol/L glutamate. (
The N-Methyl-d-Aspartate Receptors are Involved in Glutamate-Mediated Alterations of Occludin Tyrosine Phosphorylation
The evident effects of MK-801 and DNQX on occludin immunoreactivity (Figures 4B and 5B) indicate that the NMDA and AMPA/KA receptors can influence the rearrangement and redistribution of occludin. Such alterations of immunoreactivity patterns can result from changes in occludin phosphorylation. Therefore, we examined the effects of a transient glutamate exposure on occludin phosphorylation on tyrosine and threonine residues by immunoprecipitation of occludin, followed by Western blots for phosphotyrosine or phosphothreonine.
As illustrated in Figure 6A, exposure of BMEC to glutamate resulted in early and dramatic increase in occludin tyrosine phosphorylation. A significant increase in occludin tyrosine phosphorylation was observed after only 30 mins of glutamate exposure. These effects were further intensified 24 h after a transient exposure to glutamate.
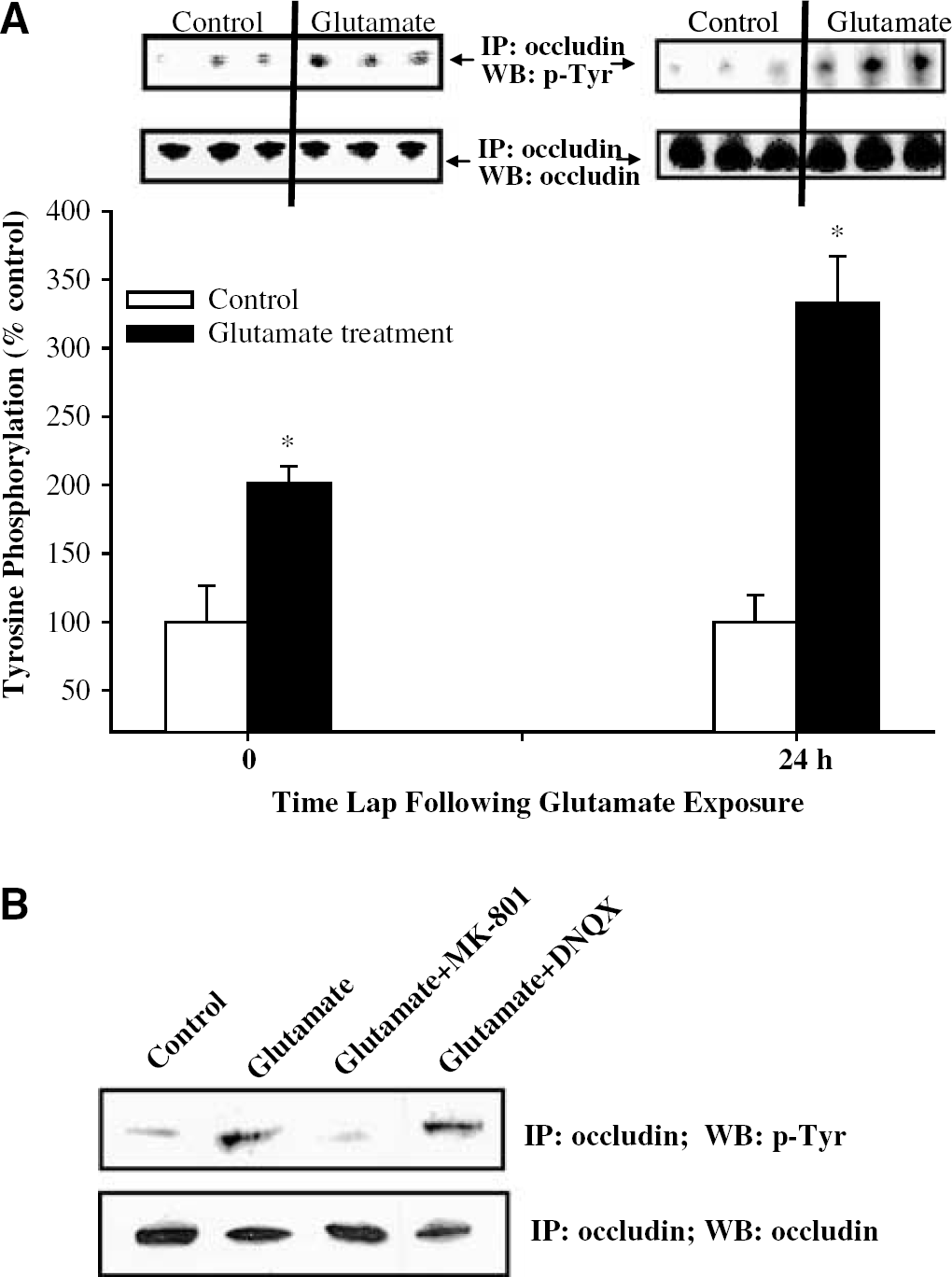
The NMDA receptors are involved in glutamate-induced tyrosine phosphorylation of occludin. Immunoprecipitation (IP) was performed using antioccludin antibody, followed by Western blot (WB) with antiphosphotyrosine antibody or antioccludin antibody (control). The blots are representative data from three different experiments and the bar graph represents quantified results from these experiments. *Statistically significant as compared with control. (
To investigate the possible involvement of the ionotropic glutamate receptors in these effects, confluent cultures were pretreated with MK-801 or DNQX for 15 mins before a 30 mins treatment with 1 mmol/L glutamate. Immunoprecipitation of occludin followed by Western blot for phosphotyrosine was performed 24 h after such a transient exposure. MK-801 markedly prevented glutamate-induced elevation of tyrosine phosphorylation (Figure 6B), indicating the role of the NMDA receptors in these effects. In contrast, inhibition of the AMPA/KA receptors by DNQX did not affect glutamate-induced effects on occludin tyrosine phosphorylation.
