
Abstract
Ischemia not only activates cell death pathways but also triggers endogenous protective mechanisms. However, it is largely unknown what is the essence of the endogenous neuroprotective mechanisms induced by preconditioning. In this study we demonstrated that systemic injection of JZL195, a selective inhibitor of eCB clearance enzymes, induces in vivo long-term depression at CA3-CA1 synapses and at PrL-NAc synapses produces neuroprotection. JZL195-elicited long-term depression is blocked by AM281, the antagonist of cannabinoid 1 receptor (CB1R) and is abolished in mice lacking cannabinoid CB1 receptor (CB1R) in astroglial cells, but is conserved in mice lacking CB1R in glutamatergic or GABAergic neurons. Blocking the glutamate NMDA receptor and the synaptic trafficking of glutamate AMPA receptor abolishes both long-term depression and neuroprotection induced by JZL195. Mice lacking CB1R in astroglia show decreased neuronal death following cerebral ischemia. Thus, an acute elevation of extracellular eCB following eCB clearance inhibition results in neuroprotection through long-term depression induction after sequential activation of astroglial CB1R and postsynaptic glutamate receptors.
Introduction
Brain ischemia, including ischemic stroke, is the most common cause of brain injury, 1 and the third leading cause of mortality and the leading primary cause of adult disability worldwide. 2 Ischemic stroke is predominantly caused by a blood clot-induced blockage of a cerebral artery, 3 and thrombolysis is used for treatment within 4.5 h of stroke occurrence. However, this treatment only benefits approximately 5% of patients with acute ischemic stroke, and many new therapeutic strategies have culminated in failure during clinical trials. 4 To develop more efficacious therapeutic approaches, a novel conceptual framework that recognises the ability of the brain to protect itself is required. 5
In addition to inducing cell death, ischemia also triggers protective mechanisms to counteract injury induced by ischemia. Ischemia preconditioning in the form of a mild ischemic insult insufficient to cause lethal damage shows the potential for self-neuroprotection (protection of many neurons against subsequent lethal ischemia).6,7 We observed that electroacupuncture preconditioning reduced middle cerebral artery occlusion (MCAO)-induced injury, and this effect was blocked by cannabinoid CB1 receptor (CB1R) antagonism and CB1R knockdown,8,9 indicating a key role for endocannabinoid (eCB) signaling in protection of neurons. However, it is unknown whether and how eCB actively produced by brain cells in living animals participates in neuroprotection.
There are two well-characterized eCBs, anandamide or N-arachidonoylethanolamine (AEA) and 2-arachidonoylglycerol (2-AG).10–12 AEA and 2-AG are primarily hydrolyzed by fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL), respectively. 13 JZL195 selectively inhibits both FAAH and MAGL to significantly increase brain levels of both AEA and 2-AG. 13 Whereas eCBs can activate presynaptic CB1R,14,15 the most abundant G protein-coupled receptor in the brain, 16 to inhibit release of glutamate and GABA from glutamatergic and GABAergic axon terminals, respectively.17,18 We have previously observed that CB1R is present in astroglial cells. 19 We found that synthetic cannabinoids induced in vivo long-term depression (LTD) at hippocampal CA3-CA1 synapses through sequential recruitment of astroglial CB1R, postsynaptic glutamate N-methyl-D-aspartate receptor (NMDAR) and glutamate α-amino-3-hydroxy-5-methyl-isoxazole propionic acid receptor (AMPAR). 19 Furthermore, we recently observed that both MAGL and FAAH inhibitors, as well as both 2-AG and AEA, were able to induce similar in vivo LTD at hippocampal CA3-CA1 synapses through sequential recruitment of astroglial CB1R and postsynaptic NMDAR and AMPAR. 20 In addition to the physiological relevance, LTD may have important clinical application for treating a variety of neurological conditions, including stroke. Synaptic LTD is likely involved in ischemic neuronal death caused by ischemic stroke,21,22 it is possible that in vivo LTD alone could mimic ischemic preconditioning to induce the neuroprotection. Therefore, we tested the hypothesis that LTD, induced by an acute elevation of interstitial 2-AG/AEA with JZL195, produces neuroprotection. JZL195 sequentially activates astroglial CB1R and postsynaptic glutamate receptors resulting in LTD and neuroprotection.
Materials and methods
Animals
Male C57BL/6 mice and Sprague Dawley rats were purchased from Charles River (Charles River Laboratories). For all experiments, 8–10-week-old age-matched littermate mice and rats weighing 20–25 g and 250–300 g were used, respectively. Mice and rats were maintained in groups of 4 and 2, respectively, in standard cages under standard laboratory conditions (12/12 h light/dark cycle, 22 ± 2℃, food and water ad libitum). Animals were randomly assigned to experimental groups based on the random number generator function in SPSS. All animal procedures were performed in keeping with the guidelines established by the Canadian Council on Animal Care as approved by the Animal Care Committee (ACC) of the University of Ottawa Institute of Mental Health Research (IMHR). Specifically, the IMHR ACC approved the present study (ACC-2012-004). Similar procedures approved by the IMHR ACC for conducting mouse global ischemia and mouse electrophysiology were also approved by the Fourth Military Medical University and Shaanxi Normal University, respectively, and in accordance with the ARRIVE (Animal Research: Reporting In Vivo Experiments) guidelines.
Generation of mutant mice
GFAP-CB1R-KO mice were generated using the Cre/loxP system as described in our previous studies.19,20 The CB1R gene was successfully deleted in mice (8 weeks) following eight daily injections of tamoxifen (Sigma, 1 mg, intraperitoneal injection, i.p.) dissolved in 90% sunflower seed oil and 10% ethanol to a final concentration of 10 mg/mL. CB1R-floxed mice were crossed with Dlx5/6-Cre transgenic mice that have Cre recombinase expression directed by the regulatory sequences of the zebrafish dlx5a/dlx6a genes to obtain the GABA-CB1R-KO mouse line.19,20 We also crossed CB1R-floxed mice with the CaMKIIa-iCre transgenic mice expressing the improved Cre recombinase in adult forebrain projecting neurons. 20 The mutant mice were referred to as the Ca-CB1R-KO mouse line in this study.
Electrophysiological analysis
Electrophysiological studies in anesthetized rats and mice were performed as previously described.19,20 Briefly, under anesthesia, a stimulating electrode was placed onto the CA3 (AP −3.9 mm, ML ±2.6 mm, D/V −2.45 mm in rats, AP −1.9 mm, ML ±1.4 mm, D/V −1.5 mm in mice) and a recording electrode into the CA1 (AP −3.9 mm, ML ±2.6 mm, D/V −2.45 mm in rats, AP −1.9 mm, ML ±1.4 mm, D/V −1.5 mm in mice), or a stimulating electrode was placed onto the prelimbic cortex, PrL (angle, 20°, AP +2.65 mm, ML ±0.5 mm, D/V −1.67 mm in mice) and a recording electrode into the nucleus accumbens shell, NAc (AP +1.94 mm, ML ±0.75 mm, D/V −3.7 mm in mice). Field excitatory postsynaptic potentials (fEPSPs) were induced by applying single pulses of stimulation at 0.067 Hz in CA1 and 0.33 Hz in PrL. Stimulus pulse intensities were typically 20–60 μA with a duration of 500 μs in CA3 and 0.1–1 mA with a duration of 0.3 ms in PrL. A stimulus intensity that yielded approximately 60% of the maximal response was selected for baseline measurements. Once the ideal placement of the electrodes was established, baseline fEPSPs were recorded for 20 min, followed by i.p. injection of JZL195 (Sigma, St. Louis, MO, USA; 20 mg/kg, i.p.) or vehicle before a continuous recording of fEPSPs for 120 min. Other groups of animals received different treatments before fEPSP recording for 120 min: (I) AM281 (Tocris Bioscience, Ellisville, USA; 3 mg/kg, i.p.; dissolved in 5% DMSO: 5% Tween 80: 90% physiological saline) or vehicle 10 min before or after a JZL195 injection (20 mg/kg, i.p.); (II) ceftriaxone (200 mg/kg, i.p., once per day for 5 days) or vehicle, followed 2 h later by a JZL195 (20 mg/kg, i.p.) or vehicle injection; (III) Ro25-6981 (Sigma; 6 mg/kg, i.p.), ifenprodil (Sigma; 5 mg/kg, i.p.) or vehicle 30 min prior to JZL195 injection (20 mg/kg, i.p.); (IV) Tat-GluR2 or Tat-GluR2S (GL Biochem, Shanghai, China) injection (1.5 µmol/kg, i.p.) 2 h before JZL195 injection (20 mg/kg, i.p.).
