
Abstract
Acid-sensing ion channels (ASICs) are ligand-gated cation channels that respond to acidic stimuli. They are expressed throughout the mammalian nervous system. In the peripheral nervous system, ASICs act as nociceptors, responding to the tissue acidosis that accompanies ischemic and inflammatory conditions. The function of ASICs in the central nervous system is not known. In this article, the authors present evidence that transient global ischemia induces ASIC 2a protein expression in neurons that survive ischemia. Western blot analysis with an anti-ASIC 2a antibody revealed up-regulation of an 80 kD protein in ischemic rat brain. Immunohistochemical analysis showed that ASIC 2a protein expression increased in neurons of the hippocampus and cortex. Klenow fragment-mediated labeling of DNA strand breaks determined that ASIC 2a induction did not occur in cells with detectable DNA damage. The current results suggest a possible role for ASICs in mediating a cellular response to ischemia.
Acid-sensing ion channels (ASICs) are members of the Deg/ENaC superfamily of amiloride-sensitive ion channels. Members of this family include the constitutively active epithelial sodium channels (ENaC) in mammals, the stretch activated degenerins (Mec-4, −10; Unc-105) in C. elegans, and the ligand-gated FMRF-amide channel in H. aspersa. Gated by the proton, ASICs are activated to conduct an inward directed cation current when extracellular pH decreases (Horisberger, 1998; Waldmann et al., 1999; Waldmann and Lazdunski, 1998).
To date, six ASIC subunits have been identified in mammalian systems. Four of these—ASIC 1a (Waldmann et al., 1997b), ASIC 1b (Chen et al., 1998), ASIC 2a (Price et al., 1996; Waldmann et al., 1996), and ASIC 3, Waldmann et al., 1997 a)—form functional channels with distinct kinetics and conductance properties, pH sensitivities, and expression patterns. A modulatory subunit—ASIC 2b (Lingueglia et al., 1997)—does not form a functional channel, but alters the properties of other subunits. The most recently cloned subunit—ASIC 4 (Akopian et al., 2000; Grunder et al., 2000)—not activated by any known ligand, may also play a modulatory role. Several subunits when coexpressed heterologously have been shown to associate into heteromultimeric channel complexes with properties distinct from those of homomultimeric channel complexes (Babinski et al., 2000; Bassilana et al., 1997; Lingueglia et al., 1997).
Acid-sensing ion channels are expressed throughout the mammalian nervous system. Those in sensory neurons in the periphery have been implicated in the perception of pain during tissue acidosis (Bevan and Yeats, 1991; Krishtal and Pidoplichko, 1981). The presence of ASICs in the brain suggests that these channels may have functions beyond nociception. Transcripts for ASIC 2a have been detected predominantly in the brain. When expressed in heterologous systems, the homomultimeric channel is activated half-maximally at pH0.5 = 4.4 and conducts a transient, sodium-selective current. Mutation of residue Gly 430 to a bulky amino acid increases pH0.5 to 6.7, abolishes inactivation, and causes cell death (Adams et al., 1998; Champigny et al., 1998; Waldmann et al., 1996). Mutation of the same residue in C. elegans degenerins causes neurodegeneration.
Because tissue acidosis accompanies ischemia, the authors hypothesize that ASICs may play a roll in mediating the cellular response to an ischemic insult. To test this hypothesis, Western blots and immunohistochemistry were used to assess the expression of ASIC 2a after global ischemia. The current results show that ASIC 2a expression increases in neurons of the cortex and hippocampus after global ischemia, but not in those neurons with detectable DNA damage.
MATERIALS AND METHODS
Ischemic model
Animal experiments complied with National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by a review board at Legacy Health System. Global ischemia was induced in adult male Sprague-Dawley rats as previously described (Pulsinelli et al., 1982; Simon et al., 1991). Briefly, vertebral arteries were coagulated, transected between the first and second cervical vertebrae, and ligated. Ischemia was induced for 15 minutes by occlusion of the common carotid artery. Brain temperature was monitored with an intraparenchymal electrode inserted into the striatum and maintained at 36°C with heating lamps. Rats were reperfused and killed 4, 8, 24, or 72 hours thereafter. In sham animals killed 6 hours after surgery, common carotid artery occlusion was omitted.
