
Abstract
Activation of ATP-sensitive potassium (KATP) channels in arterial smooth muscle (ASM) contributes to vasodilation evoked by a variety of endogenous and exogenous compounds. Although controversial, activation of KATP channels by neuropeptides such as calcitonin gene-related peptide (CGRP) and pituitary adenylate cyclase activating peptide (PACAP) in the trigeminovascular system, including the middle meningeal artery (MMA), has been linked to migraine headache. The objective of the current study was to determine if ongoing KATP channel activity also influences MMA diameter. In the absence of other exogenous compounds, the KATP channel inhibitors glibenclamide and PNU37883A induced constriction of isolated and pressurized MMAs. In contrast, KATP channel inhibition did not alter cerebral artery diameter. Consistent with tonic KATP activity in MMA, glibenclamide also induced ASM membrane potential depolarization and increased cytosolic Ca2+. Inhibitors of cAMP-dependent protein kinase (PKA) abolished basal KATP activation in MMA and caused a marked decrease in sensitivity to the synthetic KATP channel opener, cromakalim. In vivo MMA constriction in response to gibenclamide was observed using two-photon imaging of arterial diameter. Together these results indicate that PKA-mediated tonic KATP channel activity contributes to the regulation of MMA diameter.
Introduction
Migraine headaches are intense recurring headaches that can last for hours to days and are often accompanied by nausea, visual disturbances (aura), sensitivity to light and sound, and other neurological symptoms. 1 Migraine is one of the most prevalent contributors to the world-wide burden of mental and neurological disorders. 2 However, despite the human and societal costs, treatments for acute migraine and prophylaxis are limited. 3 A major hurdle to the development of effective anti-migraine therapies is the complexity and heterogeneity of this disorder, which likely involves components within the trigeminovascular system and the central nervous system.2–5
Two neuropeptides, pituitary adenylate cyclase-activating polypeptide (PACAP) and calcitonin gene-related peptide (CGRP), are among the multitude of players implicated in the pathophysiology of migraine headache.3,5–10 Several studies have demonstrated that the administration of exogenous CGRP or PACAP resulted in vasodilation of meningeal arteries, which coincided with the onset of migraine-like headache.6–12 The role of meningeal arteries in the etiology of migraine headache has long been controversial, with recent evidence suggesting that vasodilation within this vasculature may not be the causative factor in the initiation of headache pain.13,14 However, neurovascular signaling mechanisms involving the dural vasculature may contribute to the complex pathobiology of migraine, as well as cluster headache.4,15,16
Vascular smooth muscle potassium channels play a crucial role in the regulation of arterial diameter. Activation of these channels results in membrane potential hyperpolarization promoting a decrease in the activity of voltage-dependent calcium channels (VDCCs) that ultimately leads to decreased intracellular calcium concentration ([Ca2+]i), smooth muscle relaxation, and vasodilation. One key potassium channel in the vasculature is the ATP-sensitive potassium (KATP) channel. 17 These channels were initially described in cardiac myocytes by Noma 19 and have since been identified in numerous tissues including pancreatic beta cells and vascular smooth muscle.17,18 Hallmarks of KATP channels include their inhibition by elevated levels of intracellular ATP and by sulphonylurea compounds such as the anti-diabetic agent, glibenclamide.17,20 Functional KATP channels are composed of an octamer of four inward rectifying (Kir) subunits (either Kir6.1 or Kir6.2) and four auxiliary sulphonylurea receptors subunits (either SUR1, SUR2A or SUR2B). 20
In vascular smooth muscle, the predominant isoform of KATP channels consists of pore forming Kir 6.1 subunits and SUR2B sulphonylurea receptor subunits.20–22 In many arterial beds, KATP channels represent important targets for endogenous vasodilator substances, e.g. CGRP and PACAP that act via Gs protein-coupled receptors to increase cyclic adenosine monophosphate (cAMP) and in turn cAMP-dependent protein kinase (PKA). 23 PKA has been shown to phosphorylate serine and threonine residues located on Kir 6.1 and SUR2B isoforms and to increase channel activity. 24 KATP channels, composed of Kir6.1 and SUR2B subunits, have been shown to underlie meningeal artery vasodilation induced by CGRP, PACAP, as well as synthetic KATP channel activators such as pinacidil and cromakalim.25–28 However, the role of ongoing KATP channel activity in the control of meningeal artery diameter is unclear.
In the present study, we investigated the role of basal KATP channel activity in the diameter regulation of rat middle meningeal arteries (MMA). We provide evidence that tonic KATP activity provides an ongoing contribution to the regulation of MMA diameter—a phenomenon dependent upon steady state activation of PKA. In marked contrast, KATP channels are not tonically active in cerebral arteries. These findings indicate that KATP channels play a unique role in the regulation of MMA diameter.