Glutamate transporter Western blotting
Six groups of four adult male Sprague–Dawley rats (Charles River) each received the following treatment: (1) a single injection of JZL195 (20 mg/kg, i.p.) or vehicle; (2) once daily injection of ceftriaxone (200 mg/kg, i.p.), followed 2 h later by a JZL195 (20 mg/kg, i.p.) or vehicle injection; (3) once daily injection of vehicle of ceftriaxone (0.9% saline, i.p.), followed 2 h later by a JZL195 (20 mg/kg, i.p.) or vehicle injection. Two hours later, rats were killed and tissues containing the bilateral CA1 area of the hippocampus were dissected and then prepared with the standard Western blotting protocol (Molecular Clone, Edition II). The tissues were lysed for the total protein extracts in 300 μl of lysis buffer containing 10 mM Tris, 150 mM NaCl, 1% Triton X-100 and 0.5% NP-40, 1 mM EDTA at pH 7.4. Samples were mixed with a 100:1 (v/v) ratio of protease inhibitor cocktail and phosphatase inhibitor cocktail (Roche, Tucson, AZ, USA). The samples were stored at −80℃ for Western blot analysis. Anti-GLT1 (1:5000, rabbit), anti-GLAST (1:500, rabbit) and anti-EAAC1 (1:400, rabbit) were generous gifts from J.D. Rothstein (John Hopkins University, Baltimore) and have been widely used for Western blotting. Glutamate transporter immunoblots often show monomers (62 kDa), dimers (120 kDa) and sometimes tetramers (250 kDa), as previously described (Rothstein et al., 1996; Danbolt, 2001). All monomers, dimers and tetramers (if present) were used to quantify the transporter expression levels. The anti-β-actin antibody (1:2000, mouse, Sigma-Aldrich) was used as the positive control. A total of 2 µg (GLT1) or 50 µg (GLAST/EAAC1) of lysate was loaded onto 4–15% gradient SDS-PAGE gels after incubating at 37℃ in a waterbath for 30 min. Separated proteins were transferred onto a PVDF membrane (Bio-Rad) for 1 h. The membrane was blocked with 3% BSA in TBST (Tris-buffered saline with 0.1% Tween 20) and then incubated with an appropriate primary antibody overnight at 4℃. On the following day, the membrane was exposed to HRP-conjugated goat anti-rabbit or goat-anti-mouse secondary antibodies (1:5000) diluted in TBST. Bands were visualized on CL-XPosureTM film (Thermo Scientific) by ECL Plus chemiluminescent substrate (Thermo scientific). Different exposure times were used to detect different proteins. The grey density of scanned images was quantified with Image J (version 1.47).
Synaptosomal surface AMPAR measurement
Male Sprague–Dawley rats (Charles River) weighing about 250 g were killed for preparation of hippocampal slices (400 µm in thickness) 4 h after injection of vehicle (i.p.) or JZL195 (20 mg/kg, i.p.). Biotinylation and Western blot experiments were performed as previously described. 19 Briefly, protein fractions were transferred onto nitrocellulose membranes, which were subsequently incubated overnight with primary antibodies to GluR2 (1:500, Millipore, Billerica, MA, USA) at 4℃. Bands were analyzed by densitometry, and surface/total ratios for GluR2 were determined by dividing the surface intensity by the total intensity.
Induction of transient global cerebral ischemia
Male C57BL/6 or mutant mice (weighing 20–25 g) were allowed free access to water and foods under a 12:12 light–dark cycle with room temperature maintained at 22 ± 1℃. To induce transient global cerebral ischemia, surgery was performed under isoflurane anesthesia (3% for induction anesthesia and 1.4% for maintenance anesthesia) for maintenance by 100% O2. After dissection of the neck skin, the bilateral common carotid arteries were exposed and then isolated from the adjacent nerve and tissue. The bilateral common carotid arteries were occluded for 20 min, using microclips to induce transient global cerebral ischemia, which was followed by reperfusion.23,24 To prevent the occurrence of hypothermia during surgery, the rectal temperature was maintained at 37 ± 0.5℃ using a heating pad (CMA 150, CMA Microdialysis, Sweden). Seventy-two hours following the induction of ischemia, experimental animals were sacrificed for further experimental procedures.
Assessment of surviving and dying neurons
Five mice from each group were anesthetized and transcardially perfused with physiologic saline followed by 4% paraformaldehyde. Coronal sections (12 µm thick) were cut on a cryostat (Themo Scientific) to cover the part of the brain between −1.7 mm to −2.5 mm posterior to the bregma. The sections were subsequently mounted on Poly-Prep glass slides (Sigma Aldrich, USA) and stored at −20℃ for further staining. As described in our previous study, 25 NeuN and Fluoro-Jade B (FJB) staining was conducted to reveal surviving or dying neurons in the hippocampal CA1 area. For NeuN staining, slides were immersed in 0.3% H2O2 in methanol for 15 min and then rinsed three times (10 min per rinse) in distilled water. Next, the slides were incubated sequentially with 10% normal goat serum (for 30 min at room temperature), anti-NeuN rabbit polyclonal antibody (1:1000; Abcam, USA) in 1% BSA-PBS overnight at 4℃, and anti-rabbit FITC-tagged secondary antibody (1:1000; Abcam, USA) for 1 h at room temperature. Finally, the slides were mounted with mounting media (Sigma-Aldrich Co., St. Louis, MO, USA) for examination under a fluorescence microscope (BX51; Olympus, Tokyo, Japan), and images were captured with Image-Pro software 5.0.
For FJB staining, slides containing brain sections were immersed sequentially in a solution containing 1% sodium hydroxide in 80% alcohol for 5 min, 70% alcohol for 2 min and distilled water for 2 min. The slides were subsequently transferred to a 0.06% potassium permanganate solution for 10 min on a shaker. Next, the slides were rinsed in distilled water for 2 min. FJB staining solution was prepared from a 0.01% stock solution of Fluoro-Jade B within 10 min of use and a final dye concentration of 0.0004% (EMD Millipore Corporation, Billerica, MA, USA) in 0.1% acetic acid was used. After immersion in the staining solution for 20 min, the slides were rinsed three times with distilled water (1 min each per rinse). After excess water was removed, the slides were covered with mounting media (Sigma-Aldrich Co.).
Cell counts were performed by an experimenter blinded to the treatment sections (12 µm thick) on three sections (distanced 48 µm apart) per hippocampus. Images of the hippocampal CA1 region were viewed and analyzed using the 40× lens of a fluorescence microscope (Olympus BX51) equipped with Digital imaging software (DP2-BSW). All of the NeuN-positive or FJB-stained neurons in the CA1 pyramidal cell layer of each side of the hippocampus were counted. Cell counts from both sides were averaged to provide the mean value. A mean ± SEM was calculated from the data in each group and statistical analysis was performed as described below.
Induction of transient focal cerebral ischemia
Male C57BL/6 mice (weighing 22–25 g) were allowed a free access to water and food under a 12:12 light–dark cycle with room temperature maintained at 22 ± 1℃. Under isoflurane anesthesia (3% for induction and 1.4% for maintenance, with 100% O2), Focal cerebral ischemia was induced by middle cerebral artery occlusion in mice using an intraluminal filament technique as described previously. 26 Briefly, the right middle cerebral artery was occluded by an insertion of a monofilament suture (0.23 ± 0.02 mm × 0.126 mm/3 cm, RWD Inc. Shenzhen, China) with its tip rounded by the heat from a nearby flame through the right common carotid artery to induce transient focal cerebral ischemia. Reperfusion was accomplished by withdrawing the suture after 1 h. To prevent the occurrence of hypothermia during surgery, the rectal temperature was maintained at 37 ± 0.5℃ using a heating pad (Spacelabs Medical Inc, USA). Seventy-two hours after reperfusion, the neurological assessment was performed and then animals were sacrificed for further experimental procedures.
Neurological evaluation and 2,3,5-triphenyltetrazolium chloride staining
Seventy-two hours after reperfusion, the neurological assessment was performed by a blind investigator using the 18-point scoring system reported by Gracia et al. 27 The system consisted of the spontaneous activity, side stroking, vibrissa touch, limb symmetry, climbing and forelimb walking. After neurological evaluation, mice were decapitated and the brains were rapidly removed and mildly frozen to keep the morphology intact during slicing. Infarct volume was measured as described previously.28,29 In brief, the brain was rapidly dissected and sectioned into six coronal blocks in brain matrix with an approximate thickness of 1 mm and stained with 2% (w/v) 2,3,5-triphenyltetrazolium chloride (TTC) (Sigma, USA) for 30 min at 37℃ followed by overnight immersion in 4% (w/v) paraformaldehyde. The infarct tissue area remained unstained (white), whereas normal tissue was stained red. The infarct areas on each slice were demarcated and analyzed by Adobe Photoshop CC 2014(Abode, San Jose, CA, USA). The infarct volumes were calculated via the method according to the following formula: ((total contralateral hemispheric volume) – (total ipsilateral hemispheric stained volume))/(total contralateral hemispheric volume) × 100%.
Statistics
All results were analyzed by investigators blinded to the treatment. Results except neurological behavioral scores were reported as means ± SE. Statistical analysis of the data was performed using the Student’s t-test or one-way ANOVA, followed by Bonferronni or LSD post hoc test. The neurological behavioral scores were expressed as median (range) and analyzed with Kruskal–Wallis test followed by Mann–Whitney U test. Statistical significance was set at p < 0.05. Statistical analyses were performed using GraphPad Prism 6.0 software.