Western blotting
Hippocampi (n = 3 at each time point) were isolated from sham or ischemic brains, frozen in liquid nitrogen, and lysed in RIPA buffer (150 mmol/L NaCl, 1.0% Nonidet P-40, 0.5% DOC, 0.1% sodium dodecyl sulfate, 50 mmol/L Tris pH 8.0 with 1 μg/mL aprotinin and 100 μg/mL phenylmethylsulfonyl fluoride). Protein concentration was determined by Bio-Rad Protein Assay (Bio-Rad Laboratories, Hercules, CA, U.S.A.). Lysates (50 μg) were resolved by denaturing 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. Membranes were probed with primary affinity-purified rabbit polyclonal antibody against ASIC 2a (Chemicon International, Temecula, CA, U.S.A.), followed by secondary goat anti-rabbit horseradish peroxidase-conjugated antibody (Amersham Life Science, Piscataway, NJ, U.S.A.). Protein was visualized using NEN Western Blot Chemiluminescence Reagent (NEN Life Science Products, Boston, MA, U.S.A.). As controls for nonspecific binding, membranes were incubated in the absence of primary antibody or in primary antibody preabsorbed to peptide antigen.
Northern blotting
Total RNA was isolated from rat hippocampal tissue using the PolyATract System 1000 (Promega, Madison, WI, U.S.A.). Samples standardized to 20 μg were electrophoresed on a 1.2% agarose-formaldehyde gel and transferred to a Zeta-probe GT blotting membrane (BioRad). The membrane was prehybridized for 4 hours at 37°C, and then hybridized with a 32P-lableled rat ASIC2a cDNA probe for 24 hours at 42°C.
Immunohistochemistry
After perfusion with 0.9% saline then 4% paraformaldehyde in phosphate-buffered saline (PBS), ischemic or sham brains were embedded in paraffin, sectioned, deparaffinized with xylene, and rehydrated with ethanol. Staining was performed as described previously (Jin et al., 1996). Briefly, sections were incubated first in 1% H2O2 in PBS for 15 minutes to block exogenous horseradish peroxidase activity, then in blocking buffer (2% horse serum, 0.2% Triton X-100, 0.1% bovine serum albumin in PBS) for 1 hour at room temperature. Sections were probed with primary affinity-purified rabbit polyclonal antibody against ASIC 2a overnight at 4°C, washed with PBS, labeled with secondary biotinylated goat anti-rabbit IgG (Vectastatin Elite ABC; Vector, Burlingame, CA, U.S.A.), and processed according to the manufacturer's instructions with Vector ABC Kit (Vector). Control sections were processed in absence of primary antibody.
Triple-labeling fluorescence experiment
Brain sections were deparaffinized with xylene, rehydrated with ethanol, and treated with unmasking solution (Vector). After incubation in blocking buffer for 1 hour at room temperature, sections were incubated overnight at 4°C in blocking buffer containing 1:50 dilutions of 2 primary antibodies: affinity-purified rabbit polyclonal antibody against ASIC 2a and either mouse monoclonal antibody against glial fibrillary acidic protein for astrocytes (GFAP; Sigma Chemical, St. Louis, MO, U.S.A.) or mouse monoclonal antibody against neuron-specific nuclear protein (Neu N; Chemicon). Sections were washed in PBS, incubated in 1:200 dilutions of secondary fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit antibody (Vector) and rhodamine (TRITC)-conjugated rat-adsorbed donkey anti-mouse antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, U.S.A.), and counterstained with the nuclear dye Hoechst 33258 (2′-(4-hydroxyphenyl)-5-(4-methyl-1-piperazinyl)-2.5′-bi-1H-benzimidazole; Sigma; 20 μg/mL). A Nikon E800 epifluorescence microscope (Nikon, Melville, NY, U.S.A.) was used to detect fluorescence signals at the following excitation/emission wavelengths: 535/565 nm for rhodamine (red), 470/505 nm for FITC (green), and 360/400 for Hoechst 33258 (blue). Images were photographed with a Magnifire digital color camera (ChipCoolers, Warwick, RI, U.S.A.).