Materials and methods
Animal models
Animal housing and all experimental procedures were conducted in accordance with the Guide for the Care and Use of Laboratory animals (eighth edition, 2011), ARRIVE (Animals in Research: Reporting In Vivo Experiments) guidelines and followed protocols approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Vermont. Sprague–Dawley rats (males, 300–350 g; Charles River Laboratories, Saint Constant, QC, Canada) were euthanized by decapitation under deep pentobarbital anesthesia (60 mg/kg). The brain and calvaria were carefully dissected and placed in ice-cold artificial cerebral spinal fluid (aCSF) of the following composition (in mM): 125 NaCl, 3 KCl, 18 NaHCO3, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl2, and 5 glucose (pH 7.4). The dura mater was removed from the calvaria and the MMA dissected out using iris scissors. Superior cerebellar (cerebral) arteries were dissected from the brain in a similar fashion. Eight-week-old smooth muscle reporter (SMMHC-tdTomato) mice were used for in vivo imaging of meningeal arterial diameter. These mice, expressing the red fluorescent protein, tdTomato, under control of the smooth muscle myosin heavy chain (SMMHC) gene promoter were obtained by cross-breeding B6.FVB-Tg(Mhg11-cre/ERT2)1Soff/J (Jackson Lab, Stock No: 019079) mice and B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J (Jackson Lab, Stock No: 007914) mice. SMMHC-tdTomato mice were administrated tamoxifen (40–80 mg/kg/day, Harlan Laboratories) in their food for 7 days starting at the age of 21 days.
Ex vivo measurements of arterial diameter
Middle meningeal and cerebral arteries were cannulated by glass micropipettes mounted in a 5 ml myograph chamber perfused with aCSF at 37℃ that was aerated with 5% CO2, 20% O2, 75% N2 (pH 7.35–7.40). Intravascular pressure was adjusted using a pressure servo-controller and a peristaltic pump as previously described. 29 Changes in arterial diameter were measured by video edge detection (Living Systems Instrumentation, St Albans, VT) using WinDaq data acquisition software (Dataq Instruments; Akron, OH). After allowing the arteries to equilibrate for 1 h, viability of the artery was determined by brief superfusion with aCSF containing 60 mM KCl (aCSF isosmotic replacement of NaCl with KCl). Arteries that exhibited a constriction representing less than a 50% decrease in diameter were discarded. Unless noted, isolated MMA were studied at an intravascular pressure of 40 mmHg. In aCSF, arterial diameter was 54.3 ± 2.4 µm in aCSF compared to a fully dilated (passive) diameter of 90.4 ± 2.5 µm obtained at the end of experimental protocol using Ca2+-free aCSF containing 100 µM diltiazem and 1 µM forskolin. This pressure-induced constriction or “myogenic tone” represented a 39.9 ± 2.4% decrease in lumen diameter (n = 30 arteries; N = 30 animals), similar to our previous findings that examined MMA responses over a range of intravascular pressures. 30 Constriction to pharmacological agents is expressed as a percent decrease in arterial diameter relative to the arterial diameter obtained prior to the addition of the test compound. Percent vasodilation to cromakalim was normalized to maximal dilation and calculated from the equation: ((Dcromakalim-Dstart)÷(DP-Dstart)) × 100, where Dcromakalim is the lumen diameter at a specific concentration of cromakalim, Dstart is the diameter prior to giving the first concentration of cromakalim and DP (passive diameter) is the lumen diameter of the artery in Ca2+-free aCSF containing the vasodilators diltiazem (100 μM) and forskolin (1 μM). 31
Membrane potential measurements
Arteries were cannulated as described above and pressurized to 40 mmHg. Smooth muscle membrane potential was measured by insertion of a sharp glass microelectrode (∼100 MΩ) containing 0.5 M KCl into smooth muscle cells from the adventitial side of the arterial wall. The criteria for successful impalement were (1) an abrupt negative potential deflection upon entry, (2) a stable membrane potential for ≥30 s, and (3) an abrupt positive potential deflection upon microelectrode removal. 29 Membrane potential measurements were made with an electrometer (World Precision Instruments, Sarasota, FL) and recorded via computer with Axotape software (Molecular Devices, Sunnyvale, CA).
Ratiometric measurements of arterial smooth muscle intracellular Ca2+ concentration ([Ca2+]i)
Freshly isolated MMA were cannulated on glass micropipettes mounted in a 5 ml myograph chamber, as described above. After cannulation, arteries were loaded with the ratiometric Ca2+-sensitive dye fura-2 acetoxymethyl ester (fura-2 AM, 5 µM, Invitrogen, Carlsbad, CA) for 1 h in aCSF containing pluronic acid (0.1%) at room temperature (∼ 22℃). Next, the myograph chamber was placed on the stage of a Nikon TE2000-S inverted fluorescence microscope and superfused with aerated aCSF (37℃, 30 min) to allow de-esterification of intracellular fura-2 AM, and then pressurized to 40 mmHg for study. Fluorescence ratio was obtained from background corrected ratio of the 510 nm emission from arterioles alternately excited at 340 and 380 nm with hardware and software developed by IonOptix (Milton, MA). 32
In vivo two-photon imaging of meningeal blood vessels
SMMHC-tdTomato mice were initially anesthetized with isoflurane (5% induction and 2% maintenance), and a dura-intact cranial window superfused with aCSF (37℃) aerated with 5% CO2, 20% O2, 75%N2 placed over the MMA. Anesthesia was then switched to a combination of urethane (750 mg/kg, i.p.) and α-chloralose (50 mg/kg, i.p.), a change necessitated by the fact that isoflurane causes vasodilation and impaired cerebral blood flow autoregulation.33,34 Animals were head-fixed to a stereotactic frame and meningeal blood vessels were visualized by intravenous injection of FITC-dextran (∼500 kDa). Fluorescent images (512 × 512 pixels, 0.5-1.0 µm/pixels) were acquired using a Zeiss LSM-7 multiphoton imaging system (excitation, 820 nm; fluorescent bandpass filter, 525/50 nm and 675/70 nm). Intraluminal diameter (i.e. the width of intravascular FITC green fluorescence line) was measured using custom software SparkAn (written by Dr. Adrian D. Bonev, University of Vermont). 35 Meningeal arteries and veins were differentiated based on the presence and absence of vascular smooth muscle cells (identified by tdTomato red fluorescence), respectively. Drugs were applied by superfusion of the cranial window. Upon completion of the experimental protocol, animals were euthanized by decapitation while under a surgical plane of anesthesia.