Results
JZL195 induces in vivo LTD at CA3-CA1 synapses
To explore if brain eCB is able to induce LTD at CA3-CA1 synapses, we injected JZL195 (20 mg/kg, i.p.) that produces a near complete loss of mouse brain AEA and 2-AG hydrolysis activity and an over 10-fold increase of brain AEA and 2-AG for over 10 h.
13
Following in vivo recordings of fEPSP at hippocampal CA3-CA1 synapses in anesthetized mice,
19
we observed that injection with the dual FAAH/MAGL inhibitor, JZL195, resulted in a rapid decrease in the fEPSP slope to approximately 60% of the baseline levels at 2 h after injection (Figure 1(a) and (e)). To examine whether the JZL195-induced decrease in the fEPSP slope was CB1R dependent, we injected the same dose of JZL195 to anesthetized rats treated with the selective CB1R antagonist AM281 (3 mg/kg, i.p.).19,30 The JZL195-dependent reduction in the fEPSP slope was completely blocked by AM281 10 min before, but not 10 min after JZL195 injection (Figure 1(b) and (e)), thus suggesting an in vivo synaptic depression following acute eCB accumulation. This idea is further supported by our recent findings that intra-CA1 iontophoretic application of AEA and 2-AG in anesthetized rats induced similar synaptic depression characterized as in vivo LDT at CA3-CA1 synapses.
20
Therefore, JZL195-induced LTD is referred to as eCB-LTD hereafter.
JZL195 induces in vivo LTD at CA3-CA1 synapses through astroglial CB1R. (a)–(d) Plots of normalized fEPSP slopes show that i.p. injection of JZL195 at 0 min elicits CA1 LTD lasting for >2 h in naive mice (a) and rats (b). The resultant CA1 LTD was blocked following AM281 administration 10 min before, but not 10 min after, JZL195 injection (b) in wild-type (WT), Ca-CB1R-KO and GABA-CB1R-KO mice (c) and (d), but not in GFAP-CB1R-KO mice (c). Representative fEPSP traces before (1) and after (2) vehicle or JZL195 injection are shown above each plot. (e) Bar charts summarizing the average percent change of fEPSP slope before (1) and after (2) vehicle or JZL195 injection as depicted in panels (a) to (d). All summary graphs show means ± SEMs; n = numbers of animals recorded in each group (a) to (d) *p < 0.01 vs. vehicle control, Bonferronni post hoc test after one-way ANOVA ((b): F3,12 = 60.671, p < 0.01; (d): F2,7 = 60.030, p < 0.01) or t test (a) and (c).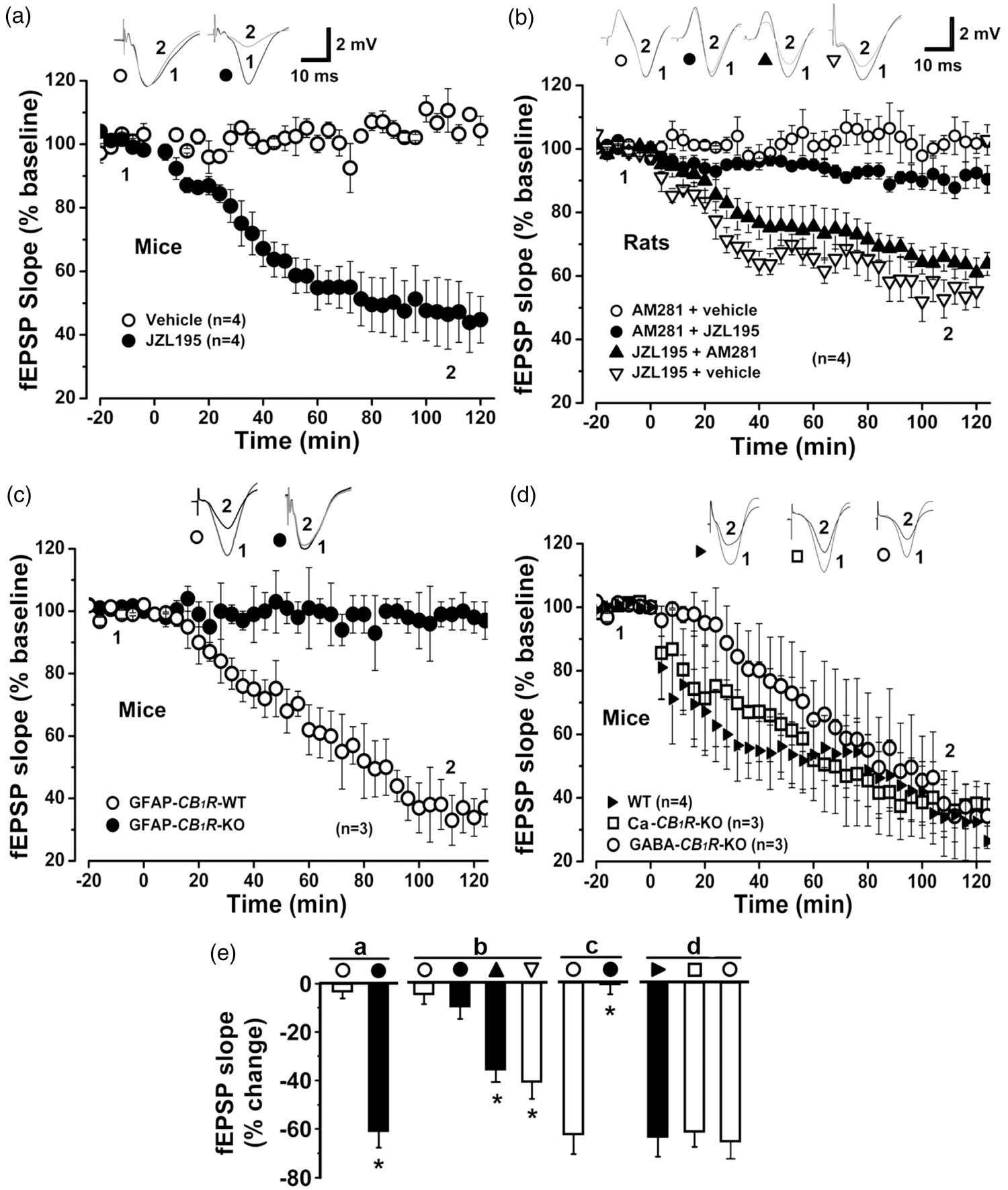
Astroglial CB1R, but not neuronal CB1R, mediates CA1 eCB-LTD
We recently demonstrated that the cannabinoids, 2-AG and AEA, activated astroglial CB1R to induce in vivo LTD at CA3-CA1 synapses.19,20 Therefore, CA1 eCB-LTD may be induced by the activity of acutely accumulated interstitial eCB on astroglial CB1R. To examine this hypothesis, we employed conditional mutant mice with a tamoxifen-inducible deletion of the CB1R gene from astroglial cells. 19 JZL195 (20 mg/kg, i.p.) consistently induced eCB-LTD at CA3-CA1 synapses from tamoxifen-treated control GFAP-CB1R-WT mice but not from tamoxifen-treated GFAP-CB1R-KO littermates (Figure 1(c) and (e)).
To exclude the possibility that JZL195 induces in vivo CA1 eCB-LTD via activation of CB1R in GABAergic or glutamatergic neurons, we examined the effects of systemic JZL195 on CA1 LTD induction in mutant mice lacking the CB1R gene selectively from forebrain GABAergic neurons (GABA-CB1R-KO mouse line) or hippocampal glutamatergic neurons (Ca-CB1R-KO mouse line), as described in detail in our recent study. 20 JZL195-elicited CA1 eCB-LTD was similar between wild-type mice and GABA-CB1R-KO or Ca-CB1R-KO littermates (Figure 1(d) and (e)). Thus, acutely accumulated interstitial eCB in vivo elicit eCB-LTD at CA3-CA1 synapses via the expression of CB1R in astroglial cells but not the expression of CB1R in GABAergic or glutamatergic neurons (thus referred to as astroglial eCB-LTD hereafter).
Glutamate transporter is required for astroglial eCB-LTD production
We recently showed that cannabinoid-, 2-AG- and AEA-induced CA1 LTD was produced via sequential activation of astroglial CB1R and postsynaptic glutamate receptors.19,20 If astroglial eCB-LTD is induced through the same mechanism, interstitial glutamate levels should be increased following eCB activation of astroglial CB1R. Because glutamate transporters are responsible for the rapid removal of glutamate from the interstitial space, we explored if glutamate transporters are required for astroglial eCB-LTD induction.
31
JZL195 (20 mg/kg, i.p.) significantly reduced glutamate transporter 1 (GLT1) (Figure 2(a)), but not glutamate/aspartate transporter (GLAST) (Figures 2(c)) or excitatory amino acid carrier 1 (EAAC1) protein expression (Figures 2(d)). Treatment with ceftriaxone (200 mg/kg, i.p., once per day for five days), a β-lactam antibiotic, known to upregulate GLT1 levels,
32
significantly increased GLT1 (Figure 2(b)), but not GLAST (Figures 2(e)) or EAAC1 (Figures 2(f)) protein expression. Ceftriaxone pretreatment not only antagonized JZL195-mediated effects on GLT1 expression (Figure 2(b)), but also prevented JZL195-induced eCB-LTD (Figure 3(a) and (e)). GLT1, expressed mainly in mature astrocytes, plays a principal role in removing excessive amounts of glutamate from the extracellular space.32,33 Thus, our findings indicate that eCB activation of astroglial CB1R following JZL195 injection suppresses GLT1 expression and then increases interstitial glutamate levels prior to LTD induction.