Klenow labeling assay
Brain sections were deparaffinized with xylene and rehydrated with ethanol. Immunohistochemistry was performed with ASIC 2a antibody as described above. DNA single and double strand breaks with protruding 5′termini were detected using the Klenow fragment of DNA polymerase I-mediated labeling technique. Sections were incubated for 1 hour at 37°C in PBS containing 10 μmol/L each dNTP, 10 μmol/L biotinylated-dATP, and 40 U/mL of E. coli Klenow fragment, or in the same reaction mix lacking enzyme to control for nonspecific labeling. Slides were washed twice in PBS to terminate the reaction, and then incubated for 1 hour at room temperature in rhodamine-avidin D (Vector) and in DAPI (4′,6-diamidino-2-phenylindole; Vector) to counterstain nuclei. Sections were washed in PBS and coverslipped with fluorescent mounting medium (Vector).
RESULTS
Global ischemia induces ASIC 2a expression in the hippocampus and cortex
Western blotting was used to assess the effect of global ischemia on ASIC 2a protein expression (Fig. 1). Lysates prepared from the hippocampi of sham and ischemic rats were probed with an antibody to ASIC 2a. Little protein was detected in sham tissue. An 80-kD band was up-regulated after ischemia. Expression was maximal at 8 hours and persisted at 72 hours.
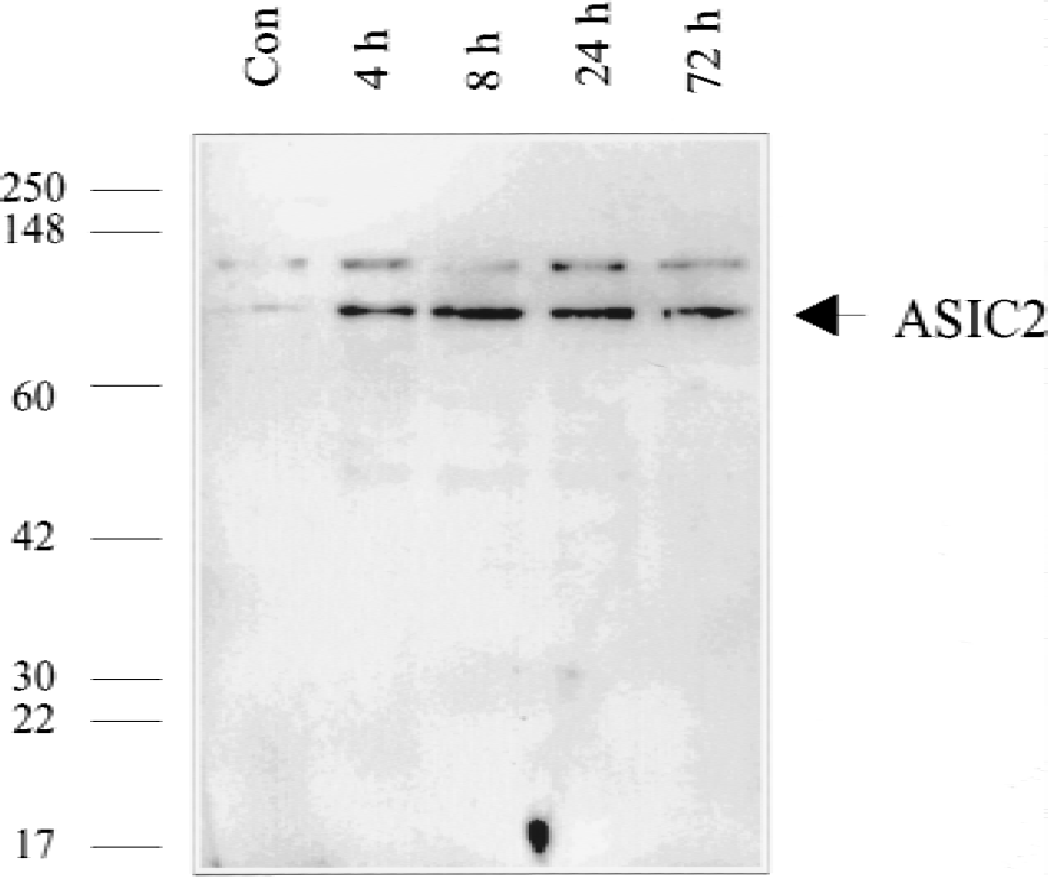
Western blot analysis of acid-sensing ion channel 2a (ASIC 2a) levels in lysates from the hippocampi of rats killed after 4, 8, 24, or 72 hours of reperfusion from global ischemia or 6 hours after a sham operation. An affinity-purified rabbit polyclonal antibody against ASIC 2a shown by manufacturer to detect a band of 80kD was used. ASIC 2a is undetectable in sham brain, but is up-regulated after global ischemia. Expression is maximal at 8 hours and persists at elevated levels at 72 hours.