Statistical analysis
Data are expressed as mean ± SEM (n: the number of observations, N: the number of animals). Analysis was performed by investigators that perform the studies; no blinding was done. Student’s two-tailed paired t test was used for comparisons between two groups.
Reagents
Fura-2 AM and pluronic acid were obtained from Invitrogen (Life Technologies, Eugene, OR). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Results
Inhibition of basal KATP channel activity causes vasoconstriction via membrane potential depolarization and elevated [Ca2+]i in MMA smooth muscle
Glibenclamide caused a concentration-dependent constriction of isolated MMA with an EC50 of ∼11 nM, consistent with an action mediated through inhibition of KATP channels.18,20 In marked contrast, glibenclamide (1 nM to 10 µM) had no effect on the diameter of isolated cerebral arteries (Figure 1(a) to (c)). PNU37883A (10 µM), a purported vascular-selective KATP channel inhibitor structurally distinct from sulfonylurea compounds,
36
induced an ex vivo constriction of MMA similar in magnitude to that observed with glibenclamide (i.e. ∼20% decrease in diameter), but did not alter cerebral artery diameter (Figure 1(d)). Using two-photon imaging on dura-intact cranial windows, we also observed glibenclamide-induced MMA constriction in vivo (Figure 1(e) and (f)). These results suggest that basal KATP channel activity in MMA impacts arterial function by providing a tonic vasodilator influence.
Tonic KATP channel activity contributes to MMA diameter regulation. (a and b) Representative diameter recordings demonstrating that glibenclamide (Glib) caused a concentration-dependent constriction of isolated, pressurized MMA, but was without effect on cerebral arteries. Passive diameter (maximally dilation) was induced using Ca2+-free aCSF containing 100 µM diltiazem and 1 µM forskolin. (c) Glibenclamide concentration-response curves obtained from MMA (n = 4) and cerebral arteries (n = 4). (d) Summary data of glibenclamide- and PNU37883-induced constrictions. **p < 0.01 MMA (n = 5) vs. cerebral artery (n = 5) using unpaired t-tests. (e) In vivo two-photon imagining of middle meningeal artery and vein in an SMMHC-tdTomato mouse in the presence and absence of glibenclamide (10 µM) applied via superfusion of the cranial window. (f) Summary data of glibenclimide-induced changes in blood vessel diameter in vivo.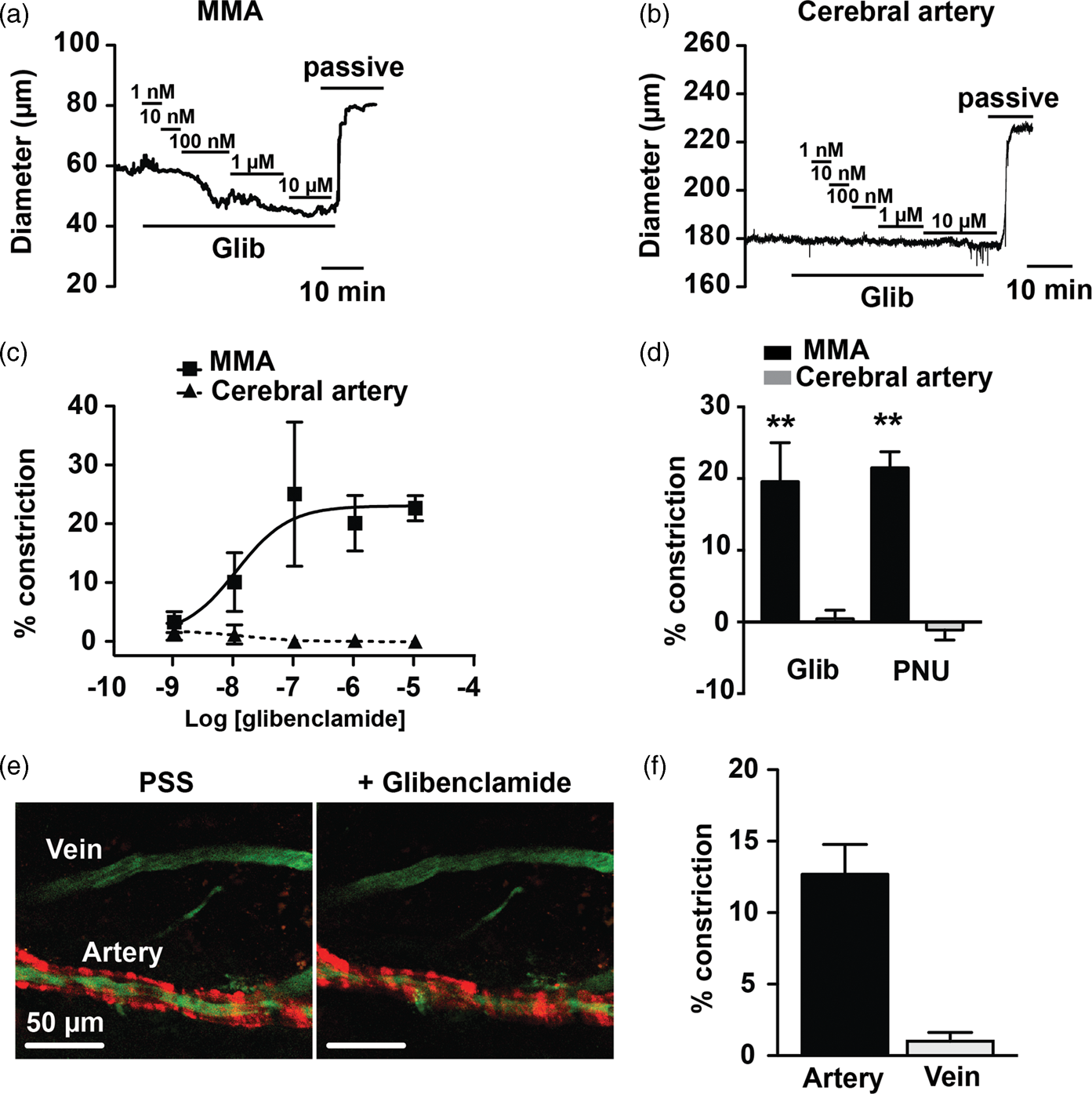
Activation of arterial smooth muscle K+ channels promotes membrane potential hyperpolarization, decreased VDCC-mediated Ca2+ influx and vasodilation.