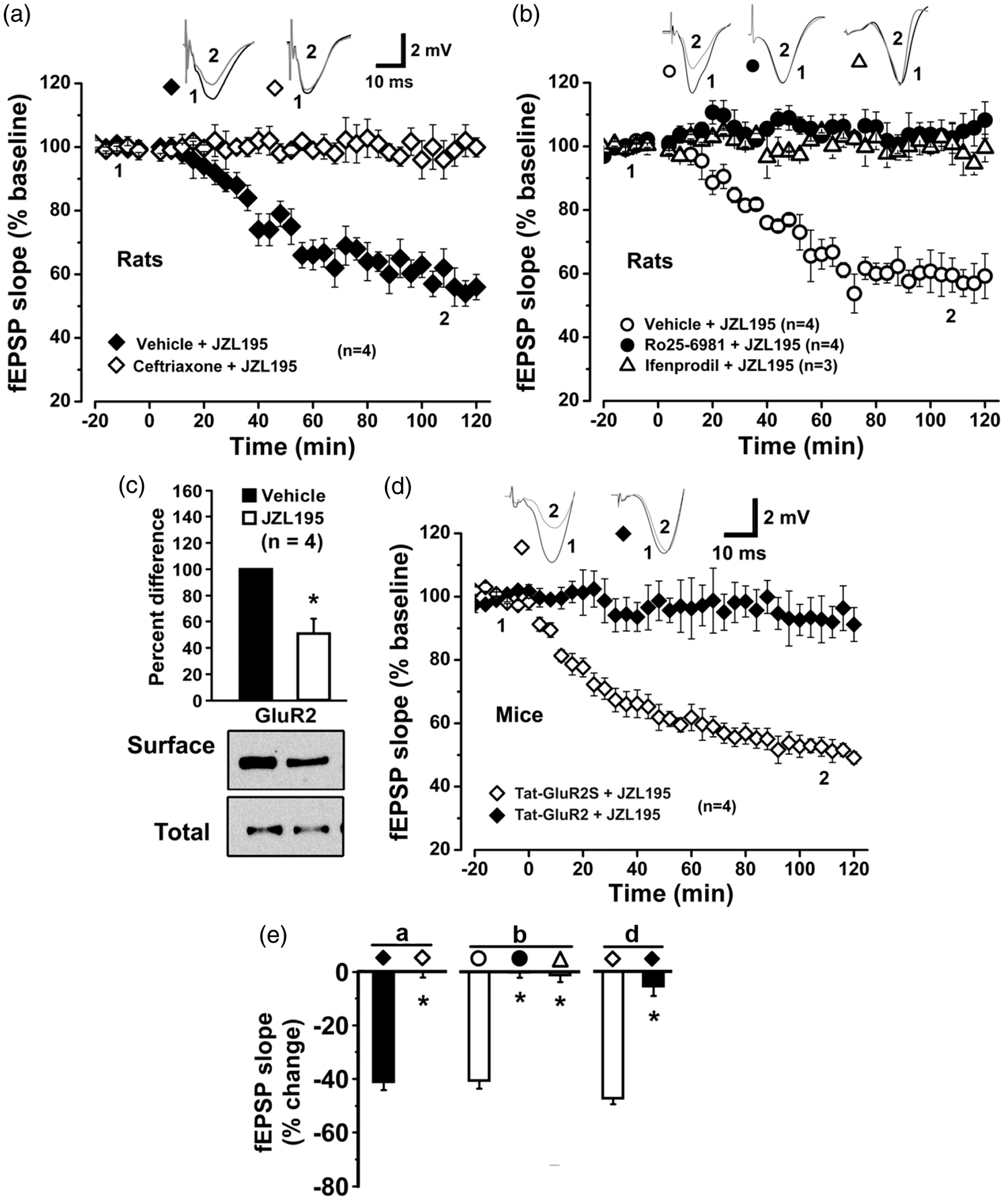
Ceftriaxone antagonizes JZL195-induced reduction in GLT1 tetramer expression. (a) Left photos show expression levels of GLT1 tetramer, dimer and monomer in the hippocampal CA1 extracts of four rats receiving vehicle or JZL195 injection (20 mg/kg, i.p.). Graphs show that JZL195 significantly reduced expression levels of tetramer, but not dimer or monomer. (b) Left photos show expression levels of GLT1 tetramer, dimer and monomer in the hippocampal CA1 extracts of one rat receiving vehicle for ceftriaxone (V1), ceftriaxone (c), ceftriaxone + vehicle for JZL195 (C + V2) or ceftriaxone + JZL195 (C + J). Graphs show that ceftriaxone increases expression levels of tetramer, but not dimer or monomer; these expression level changes were antagonized by JZL195 injection. Neither JZL195 nor ceftriaxone induces significant changes in GLAST and EAAC1 expression. (c–f) Photos show expression levels of GLAST (c, e) and EAAC1 (d, f) in the hippocampal CA1 extracts of four rats receiving vehicle or JZL195 injection (20 mg/kg, i.p.), ceftriaxone (C) or its vehicle (V1) pretreatment prior to JZL195 (J) or its vehicle (V2) injection (20 mg/kg, i.p.). Summary graphs show that JZL195 does not significantly induce expression changes in GLAST (c, e) and EAAC1 (d, f). The summary graphs show means ± SEMs; n = 4 in each group. *p < 0.05 vs. vehicle control, t-test (a) or LSD post-hoc test (b) after one-way ANOVA (tetramer: F3,12 = 3.691, p < 0.05; dimer: F3,12 = 0.070, p = 0.975; monomer: F3,12 = 0.283, p = 0.837); t-test ((c): p = 0.582; (d): p = 0.7342) or Bonferronni post hoc test after one-way ANOVA ((e): F3,12 = 1.284, p = 0.6810; (f): F3,12 = 1.209, p = 0.5700). JZL195 elicits CA1 LTD via glutamate receptors. Plots of normalized fEPSP slopes in anesthetized rats (a, b) or mice (d) show that JZL195 injection at 0 min elicits CA1 LTD in animals, which is blocked by pretreatment with ceftriaxone (a), Ro25-6981, ifenprodil (b) or Tat-GluR2 (d). Representative fEPSP traces before (1) and after (2) treatment are shown above each plot. (c) Graphs and immunoblotting (bottom photos) show a decrease in GluR2 at the synaptic surface of CA1 neurons after JZL195 injection. (e) Bar charts summarizing the average percent change of fEPSP slope before (1) and after (2) vehicle or JZL195 injection as depicted in panels a, b and d. All summary graphs show means ± SEMs; n = numbers of animals assessed in each group (a) to (d). *p < 0.01 vs. vehicle (a) and (b) or Tat-GluR2S (d), Bonferronni post-hoc test after one-way ANOVA ((b): F2,8 = 41.090, p < 0.01) or t test (a), (c) and (d).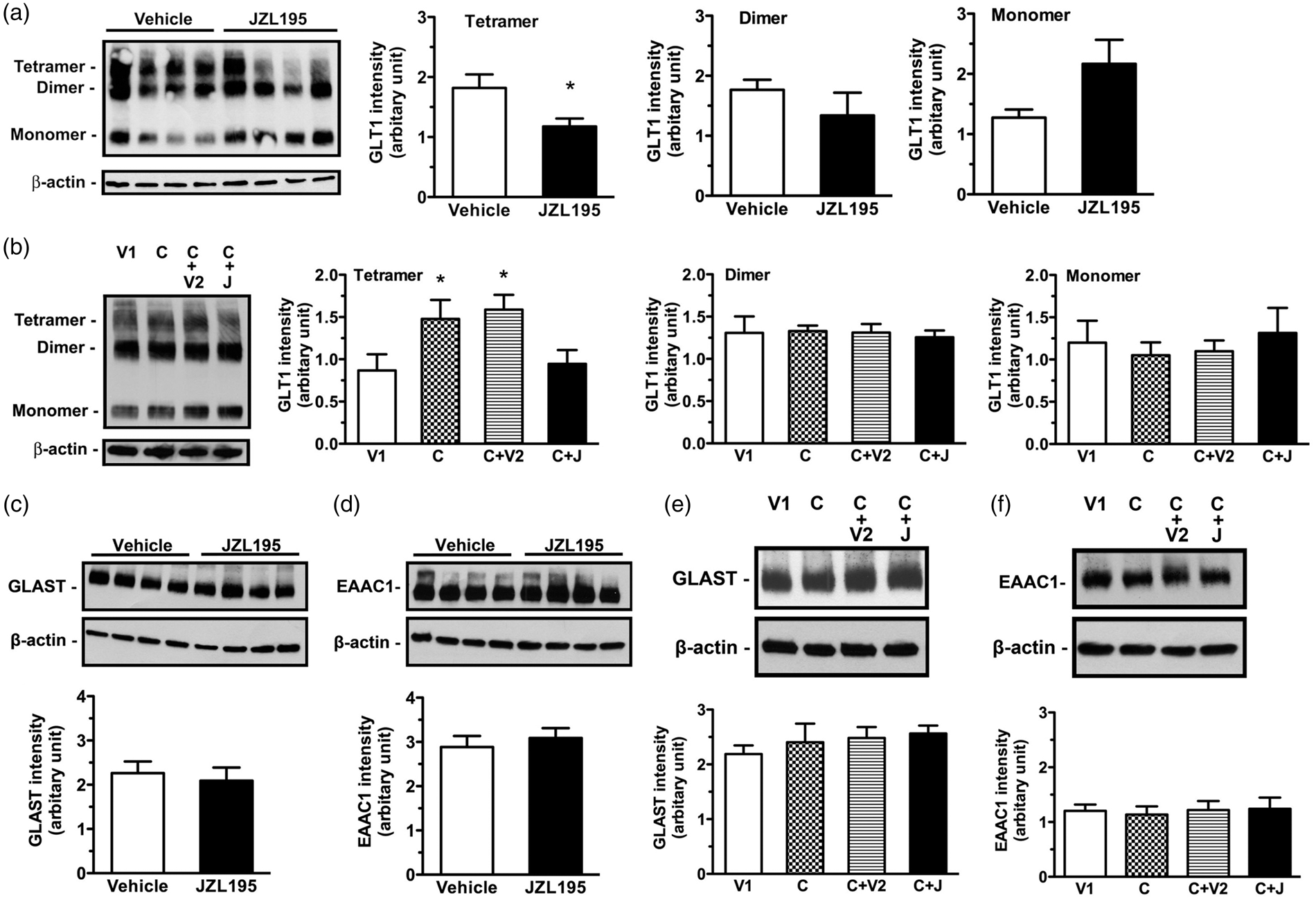
Glutamate receptors are required for astroglial eCB-LTD production
We recently demonstrated that cannabinoid-, 2-AG- and AEA-induced astroglial-LTD at CA3-CA1 synapses requires NR2B activation.19,20 Thus, increased interstitial glutamate levels could activate extracellular NR2B-containing NMDAR (NR2B) to induce postsynaptically expressed LTD. Accordingly, JZL195-induced astroglial eCB-LTD may also require NR2B activation. This idea is further supported by our findings that in contrast to vehicle, the NR2B antagonists Ro25,6981 (6 mg/kg, i.p.) 34 and ifenprodil (5 mg/kg, i.p.),20,35 which have been shown to block cannabinoid-, 2-AG- and AEA-induced LTD, 19 completely blocked in vivo astroglial eCB-LTD induced by systemic JZL195 (Figure 3(b) and (e)).