Western blotting results were confirmed by Northern blot analysis of mRNA isolated from the hippocampi of sham and ischemic rats brains (Fig. 2). An ASIC2a cDNA probe detected a transcript whose levels increased within 4 hours of ischemia and diminished within 24 hours.
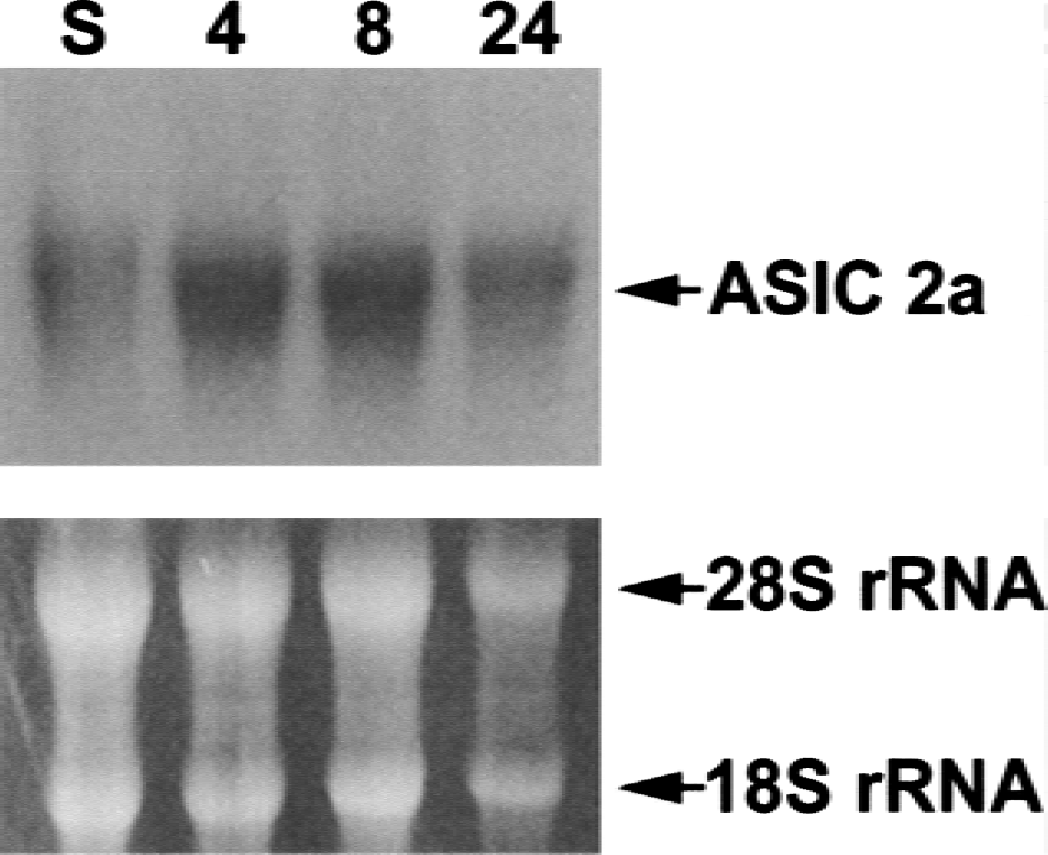
Northern blot analysis of mRNA isolated from sham and ischemic rat hippocampal tissue. Hybridization with an acid-sensing ion channel 2a (ASIC 2a) cDNA probe reveals an increase in the levels transcript after ischemia.
To examine the expression pattern of ASIC 2a in specific regions of the brain, immunohistochemistry was performed with the same antibody (Fig. 3). In sham tissue, a low level of staining was observed in the CA1 (Fig. 3A) and CA3 (Fig. 3B) regions of the hippocampus. In ischemic tissue, staining appeared more vivid and widespread in these regions. In the cortex (Fig. 3C), a single cell was boldly labeled in sham tissue, whereas numerous cells were boldly labeled 8 hours after ischemia. At 24 hours after ischemia, antibody also labeled nerve cell processes.