17
Evidence provided above (Figure 1) suggests that inhibition of KATP channels in MMA causes the inverse response, i.e. membrane potential depolarization, an increase in VDCC open-state probability, and vasoconstriction. To directly examine this pathway, intracellular microelectrodes were used to measure arterial smooth muscle membrane potential in the absence and presence of KATP channel inhibition (Figure 2(a) to (c)). In MMAs, glibenclamide (10 µM) caused a membrane potential depolarization of ∼14 mV, from −45.4 ± 1.4 mV to −31.3 ± 2.7 mV. In contrast, cerebral artery smooth muscle membrane potential was not significantly different in the absence (−37.1 ± 0.9 mV) and presence (−36.8 ± 0.8 mV) of glibenclamide. Dimethyl sulfoxide (DMSO, 1:1000 dilution in aCSF), the vehicle used to dissolve glibenclamide, did not affect the membrane potential of MMA myocytes (46.0 ± 1.2 mV, n = 5; N = 5 animals). To examine the impact of KATP inhibition on [Ca2+]i, MMA was loaded with the ratiometric Ca2+ indicator dye, fura-2. Treatment with 10 µM glibenclamide evoked a vasoconstriction of 23.4 ± 1.0% and an increase in the fura-2 340:380 fluorescence ratio from 1.4 ± 0.1 to 1.8 ± 0.2, consistent with an increase in [Ca2+]i (n = 4; N = 4 animals). We further postulated that this glibenclamide-induced increase in [Ca2+]i was the result of smooth muscle VM depolarization and enhanced VDCC gating. Thus, VDCC inhibition with diltiazem should prevent glibenclamide-induced MMA constriction. As VDCCs are the predominant Ca2+ entry pathway underlying pressure-induced myogenic tone,32,37 this experimental series was done using arteries held at 10 mmHg to minimize the confounding effects of elevated intravascular pressure on VDCC activity. In the absence of diltiazem, glibenclamide (10 µM) caused a constriction of 14.7 ± 2.1 µm representing a 21.2 ± 2.6% decrease in diameter, a response similar in magnitude to that observed at 40 mmHg. As predicted, this glibenclamide-induced vasoconstriction was abolished by diltiazem (100 µM) (Figure 2(d) to (f)). In sum, these data are consistent with basal KATP channel activity exerting a tonic hyperpolarizing/vasodilating influence on MMA, but not cerebral artery diameter.