One of the best characterized functional outputs of NR2B is the regulated endocytosis of postsynaptic AMPAR, a common mechanism responsible for the expression of various forms of LTD.36–38 While CA1 synaptic membranes contain a large amount of the AMPAR subunit GluR1/GluR2 heterodimer, 39 an acute injection of JZL195 significantly reduced surface levels of GluR2 in synaptosomes isolated from the CA1 region (Figure 3(c)), suggesting the occurrence of AMPAR endocytosis in postsynaptic CA1 pyramidal cells following the acute accumulation of eCB in vivo. The facilitated endocytosis of AMPAR can be selectively blocked by the brain-penetrating “Tat-GluR2” peptide derived from GluR2.19,37,40 An acute injection of the Tat-GluR2 peptide (1.5 µmol/kg, i.p.), but not its scrambled analogue (Tat-GluR2S),16,17 blocked JZL195-induced in vivo eCB-LTD at CA3-CA1 synapses (Figure 3(d) and (e)). Altogether, these results strongly suggest that the acute accumulation of interstitial eCB in vivo by JZL195 induces astroglial eCB-LTD at CA3-CA1 synapses via activation of postsynaptic NR2B and subsequent endocytosis of AMPAR.
Astroglial eCB-LTD is required for JZL195 to induce neuroprotection
In order to study if JZL195-induced in vivo astroglial eCB-LTD at CA3-CA1 synapses could serve as a sub-threshold cell death signal to induce neuroprotection, we employed a transient global ischemia model.
41
Mice were treated with JZL195 (20 mg/kg, i.p.) or vehicle 2 h before global ischemia. Seventy-two hours later, the mice subjected to global ischemia were perfused to facilitate the quantification of both surviving and dying CA1 pyramidal neurons using NeuN immunohistochemical and FJB staining.
25
Pretreatment with JZL195, but not its vehicle, significantly increased the number of NeuN-positive surviving neurons (Figure 4(a) and (b)) and reduced the number of FJB-stained dying neurons (Figure 4(a) and (c)), thereby suggesting that CA1 neurons exhibit neuroprotection following JZL195 preconditioning.
JZL195 preconditioning protects neurons against global ischemia and eCB-LTD is required for JZL195 to induce neuroprotection. Top representative photos (a) and bottom graphs show that JZL195 preconditioning significantly suppresses the decrease of the number of NeuN-positive surviving neurons (b) and increase of the number of FJB-stained dying neurons (c) induced by global ischemia for 20 min. The scale bar represents 50 µm. Top photos (d) and bottom graphs (e) and (f) show that JZL195-induced neuroprotection is abolished by pretreatment with Tat-GluR2 or Ro25-6981, but not by Tat-GluR2S or vehicle. The scale bar represents 50 µm. The summary graphs show means ± SEMs; n = 5 animals in each group. *p < 0.01 vs. ischemia or vehicle, Bonferronni post hoc test after one-way ANOVA ((b): F3,16 = 38.190, p < 0.01; (c): F3,16 = 41.070, p < 0.01; (e): F3,16 = 11.220, p < 0.01; (f): F3,16 = 12.490, p < 0.01).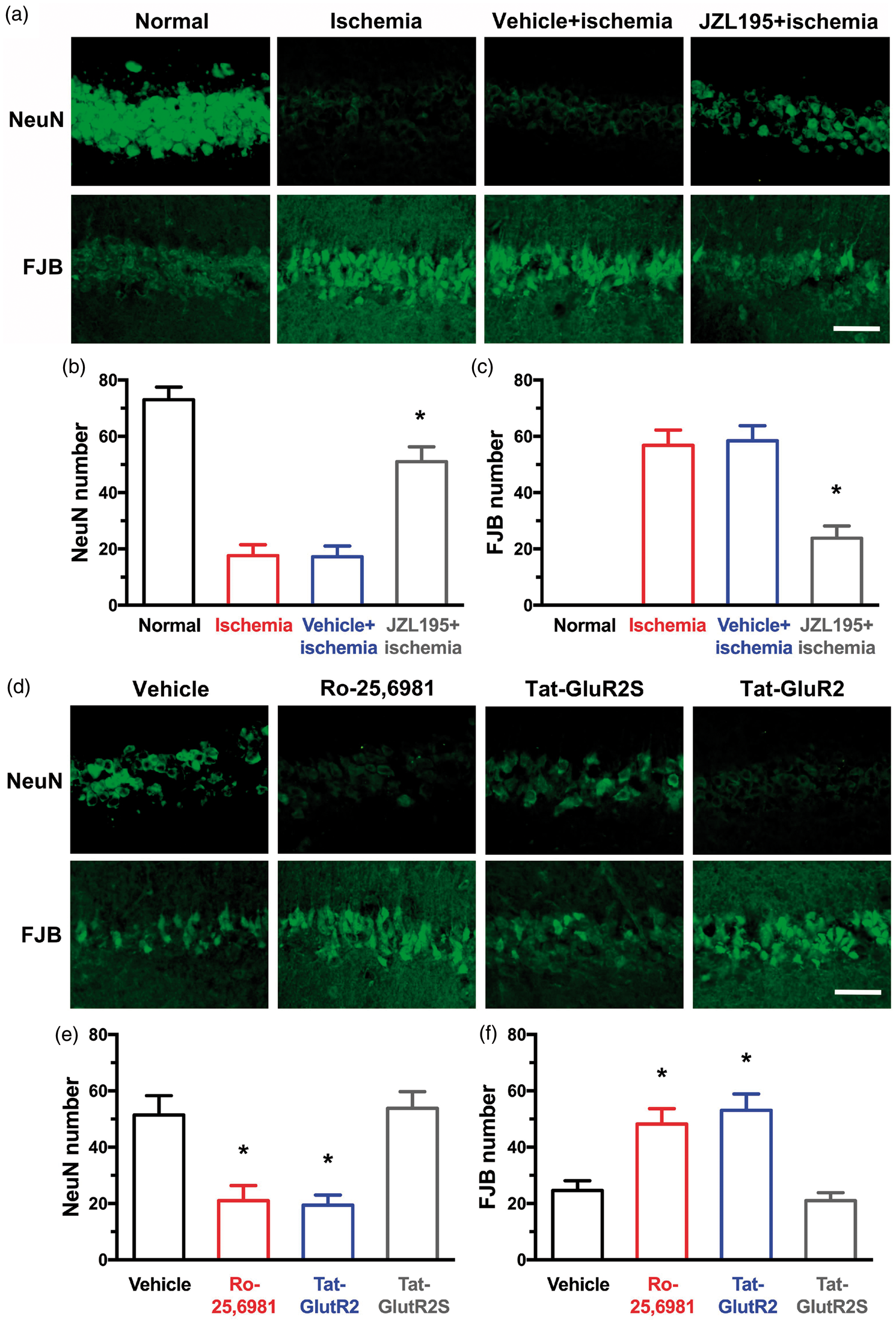
Because JZL195-induced astroglial eCB-LTD is characterized by NR2B activation and AMPAR endocytosis, we further studied whether these mechanisms underlie neuroprotection of CA1 pyramidal neurons following JZL195 treatment. To identify the role of NR2B activation in JZL195 preconditioning, we treated mice with the NR2B antagonist Ro25,6981 (6 mg/kg, i.p.) or its vehicle 30 min before JZL195 preconditioning. To avoid a possible blockade of both JZL195 preconditioning and ischemic neuronal death caused by an acute Ro25,6981 injection, global ischemia was induced 12 h after JZL195 preconditioning. This time-point for injection was chosen because the half-life of Ro25,6981 is about 5 h. 42 Mice treated with Ro25,6981 before JZL195 preconditioning showed fewer numbers of NeuN-positive cells and more FJB-stained cells compared to the numbers of NeuN-positive cells (Figure 4(d) and (e)) and FJB-stained cells (Figure 4(d) and (f)) in mice receiving vehicle before JZL195 preconditioning. Ro25,6981 abolished the neuronal protective effects associated with JZL195 preconditioning, suggesting that NR2B activation is necessary for JZL195 preconditioning.