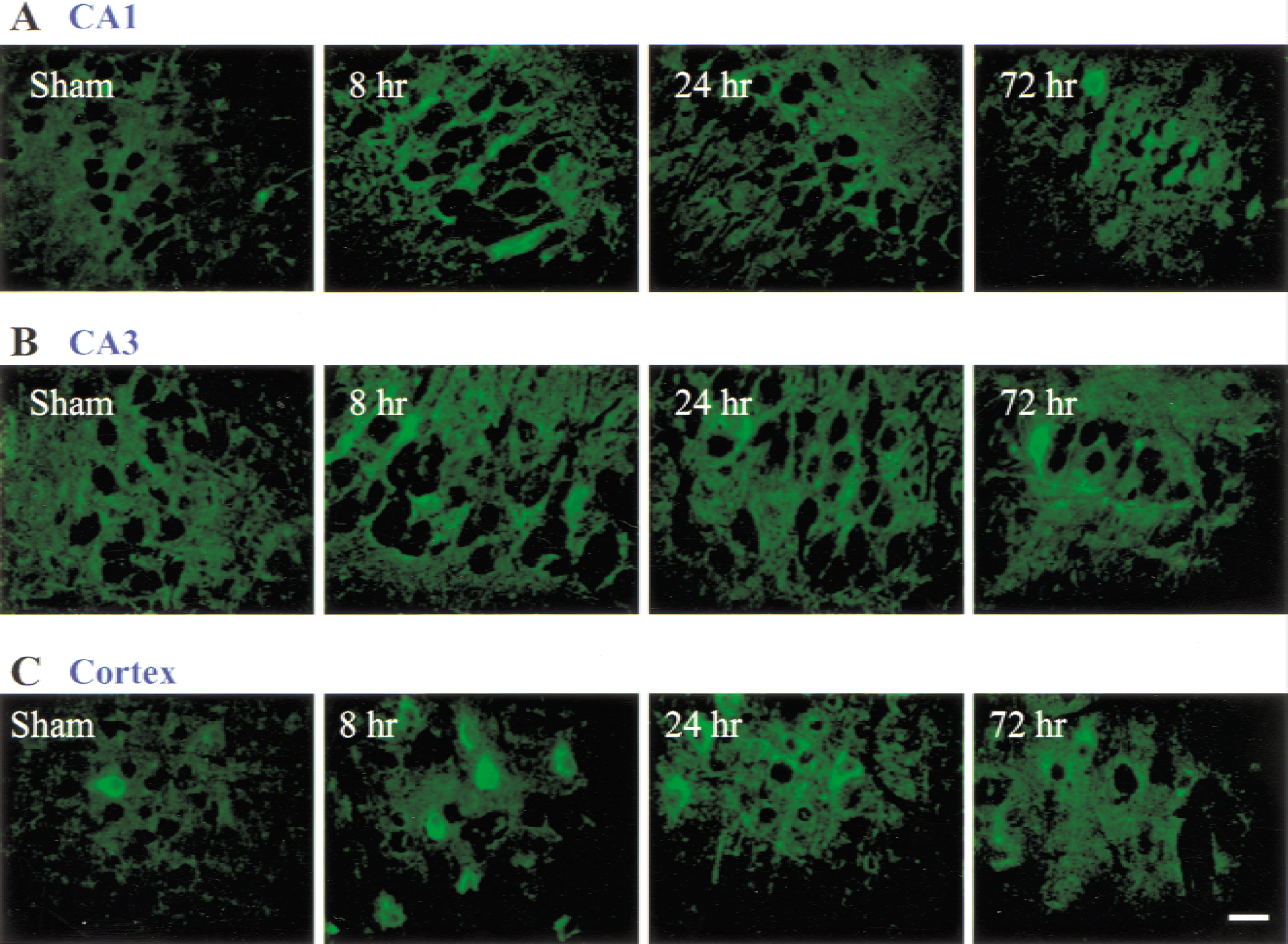
Immunohistochemical analysis of acid-sensing ion channel 2a (ASIC 2a) expression in the CA1
Global ischemia induces ASIC 2a expression in neurons but not in astrocytes
To determine the cellular distribution of ASIC 2a in ischemic tissue, triple fluorescence labeling experiments were performed with nuclear dye Hoechst 33258 and antibodies to ASIC2a and neuronal marker Neu N (Fig. 4A) or ASIC 2a and astrocytic marker GFAP (Fig. 4B). ASIC 2a and Neu N immunoreactivity colocalized, indicating that global ischemia induces ASIC 2a expression in neurons. ASIC 2a and GFAP immunoreactivity did not colocalize.