Inhibition of basal KATP channel activity causes smooth muscle membrane potential depolarization and VDCC-dependent constriction of MMA. (a and b) Membrane potential recordings obtained from an isolated, pressurized MMA (a) and cerebral artery (b) in the absence and presence of glibenclamide (Glib, 10 µM). (c) Summary data of glibenclamide treatment on membrane potential. (d and e) Glibenclamide-induced diameter changes in MMA in the absence (d) and the presence (e) of the VDCC inhibitor diltiazem (Dilt, 100 µM). (f) Summary data of glibenclamide-induced MMA constriction in the absence and presence of diltiazem (n = 5) p < 0.01 using paired t-test.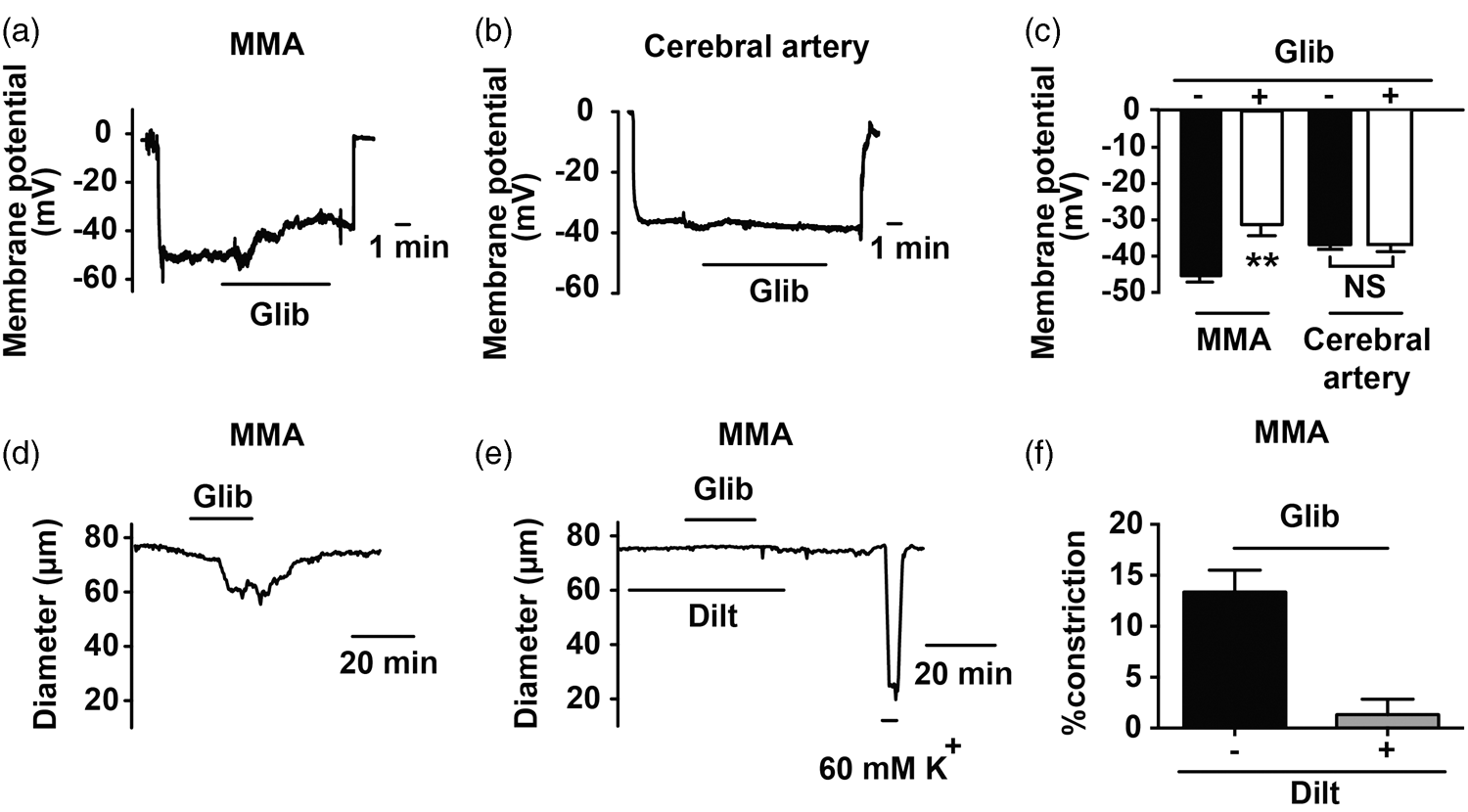
Cyclic AMP-dependent protein kinase (PKA) underlies tonic KATP channel activity in MMA
KATP channel activity is enhanced by PKA-mediated channel phosphorylation.
23
We therefore hypothesized that ongoing PKA activity may underlie basal KATP channel activity in MMA. To explore this concept, we examined if KT5720, a PKA inhibitor,
38
could mimic the actions of glibenclamide to decrease KATP channel activity promoting enhanced MMA constriction. Indeed, KT5720 (1 µM) caused constriction (25.2 ± 5.2%) of MMA comparable to that induced by glibenclamide. Further, in the presence of KT5720, glibenclamide was without effect, consistent with the two drugs acting through a common signaling pathway (Figure 3(a) to (c)). As with glibenclamide, KT5720 did not alter cerebral artery diameter (Figure 3(d)). In addition, we used a second PKA inhibitor, H89
39
(1 µM), to examine the impact of PKA inhibition on the membrane potential of MMA smooth muscle. Similar to the actions of glibenclamide, H-89 caused a membrane potential depolarization of ∼13 mV, from −46.8 ± 0.5 mV to −33.5 ± 0.6 mV. In the presence of H-89, glibenclamide no longer caused membrane potential depolarization or vasoconstriction (Figure 3(e) and (f)). These results indicate that basal PKA activity contributes to the activation of KATP channels in MMA.