To reveal the effects of a selective blockade of AMPAR endocytosis on JZL195-induced neuroprotection of CA1 pyramidal cells, we treated mice with Tat-GluR2 or Tat-GluR2S (1.5 µmol/kg, i.p.) 1 h before JZL195 preconditioning.19,40 Because the half-life of Tat-GluR2 is about 3 h, 40 we induced global ischemia 12 h after JZL195 preconditioning in order to avoid the potential inhibition by Tat-GluR2 of both JZL195 preconditioning and ischemic neuronal death. While mice treated with Tat-GluR2S before JZL195 preconditioning showed comparable numbers of NeuN-positive cells (Figure 4(d) and (e)), FJB-stained cells (Figure 4(d) and (f)) in mice receiving JZL195 preconditioning, Tat-GluR2 abolished neuronal protective effects associated with JZL195 preconditioning against global ischemia. Thus, AMPAR endocytosis is necessary for JZL195 preconditioning.
As shown above, JZL195-induced astroglial eCB-LTD required astroglial CB1R. Therefore, we also examined whether JZL195-induced neuroprotection of CA1 pyramidal neurons required astroglial CB1R. Surprisingly, JZL195 preconditioning produced comparable numbers of NeuN-positive (Figure 5(a) and (b)) and FJB-stained neurons (Figure 5(a) and (c)) in the hippocampal CA1 area of GFAP-CB1R-WT and GFAP-CB1R-KO mice. These unexpected results may have been caused by the fact that eCB-mediated activation of astroglial CB1R is required for global ischemia to injure CA1 neurons. In support of this hypothesis, we found that global ischemia for 20 min produced significantly more surviving neurons with NeuN-positive staining (Figure 5(d) and (e)) and less dying neurons with FJB-staining (Figure 5(d) and (f)) in GFAP-CB1R-KO mice than GFAP-CB1R-WT mice. Overall, these results suggest that the same mechanisms underlying JZL195-induced astroglial eCB-LTD at CA3-CA1 synapses mediate JZL195-induced neuroprotection of CA1 pyramidal neurons and that eCB activation of astroglial CB1R is required for ischemia to injure neurons.
Astroglial CB1R participates in ischemic neuronal injury. Top representative photos (a) and bottom graphs (b) and (c) show that JZL195 preconditioning results in similar neuronal death in GFAP-CB1R-WT and GFAP-CB1R-KO mice. Top representative photos (d) and bottom graphs (e) and (f) show that global ischemia results in significantly more severe neuronal death in GFAP-CB1R-WT mice than in GFAP-CB1R-KO mice. The summary graphs show means ± SEMs; n = 5 animals in each group. *p < 0.01 vs. WT mice, t-test.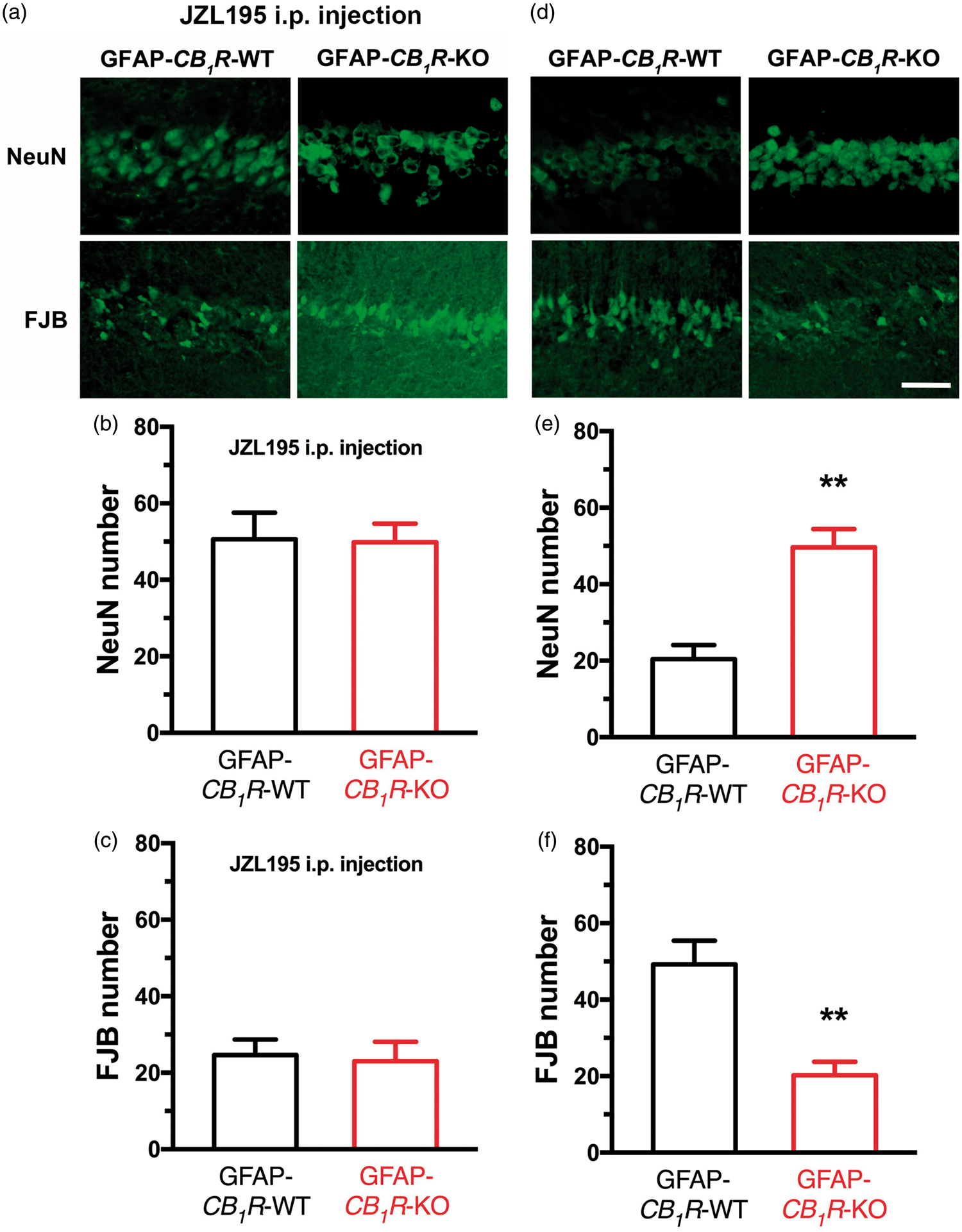
eCB-LTD is required for JZL195 to induce neuroprotection in focal cerebral ischemia
To provide further evidence, in vivo recordings of fEPSP were detected at PrL-NAc synapses in anesthetized mice. Injection of JZL195 (20 mg/kg, i.p.) resulted in a rapid decrease in the fEPSP slope to approximately 30% of the baseline levels at 2 h after injection (Figure 6(a) and (c)). To examine whether the JZL195-induced decrease in the fEPSP slope was CB1R dependent, AM281 (3 mg/kg, i.p.) was administrated 10 min before JZL195 injection. The JZL195-dependent reduction in the fEPSP slope was almost entirely blocked by AM281 10 min before JZL195 injection (Figure 6(b) and (c)), thus suggesting an in vivo synaptic depression following acute eCB accumulation at middle cerebral artery ischemic area.