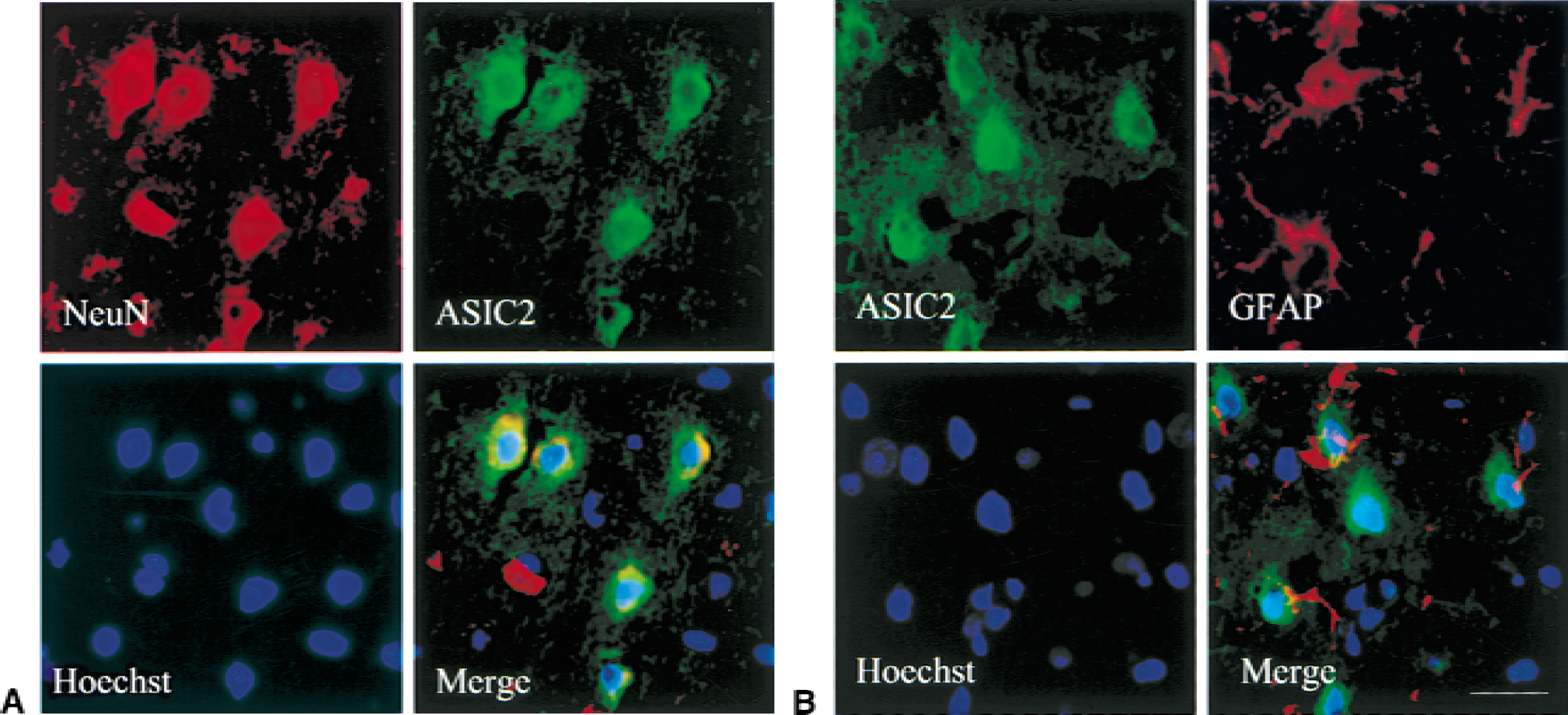
Triple-labeling fluorescence study to identify what cell types express ASIC 2a. Staining was performed on paraffin-embedded sections of brain isolated from rats killed 8 hours after global ischemia.