Basal PKA activity underlies tonic KATP channel activity in MMA. (a and b) Diameter measurements illustrating that the PKA inhibitor KT5720 (1 µM) abolished glibenclamide-induced constriction in rat MMA (c) Summary data of glibenclamide-induced MMA constriction in the absence (n = 5) and presence of KT5720 (n = 5). **p < 0.01 using unpaired t-test. (d) Summary data demonstrating that KT5702 caused marked constriction of MMA (n = 5), but not cerebral arteries (n = 5). **p < 0.01 using unpaired t-test. (e) Membrane potential measurement in rat MMA demonstrating that the PKA inhibitor H89 (1 µM) abolished glibenclamide-induced depolarization. (f) Summary data illustrating that H-89 abolished glibenclamide-induced membrane potential depolarization (right panel, n = 7) and vasoconstriction (left panel, n = 7).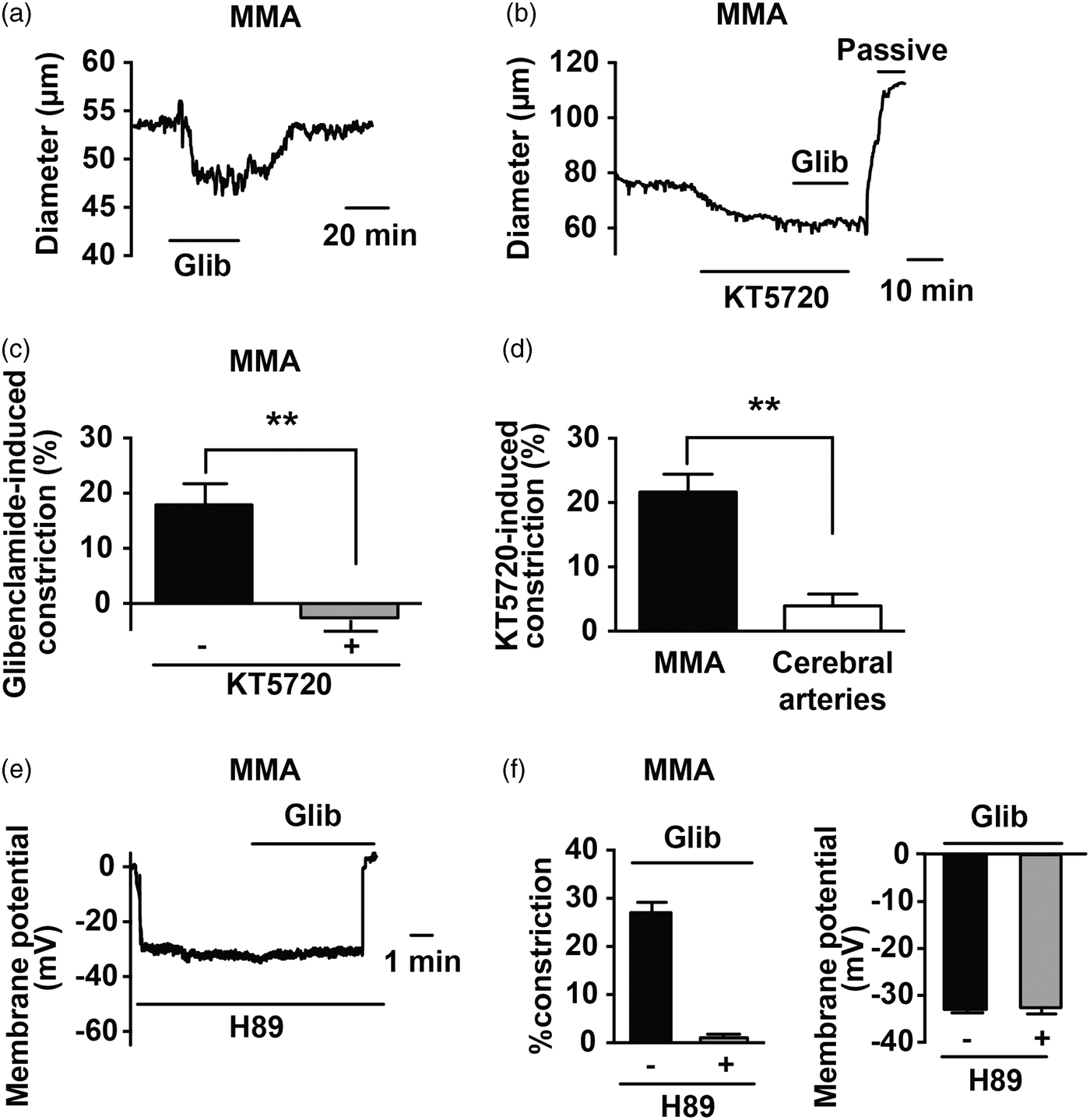
Ongoing PKA activity underlies enhanced sensitivity of MMA to the KATP channel opener cromakalim
In addition to marked differences in responsiveness to KATP channel inhibitors, MMAs and cerebral arteries exhibit a striking disparity in sensitivity to the synthetic KATP channel opener, cromakalim. As illustrated by the cumulative concentration response curves shown in Figure 4, MMA exhibited a 10-fold greater sensitivity to cromakalim (EC50 ∼ 100 nM) compared to cerebral arteries (EC50 ∼ 1 µM). Despite the difference in potency, the efficacy of cromakalim was similar between artery types with 10 µM cromaklim causing a dilation of 96.9 ± 3.1% and 89.1 ± 3.5% of tissue maximum in MMA and cerebral arteries, respectively. To examine whether basal PKA activity contributes to the greater potency of cromakalim in MMA, concentration response curves to this KATP opener were repeated in the presence of KT5720 (1 µM). As shown in Figure 4, KT5720 caused a rightward shift in the cromakalim concentration response curve in MMA and a three-fold increase in the EC50 value to ∼300 nM. These findings indicate that ongoing PKA activity not only increases basal KATP activity in MMA, but also increases the sensitivity of this tissue to KATP channel activators.