The Alpha-Amino-3-Hydroxy-5-Methylisoxazole-4-Propionate/Kainate Receptors are Involved in Glutamate-Induced Alterations of Occludin Threonine Phosphorylation
Experiments similar to those described for tyrosine phosphorylation were performed to determine possible effects of glutamate on threonine phosphorylation of occludin. Immediately after transient glutamate exposure, occludin phosphorylation on threonine residues was decreased by 30%; however, these changes did not reach statistical significance. In contrast, 24 h after glutamate exposure, a highly significant decrease (by 60%) in threonine occludin phosphorylation was determined (Figure 7A).
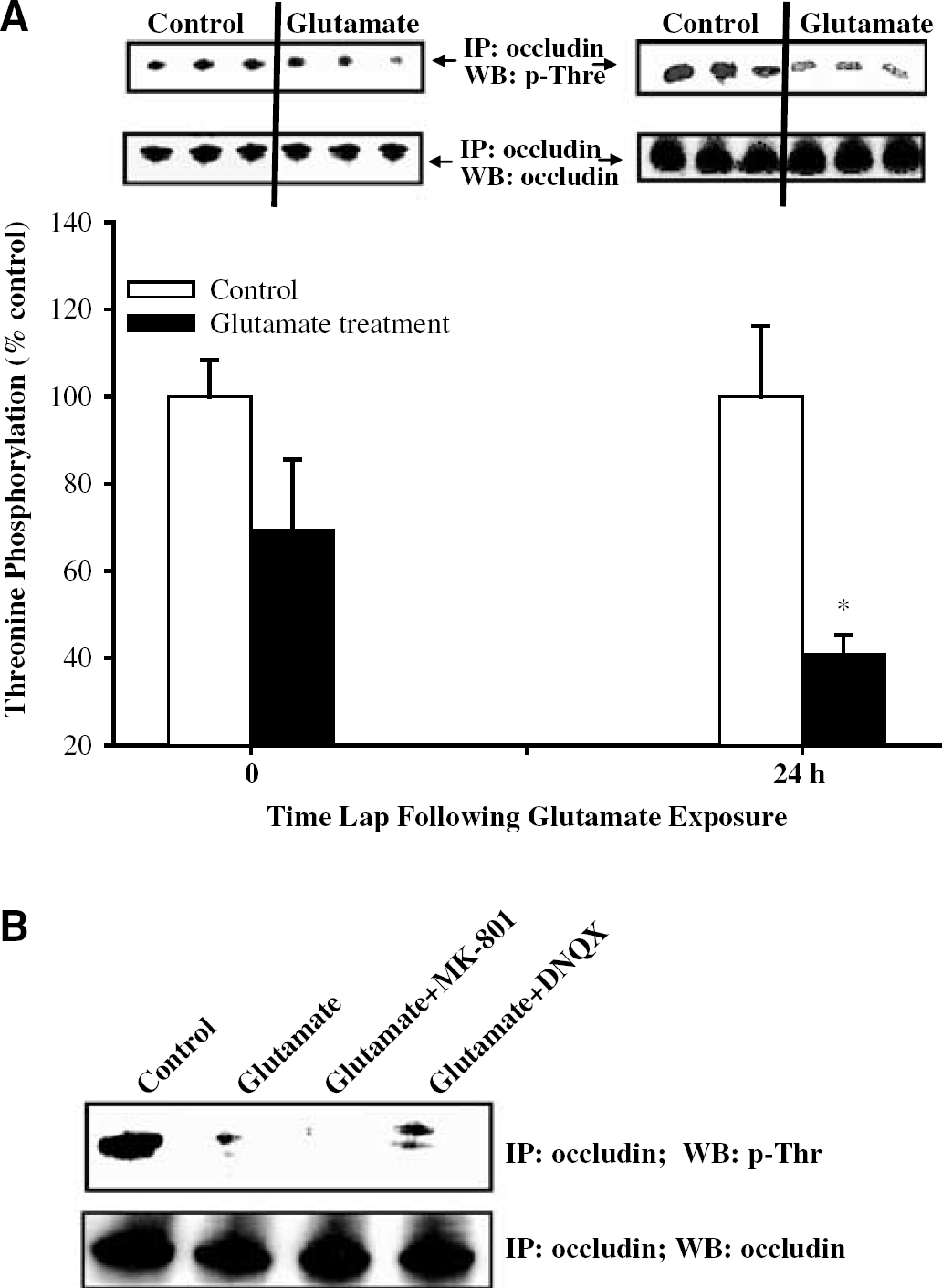
The AMPA/KA receptors are involved in glutamate-induced dephosphorylation of threonine residues of occludin. Immunoprecipitation (IP) was performed using antioccludin antibody, followed by Western blot (WB) with antiphosphothreonine antibody or antioccludin antibody (control). The blots are representative data from three different experiments and the bar graph represents quantified results from these experiments. *Statistically significant as compared with control. (
As indicated in Figure 7B, DNQX-attenuated glutamate induced decrease in occludin threonine phosphorylation. In contrast, MK-801 appeared to even further potentiate glutamate-induced occludin dephosphorylation of threonine residues.
The N-Methyl-d-Aspartate but not Alpha-Amino-3-Hydroxy-5-Methylisoxazole-4-Propionate/Kainate Receptors Participate in Glutamate-Induced Disruption of the Barrier Function of Brain Microvascular Endothelial Cells