Although Neu N refers to neuron-specific nuclear protein, the subcellular localization of Neu N immunostaining varies widely, depending on the condition and treatment of the tissue sections. However, the antibody consistently labels neurons and not glia. Neu N immunostaining in the current study appeared in the cytoplasm, as has been observed in previous studies (Ho and Blum, 1998).
ASIC 2a expression does not increase in cells with DNA damage
To determine if up-regulation of ASIC 2a in selectively vulnerable regions of ischemic brain occurs in cells destined to survive or apoptose, the authors double-labeled tissue for ASIC 2a and DNA strands breaks (Fig. 5). Klenow fragment-mediated labeling of DNA single and double strand breaks with protruding 5′ termini revealed widespread DNA damage in cells of the CA1 sector 72 hours after reperfusion from ischemia. The antibody to ASIC 2a labeled scattered cells, but did not colocalize to cells with DNA damage.
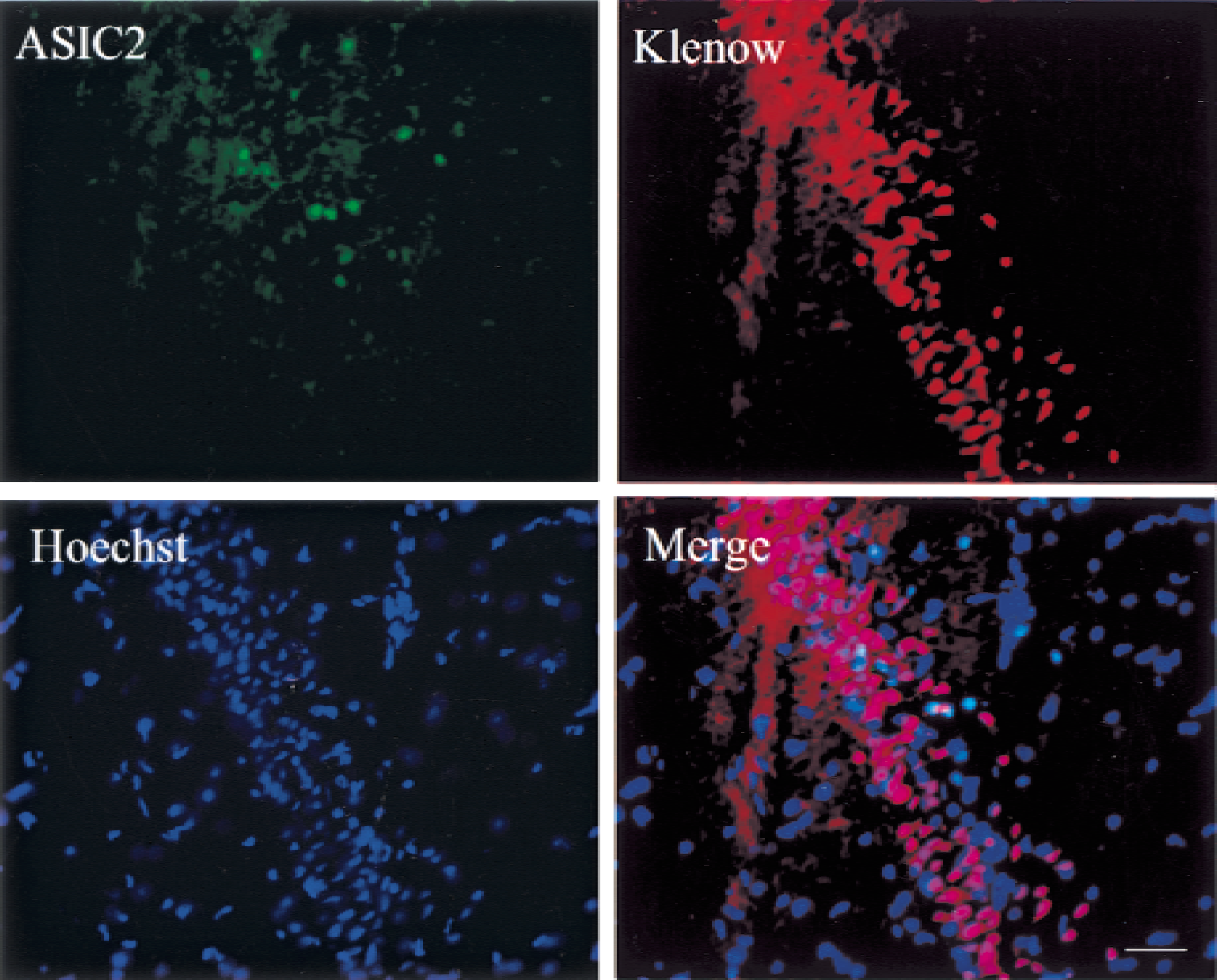
Double-labeling experiment to determine if acid-sensing ion channel 2a (ASIC 2a) expression occurs in cells with DNA damage. Staining was performed on paraffin-embedded sections of ischemic brain. DNA damage was identified by Klenow fragmentmediated labeling of single-and doublestrand breaks with 5′ protruding termini. Cells expressing ASIC 2a (green) are present in the CA1. Widespread DNA damage (red) is evident. ASIC 2a is not expressed in cells with DNA damage (merge). Scale bar = 200 μm.