PKA inhibition decreases MMA sensitivity to the KATP channel opener cromakalim. (a–c) Diameter recordings of vasodilation evoked by cumulative increases in the concentration of the KATP channel opener cromakalim in middle meningeal (a and c) and cerebral arteries (b). (d) Summary of cromakalim concentration-response curves of MMA and cerebral arteries. Cromakalim exhibited greater potency as a vasodilator of MMAs (EC50 value: ∼100 nM, n = 4) compared to cerebral arteries (EC50 value: ∼1 µM, n = 4). KT5720, a PKA inhibitor, decreased cromakalim potency in MMA (EC50 value: ∼300 nM n = 5).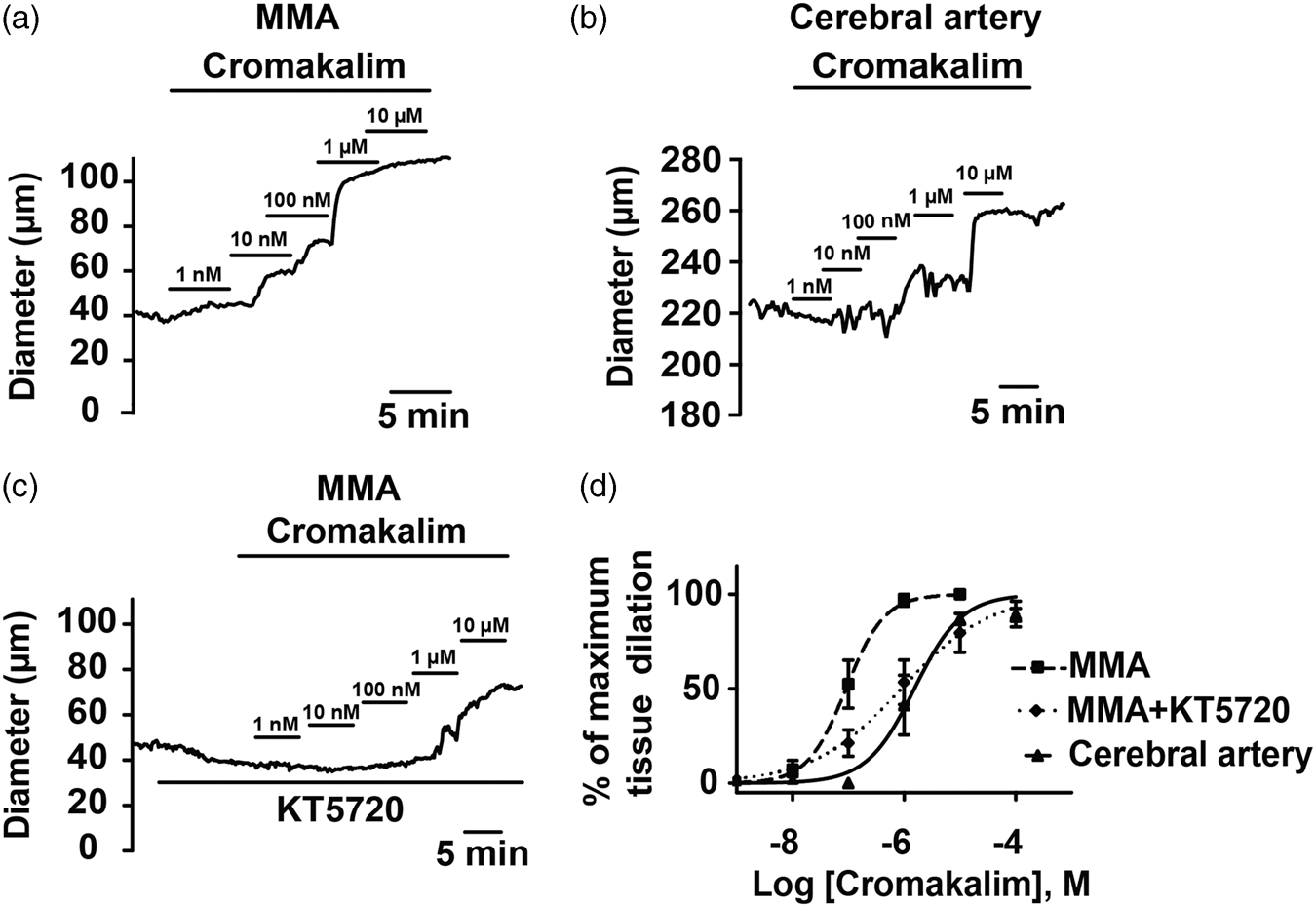
Discussion
The results presented in this study provide evidence that tonic KATP channel activity contributes to the regulation of MMA diameter. In the absence of other vasoactive stimuli, we found KATP channel inhibition caused smooth muscle membrane potential depolarization, increased [Ca2+]i and vasoconstriction of isolated MMAs consistent with an ongoing vasodilatory influence mediated by KATP channel activity. Our data also indicate that PKA underlies this persistent KATP channel activity. In marked contrast, cerebral arteries do not exhibit basal KATP channel activity and are less sensitive than MMA to the direct KATP channel activator, cromakalim. These findings signify that KATP channel activity may be uniquely tuned by PKA in the meningeal vasculature.