Tight junction proteins, such as occludin, regulate the integrity of the brain endothelium. Therefore, we determined the effects of glutamate and the NMDA and AMPA/KA receptor inhibitors on the barrier function in cocultures of BMEC and astrocytes. The cocultures constitute a better in vitro model than BMEC monocultures to perform functional assays, such as permeability measurements. As illustrated in Figure 8A, exposure to glutamate markedly increased endothelial permeability as determined by SF flux. Most interestingly, preexposure to MK-801 but not to DNQX attenuated this effect. Sodium fluorescein is a paracellular marker and its transendothelial permeability reflects the integrity of tight junctions.
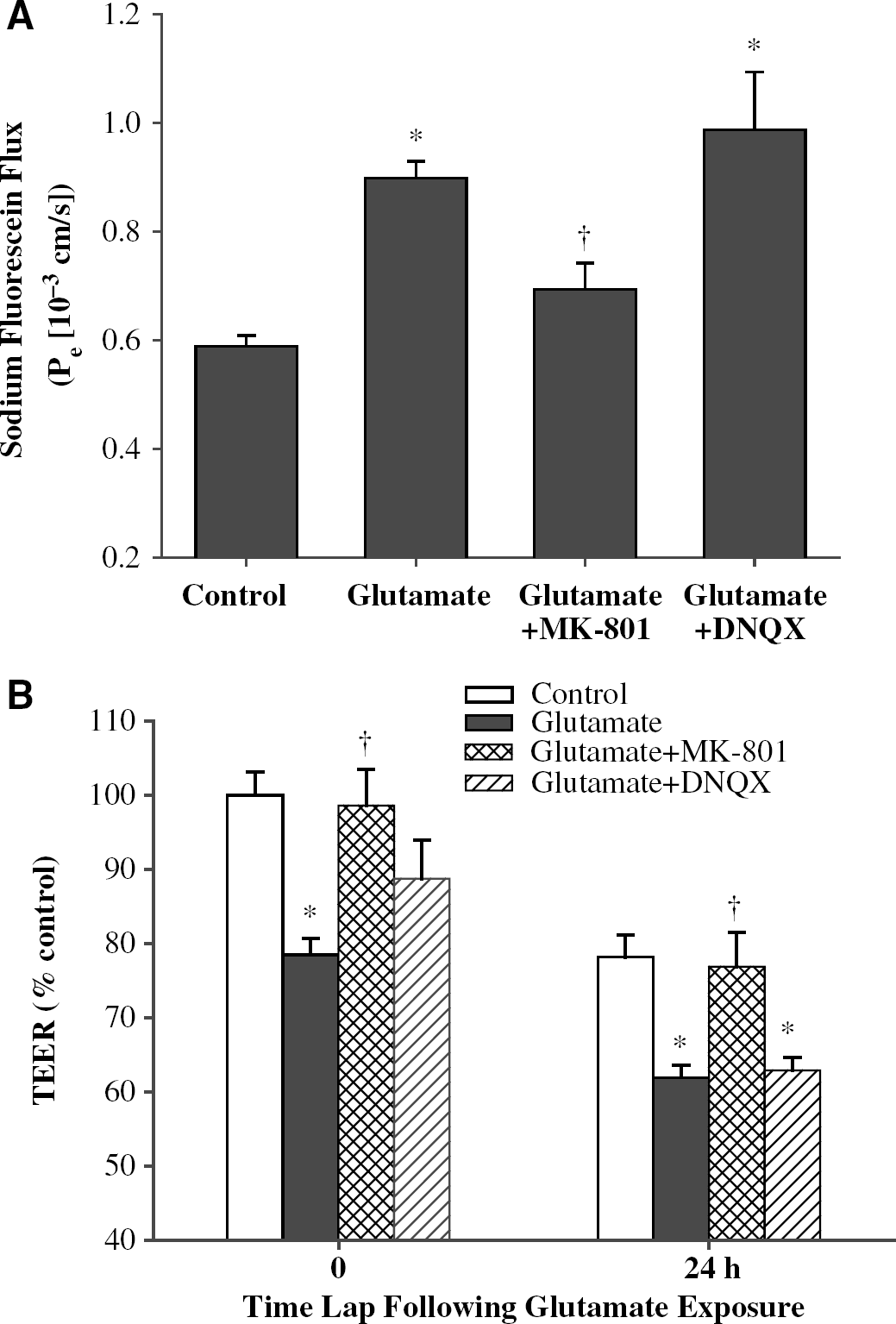
The NMDA but not AMPA/KA receptors are involved in glutamate-induced disruption of the barrier function of BMEC. (
Transendothelial electrical resistance is another marker of endothelial integrity. The baseline TEER values of BMEC monolayers in cocultures with astrocytes varied between 182 and 268Ω cm2 and decreased as a result of a 24 h maintenance in serum-free environment. However, independent of culture conditions, treatment with glutamate significantly reduced TEER values, indicating disruption of BMEC integrity (Figure 8B). Most importantly, inhibition of the NMDA receptors by MK-801 fully protected against these effects. In contrast, pretreatment with DNQX did not affect glutamate-induced alterations of TEER.
Discussion