JZL195 induces in vivo LTD at PrL-NAc synapses through CB1R and protects neurons against focal cerebral ischemia. (a) and (b) Plots of normalized fEPSP slopes show that i.p. injection of JZL195 at 0 min elicits CA1 LTD lasting for >2 h in naive mice (a). The resultant CA1 LTD was blocked following AM281 administration 10 min before. Representative fEPSP traces before (1) and after (2) vehicle or JZL195 injection are shown above each plot (b). (c) Bar charts summarizing the average percent change of fEPSP slope before (1) and after (2) vehicle or JZL195 injection as depicted in panels (a) and (b). All summary graphs show means ± SEMs; n = numbers of animals recorded in each group (a) and (b) *p < 0.01 vs. vehicle control, t test ((a): p < 0.01; (b): p < 0.01). Right representative photos (d) and left graphs show that JZL195 preconditioning significantly improved neurological score (e) and decreased infarct volume (f) induced by focal cerebral ischemia for 60 min. Right representative photos ((g),) and left graghs ((h) and (i)) show that JZL195-induced neuroprotection is abolished by pretreatment with AM281, but not by vehicle. The neurological behavioral scores graphs were expressed as median (range) values and analyzed with Kruskal–Wallis test followed by Mann–Whitney U test. *p < 0.01 vs. vehicle, U test ((e): p = 0.0010; (h): p = 0.0061). The infarct volume graphs show means ± SEMs; n = 10 animals in each group. *p < 0.01 vs. vehicle, t-test ((f): p = 0.0038; (i): p = 0.0008).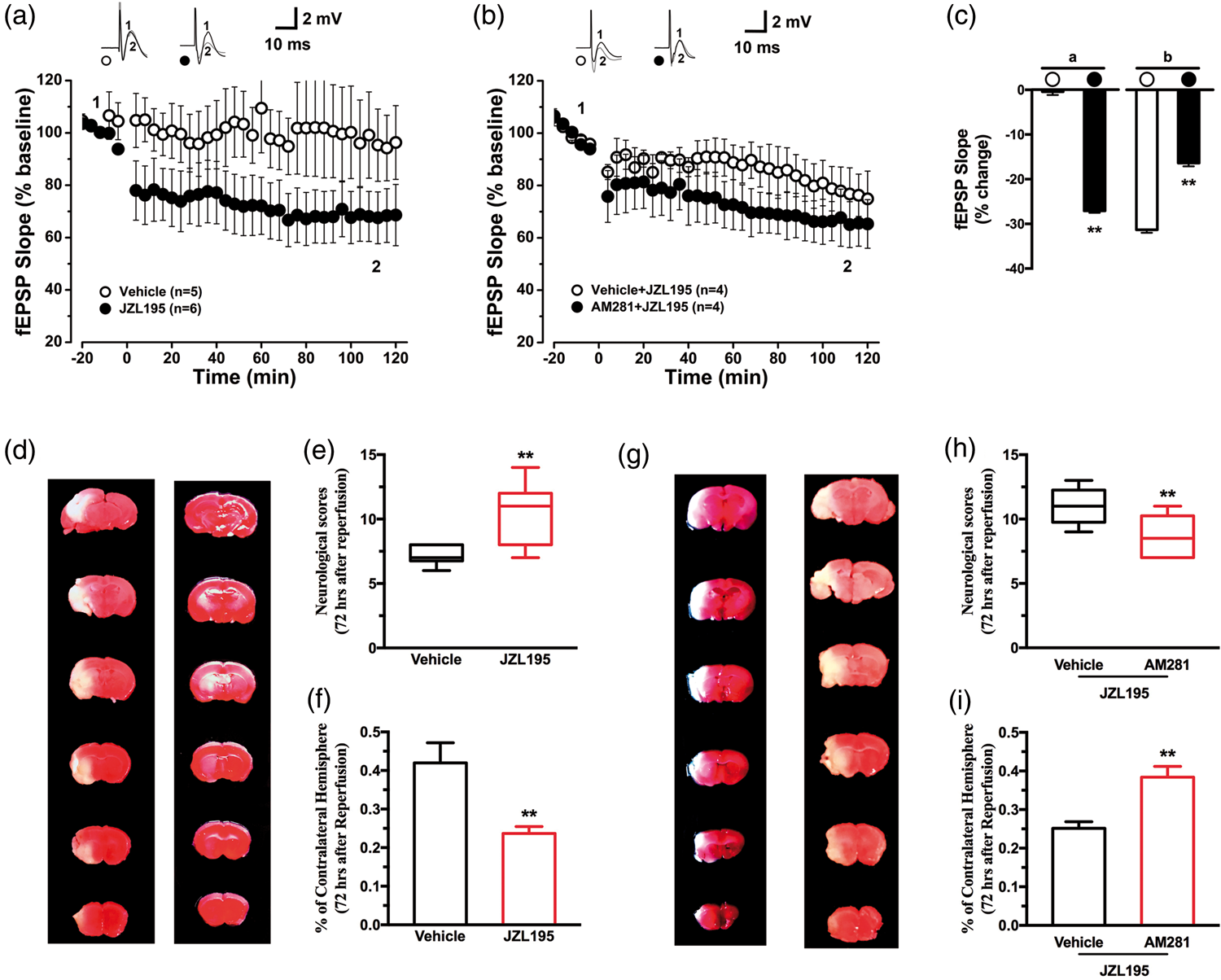
To study if JZL195-induced in vivo eCB-LTD could serve as a sub-threshold cell death signal to induce neuroprotection, we employed a focal cerebral ischemia model. Before JZL195 administration, mice received AM281 or vehicle, respectively. JZL195, but not vehicle, significantly improved neurological behavioural scores (Figure 6(e)) and reduced the infarct volume (Figure 6(d) and (f)). AM281 (Figure 6(g) and (h)) blocked the neuroprotective effect of JZL195, presented with lower neurological scores and larger infarct volumes. These results suggest that, similar to global ischemia, JZL195 preconditioning is also able to induce neuroprotection in focal cerebral ischemia, and AM281 activation is necessary for neuroprotection of JZL195 preconditioning.
Discussion
Neuronal eCBs exist in postsynaptic cytoplasm and their rapid synthesis and release into synaptic clefts is likely to occur in an activity-dependent or “on demand” manner.43,44 Then, eCBs released from the postsynaptic membrane and travel retrogradely to act on presynaptic CB1R, causing the presynaptic expression of LTD or two forms of short-term synaptic depression (i.e. depolarization-induced suppression of excitation and inhibition at excitatory and inhibitory synapses), respectively.17,45,46 In this study we demonstrated that in vivo exposure to the MAGL/FAAH inhibitor JZL195 induced LTD at CA3-CA1 synapses and PrL-NAc synapses, which was mediated by astroglial CB1R but not glutamatergic or GABAergic CB1R. We also provide the evidence suggesting that JZL195-induced LTD production requires suppressed expression of astroglial glutamate transporter GLT1 and subsequent activation of NR2B and endocytosis of AMPAR. Our findings are the first to strongly suggest that astroglial eCB-LTD requires astroglial CB1R, astroglial GLT1, synaptic NR2B and synaptic AMPAR for LTD production in living animals.
The previous in vivo microdialysis studies have shown baseline levels of interstitial AEA and 2-AG in parts of the rodent brain,47–52 including the hippocampus. These results imply the existence of constitutional eCB in brain interstitial space. The dual FAAH/MAGL inhibitor JZL195 significantly increased interstitial levels of both AEA and 2-AG. 51 These data, together with the observation of a 2-fold increase of interstitial eCB levels in mutant mice without the FAAH gene, 50 strongly support the notion that brain interstitial eCB forms a basal eCB stream with a constant supply and a continuous clearance pathway. Therefore, the acute inhibition of eCB clearance pathways rapidly increases eCB levels in brain interstitial space.
In the rodent hippocampus, CB1R has been found in both GABAergic and glutamatergic presynaptic membranes.14,15 Employing both wild-type and conditional or constitutional CB1R mutant mice, our recent study showed a low density of CB1R in hippocampal astrocytes. 19 Astrocytes are one of the major cell types in the mammalian brain that possess a highly ramified morphology and occupy at least 50% of brain volume. 53 With their unique morphology, astrocytes are extensively surrounded by interstitial space-containing interstitial fluid. 54 Therefore, our findings pertaining to astroglial CB1R 19 indicate that interstitial eCB may directly stimulate astroglial CB1R. An acute inhibition of the extracellular eCB clearance pathway would rapidly increase interstitial eCB levels, thereby activating astroglial CB1R. Indeed, as part of this analysis we observed that inhibition of 2-AG/AEA clearance caused by an acute administration of JZL195 resulted in the rapid induction of astroglial CB1R-mediated synaptic LTD at the hippocampal CA3-CA1 synapses. This observation strongly suggests that an elevated extracellular eCB stream can regulate synaptic plasticity in the hippocampus.