KATP channels are widely expressed in arterial smooth muscle throughout the vasculature, including the meningeal and cerebral circulations.17,25 In addition to responding to metabolic signals such as shifts in the intracellular concentrations of ATP and ADP and to synthetic channel openers (e.g. cromakalim, minoxidil, diazoxide), KATP channel gating is enhanced by increased PKA activity. Phosphorylation of KATP channels by PKA has been shown to decrease the sensitivity of the channel to the inhibitory action of ATP and exogenously applied PKA activators such as cAMP, forskolin and isoproterenol elicit KATP currents in isolated smooth muscle myocytes.24,40 Importantly, endogenous vasodilators, e.g. adenosine, CGRP, PACAP, Vasoactive Intestinal Peptide (VIP), which bind to Gs-coupled membrane receptors, enhance KATP channel activity via the adenylyl cyclase/PKA signaling cascade.41,42 Here, we provide evidence that in the MMA, ongoing PKA activity—in the absence of added PKA-stimulating compounds—is sufficient to cause KATP channel activation. We found that two structurally distinct KATP channel blockers, glibenclamide and PNU37883 independently caused constriction of isolated MMA (Figure 1). In stark contrast, basal KATP activity was not observed in cerebral arteries, despite the functional presence of these channels. Our findings, including our in vivo observations, are consistent with a previous in vivo study that reported i.v. glibenclamide infusion caused constriction of MMA but not pial arteries. 43 Using PNU37883A (0.5 mg/kg i.v.), Ploug et al. 25 observed a modest transient MMA constriction; however, the PNU37883A dosage used was considerably lower than the previously reported EC50 value (2 mg/kg, i.v.) for in vivo block of pinacidil-induced vasodilation. 44
Our data also demonstrate that PKA inhibition abolishes basal KATP activity (Figure 3). Further, our work indicates that basal PKA activity contributes to the enhanced sensitivity of MMA to the KATP channel opener cromakalim compared to cerebral arteries (Figure 4). A number of factors may contribute to PKA-mediated enhanced KATP activity present in MMA. Activation of KATP by PKA is likely caused by direct PKA-mediated channel phosphorylation, with subsequent channel dephosphorylation by protein phosphatase 1 (PP1) and/or protein phosphatase 2B (PP2B) terminating channel activity. 45 Thus, greater PKA expression/activity, or lesser PP1/PP2B expression/activity in MMA relative to cerebral artery myocytes would be expected to promote enhanced KATP in MMA. Similarly, it is possible that activity of phosphodiesterase 3 (PDE3) may be less in MMA smooth muscle myocytes. PDE3 is the dominant isoform in vascular smooth muscle responsible for metabolizing and inactivating cAMP. 46 It is also widely appreciated that cell signaling pathways frequently operate within defined microdomains. Proteins such as the family of A-kinase anchoring proteins (AKAPs) play an integral role in the physical assembly of many enzyme/substrate signaling complexes, enabling the phosphorylation and de-phosphorylation of specific plasma membrane targets such as ion channels. 47 Relevant to our current work, Hayabuchi et al. 48 demonstrated the requirement of AKAPs for PKA-dependent basal KATP channel currents in rat mesenteric artery myocytes. 48 It is also conceivable that ongoing neurotransmitter release (e.g. CGRP, PACAP, etc.) could contribute to basal KATP channel activity in isolated MMA arteries. In humans, the MMA is primarily innervated by the sympathetic nervous system, with a lower density of cholinergic and sensory neurons, which immunostain positively for the vasodilatory peptides, CGRP, VIP, and Substance P. 49 Recently, our immunocytochemical studies have demonstrated prevalent dual localization of CGRP and PACAP in perivascular nerves of the MMA in rat. However, we feel ongoing neurotransmitter release is unlikely to account for basal KATP activity, as the PACAP receptor antagonist, PACAP6-8, alone did not alter MMA diameter ex vivo 30 and the basal KATP currents measured by Hayabuchi et al. 48 were obtained from isolated mesenteric artery myocytes. It is also possible that constitutive Gs-protein-coupled membrane receptor activity in the absence of receptor ligands could induce KATP channel activity in MMA and that differences in KATP channel structure/function due to differences in splice variants or post-translational modifications may exist between KATP channels present in MMA and cerebral artery myocytes. Future studies are required to dissect the impact of microdomain structure/composition and the role of constitutive Gs-protein-coupled membrane receptor activity on this basal PKA-mediated KATP activation.
The contribution of meningeal artery dilation in migraine headache is controversial13,14,50 and beyond the scope of the present work. A magnetic resonance angiography study of humans undergoing migraine attacks found no association between headache pain with changes in MMA diameter, casting doubt on the causal role of meningeal artery dilation in the development of migraine headache.13,14 However, our observations that KATP channel activity uniquely tunes the diameter of MMAs are intriguing in light of the work of others implicating KATP channels and the meningeal vasculature in the development of headache pain.25,26,28,51 For example, administration of the neuropeptides CGRP and PACAP—both implicated in migraine pathophysiology and known activators of vascular KATP channels—evokes both MMA dilation and headache in healthy volunteers.7,9,26,28 Similarly, the use of synthetic KATP channel openers such as levcromakalim has been associated with a high incidence of headache during clinical trials.15,16,28 The present work indicates that ongoing PKA activity in MMA smooth muscle underlies the heightened sensitivity of this vascular bed to KATP modulating compounds.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Institutes of Health P01 HL095488, P01 HL095488 03S1, P30 RR032135, P30 GM103498 and S10 OD 010583; the American Heart Association 14SDG20150027 and 17GRNT33700280; the Totman Medical Research Trust and the Peter Martin Brain Aneurysm Endowment.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
AUS, MK and JEB contributed to the conception and design of this research, data acquisition, data analysis and interpretation, and preparing the manuscript. GCW contributed to the conception and design of this research, data analysis and interpretation, and preparing the manuscript.