Ischemic stroke is the second leading cause of death worldwide and the main disabilitating disease in the United States (Rymer and Thrutchley, 2005). One of the major complications of stroke is cytotoxic and/or vasogenic brain edema. Cytotoxic edema is caused by the loss of membrane ionic pumps and by cell swelling owing to cerebral ischemia. In addition, BBB leakage appears to be responsible for vasogenic edema. In this study, we hypothesize that alterations of occludin expression induced by high levels of extracellular glutamate may contribute to the disruption of the BBB. To support this hypothesis, changes in tight junction proteins in an in vitro BBB model were shown in hypoxia/reoxygenation modeling of transient ischemia. Specifically, hypoxia resulted in redistribution of occludin, claudin-1, ZO-1, and ZO-2. In addition, reoxygenation increased the levels of these proteins (Brown et al, 2003; Mark and Davis, 2002).
The results of this study indicate that a transient exposure to glutamate can result in progressive changes in occludin immunoreactivity. The first noted pathology observed immediately after 30 mins of glutamate exposure was occludin redistribution and an increase in occludin immunoreactivity at distinct cell–cell border segments (Figure 3B). Such alterations can markedly affect the BBB function. For example, redistribution of tight junction particles in the internal/external membrane leaflets can lead to a disturbed fence function, with the polarity change of the glucose transporter GLUT-1 (Lippoldt et al, 2000). In the present study, a 30 mins exposure to glutamate resulted in a significant decrease in TEER values (Figure 8B).