Several studies have demonstrated that NR2B activation is required for ischemia-induced neuronal injury. 55 Thus, the selective NR2B antagonists Ro25,6981, ifenprodil and related compounds, including eliprodil (SL-82.0715) and traxoprodil (CP-101,606), have been shown to effectively protect neurons against ischemic neuronal death both in in vitro and in vivo ischemic models, as well as in double-blinded placebo-controlled Phase II clinical studies. 56 Remarkably, AMPAR endocytosis has been recognized as an essential downstream step associated with NR2B-mediated neuronal death.21,57,58 Inhibition of AMPAR endocytosis by two structurally different inhibitors of clathrin-mediated endocytosis prevents NMDAR-mediated neuronal death without affecting NMDAR activity. 57 More interestingly, the specific inhibition of AMPAR endocytosis by the GluR2 peptide, whose sequence is derived from the GluR2 C-terminus that is required for the regulation of AMPAR endocytosis and LTD expression, protects neurons against excitotoxic damage in vitro. 57 Disruption of AMPAR endocytosis with another interfering peptide, GluR2NT1-3-2, also decreased ischemic neuronal death in both an in vitro model of brain ischemia 58 and in vivo rat models of global and focal ischemia. 21 These findings strongly support the notion that synaptic LTD contributes to neuronal death mechanisms. 56 In this study, we show that global ischemia-induced death of CA1 neurons also requires CB1R in GFAP-positive astrocytes, which are more resistant than neurons and GFAP-negative astrocytes to ischemic injury. Because JZL195-induced eCB-LTD at CA3-CA1 synapses is characterized by the recruitment of astroglial CB1R, postsynaptic NR2B and AMPAR (as demonstrated in the present study), all of the available evidence supports the idea that ischemia-induced death of CA1 pyramidal neurons requires LTD at CA3-CA1 synapses following the sequential recruitment of astroglial CB1R, postsynaptic NR2B and AMPAR.
However, LTD occurs in the presence of a variety of physiological conditions and thus, LTD alone is unlikely to be capable of inducing neuronal death. This hypothesis is in agreement with the recent observation that NMDAR activation triggers a cascade of intracellular activities to induce AMPAR endocytosis, ultimately resulting in neuronal death signals without inducing neuronal death.22,38 Thus, it is plausible to hypothesize that in vivo astroglial eCB-LTD at CA3-CA1 synapses that require NMDAR-mediated endocytosis of AMPAR could serve as a sub-threshold cell death signal to induce neuroprotection in living animals. This hypothesis is strongly supported by our findings that JZL195 administration rapidly induces an in vivo eCB-LTD at CA3-CA1 synapses, and that JZL195 preconditioning (with the same dose of JZL195 as was used for LTD induction) either 2 or 12 h prior to acute global ischemia resulted in the significant protection neurons against subsequent lethal ischemia. Furthermore, JZL195-elicited astroglial eCB-LTD and JZL195 preconditioning shares at least two key molecular mechanisms: NR2B activation and AMPAR endocytosis. Our findings are in agreement with the recent observation that the FAAH inhibitor URB597 produced significant neuroprotection when the compound was administered 90 min prior to occlusion but not immediately after ischemia. 59
In order to provide more evidences, we studied whether JZL195-induced in vivo eCB-LTD could also serve as a sub-threshold cell death signal to induce neuroprotection in focal cerebral ischemia model. The middle cerebral artery occlusion induced injury is related to striatum and cortex. As a part of striatum, nucleus accumbens (NAc) can be divided into two structures – the NAc core and the NAc shell. One of the major glutamatergic inputs to the NAc shell is the prefrontal cortex, particularly the prelimbic cortex (PrL). 60 Therefore, in vivo recordings of fEPSP detected at PrL-NAc synapses were used to provide further evidences to support our hypothesis. The results demonstrated that JZL195 preconditioning induces LTD at ischemia penumbra synapses, which is mediated by CB1R. Similar to global ischemia, JZL195 preconditioning is also able to induce neuroprotection in focal cerebral ischemia, and AM281 activation is essential for the induction of neuroprotection by JZL195.
In conclusion, the present study provides evidence supporting the occurrence of a functional extracellular eCB stream, astroglial eCB-LTD and LTD preconditioning for neuroprotection. Collectively, our findings support a scenario (Figure 7), in which a rapid elevation of extracellular eCB sequentially stimulates astroglial CB1R, suppresses astroglial glutamate transporter GLT1, increases ambient glutamate, activates NR2B, and induces postsynaptic AMPAR endocytosis to trigger astroglial eCB-LTD. The LTD preconditioning induced by endogenous cannabinoids protects neurons against subsequent lethal ischemia. These findings may provide a potential therapeutic target for ischemic stroke.
Diagrammatic sketch for neuroprotection through astroglial eCB-LTD. An elevation of interstitial eCB flux stimulates astroglial CB1R, suppresses GLT1, increases ambient glutamate, activates postsynaptic NR2B, and induces AMPAR endocytosis, which elicits LTD expression at glutamatergic synapses to trigger neuroprotection.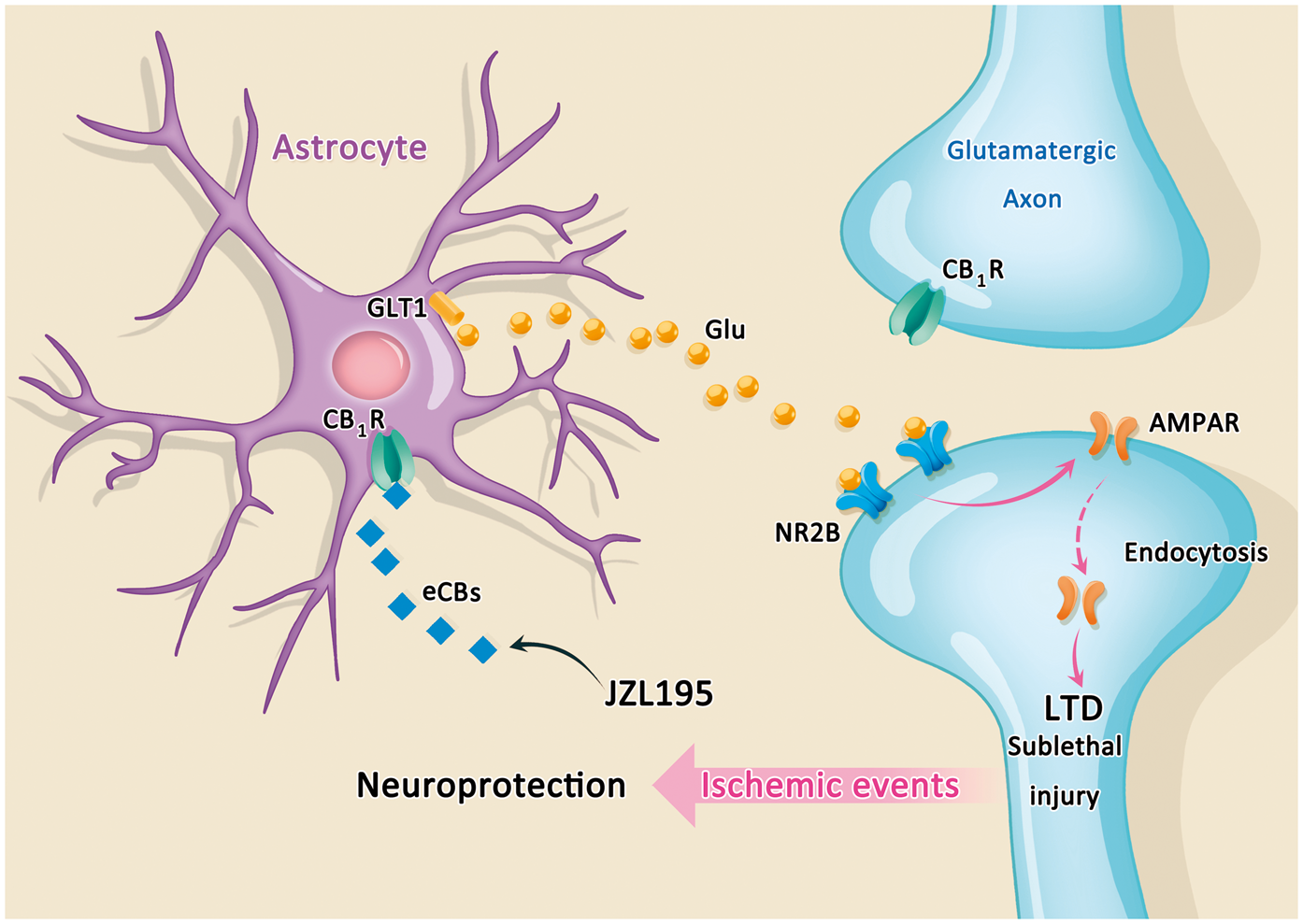
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work received support from International Cooperation and Exchange of the National Natural Science Foundation of China (grant no. 81420108013 to LX), Canadian Institutes of Health Research (MOP 123256 to XZ), Canadian Foundation for Innovation (No. 19317 to XZ), Shaanxi Province Natural Science Foundation (No. 2016JQ8024 to FW), NIH grants (1R01MH099554-01 to YY) and National Natural Science Foundation of China (No. 81771227 to JH).
Acknowledgment
We thank JC Maillet for technical assistance.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
FW, JH, HH, JW, LJ and LT conducted experiments and data analysis, and were also involved in manuscript writing. YY and HD were involved in partial experimental design and data analysis. LX and XZ designed the project and wrote the manuscript.