We observed that alterations of occludin expression progressed further even after the removal of glutamate from cell culture media after a 30 mins exposure. Indeed, occludin immunoreactivity at several cell–cell border segments was weaker or absent 24 h after the transient glutamate treatment (Figure 3B). The time course of these alterations is supported by the observation that cytotoxic and vasogenic edema can reach maximum intensity 24 to 72 h after the ischemic event (Rosenberg, 1999). Similar delayed BBB opening was described in an animal model of global cerebral ischemia in which a 20 mins of two-vessel occlusion resulted in a marked BBB permeability increase 24 h later (Preston and Webster, 2004). In addition, KA (a glutamate analog)-induced BBB breakdown with plasma leakage into brain tissue only 24 h after the intrahippocampal injection (Chen et al, 1999).
Next, we studied the involvement of the ionotropic glutamate receptors in the glutamate-induced changes in occludin expression and phosphorylation. Stimulation of glutamate receptors can induce a variety of downstream kinases, including mitogen-activated protein kinase (Sgambato et al, 1998), Src family kinases (Kalia et al, 2004), and calciumcalmodulin-dependent protein kinase II (CaMK II) (Krizbai et al, 1998). Activation of these kinases, alone or in concert, can result in alterations of tight junction phosphorylation in response to glutamate treatment. To distinguish the effects of individual ionotropic glutamate receptors on occludin expression, BMEC were exposed to glutamate in the presence of pharmacological inhibitors of these receptors, such as MK-801 (an inhibitor of the NMDA receptors) or DNQX (an inhibitor of the AMPA/KA receptors). Treatment with MK-801 or DNQX did not prevent a decrease in occludin levels induced by a transient exposure to glutamate. However, these inhibitors effectively rescued the pattern of occludin-positive staining at the cell–cell borders. Specifically, the junctional occludin immunoreactivity became stronger, continuous, and more uniform in cells treated with glutamate plus MK-801 or glutamate plus DNQX as compared with cultures exposed to glutamate alone (Figures 4B and 5B).
Occludin can be phosphorylated on tyrosine and serine/threonine residues (Sakakibara et al, 1997; Tsukamoto and Nigam, 1999; Wong, 1997). In this study, glutamate-induced alterations of occludin immunoreactivity were associated with significant disturbances in phosphorylation patterns. As compared with controls, glutamate treatment markedly increased phosphorylation of tyrosine residues; however, it decreased phosphorylation of threonine residues (Figures 6 and 7). Results of this study also indicated that phosphorylation of specific occludin amino acids can be regulated by the NMDA and AMPA/KA receptors. For example, a blockage of the NMDA receptors, but not the AMPA/KA receptors, effectively inhibited glutamate-induced tyrosine phosphorylation of occludin (Figure 6B). In contrast, DNQX protected against a decrease in threonine phosphorylation (Figure 7B). Thus, glutamate-induced phosphorylation and dephosphorylation of occludin on tyrosine and threonine residues follow distinct signaling pathways involving different types of ionotropic receptors.
Alterations of phosphorylation status of junctional proteins can result in disruption of tight junction assembly and decreased barrier function of the BBB. For example, highly phosphorylated occludin has been shown to be selectively localized at the tight junctions, whereas the less phosphorylated occludin was found in the cytoplasm of epithelial cells (Andreeva et al, 2001; Sakakibara et al, 1997; Tsukamoto and Nigam, 1999). A correlation was also shown between tyrosine phosphorylation of occludin and the disruption of tight junctions (Basuroy et al, 2006). Indeed, tyrosine kinase inhibitors prevented disruption of tight junctions in epithelial cells (Atkinson and Rao, 2001; Rao et al, 1997). Consistent with these reports, we show in this study that inhibition of the NMDA receptors, which participate in tyrosine hyperphosphorylation of occludin, selectively protect against glutamate-induced disruption of BMEC integrity (Figure 8). These results are supported by earlier observations on the involvement of the NMDA receptors in the regulation of the BBB functions and endothelial permeability (Sharp et al, 2003). However, it should be noted that disruption of tight junctions was also linked to occludin dephosphorylation at the serine/threonine residues (Andreeva et al, 2001; Clarke et al, 2000; Hirase et al, 2001).
In summary, a transient exposure to extracellular glutamate resulted in initial redistribution of occludin, followed by a marked decrease in expression of this tight junction protein. Treatment with glutamate also induced alterations of occludin phosphorylation, such as excessive phosphorylation of the tyrosine residues and a decrease in phosphorylation of the threonine residues. Glutamate-induced hyperphosphorylation of tyrosine and an increase in BMEC permeability appeared to be regulated by the NMDA receptors. In contrast, the AMPA/KA receptors were involved in glutamate-mediated hypophosphorylation of threonine residues of occludin. Overall, these findings contribute to the better understanding of the mechanisms of glutamate-induced BBB alterations in relationship to pathophysiology of cerebral ischemia and stroke.