DISCUSSION
A decrease in pH is a central metabolic consequence of ischemia (Siesjo et al., 1996). The numerous effects of acidosis in ischemic brain have received extensive study. The discovery that protons gate a new class of ion channels that are present in the brain suggests that the brain's response to ischemia may be mediated by previously unknown means. To explore this possibility, the authors used Western blotting and immunohistochemistry to assess the expression of ASIC 2a protein in normal and ischemic rat brains.
The antibody used in these experiments was an affinity-purified rabbit polyclonal antibody raised against an antigenic peptide corresponding to residues 2–18 of ASIC 2a. This sequence is unique to ASIC 2a and has no significant homology in other ASIC subunits, including the splice variant ASIC 2b.
The molecular weight predicted from the amino acid sequence of ASIC 2a is 57.7 kD. In Western blots of rat hippocampal lysates the authors detected an 80-kD band. The antibody manufacturer also detects an 80-kD band in blots of rat brain membranes. Glycosylation, for which there are several potential sites, might account for the difference between the predicted and observed sizes of the ASIC 2a protein. The higher molecular weight band observed in the current study might also be attributable to glycosylation or to aggregation of the reduced polypeptide.
Because the ASIC 2a antibody has been only partially characterized, protein results were verified at the RNA level. Northern blot analysis of hippocampal tissue showed that the level of ASIC 2a transcript increased within 4 hours of ischemia, remained elevated at 8 hours, and diminished thereafter (Fig. 2). This was entirely consistent with the changes in protein expression detected by Western blot. Immunohistochemistry results further supported findings from Western and Northern blot analyses (Fig. 3A and 3B).
Immunohistochemistry also showed a marked increase in the expression of ASIC 2a in cortical neurons (Fig. 3C). The cortex and the hippocampus are particularly susceptible to ischemic injury. Neurons in these selectively vulnerable regions of the brain develop DNA damage and die within a few days after transient global ischemia. In previous studies of ischemia, the current authors and others have observed induction of the death promoter protein Bax in neurons that die (Chen et al., 1996; Krajewski et al., 1995), and also have observed induction of the death suppressor proteins Bcl-2 and Bcl-w in neurons that survive (Chen et al., 1995; Yan et al., 2000).
The induction of ASIC 2a in selectively vulnerable regions suggested that the channel might modulate cell death or survival. To determine the fate of those cells expressing ASIC 2a, single-and double-stranded DNA breaks were used as a marker of an injured cell (Jin et al., 1999). Double labeling of CA1 neurons for DNA strand breaks and ASIC 2a expression (Fig. 5) revealed that only cells without DNA damage express ASIC 2a. This expression pattern is similar to the expression pattern of antiapoptotic proteins Bcl-2 and Bcl-w and implicates a protective role for the channel. However, cells severely damaged by an ischemic insult have a diminished capacity for protein synthesis because of a depletion of adenosine triphosphate. The absence of ASIC 2a from damaged cells might be attributable to translational block.
Within 1 minute of global ischemia, extracellular pH decreases from 7.2 (Simon et al., 1985) to 6.5 (Tombaugh and Sapolsky, 1993), a level sufficient to activate ASIC 1a and ASIC1a/ASIC 2b comprising channels (Lingueglia et al., 1997). The up-regulation of ASIC 2a in ischemic brain in response to prolonged acidosis may alter existing channel composition. The effect, when one considers studies performed in heterologous systems, may be to make those cells expressing ASIC 2a less responsive to acidosis. ASIC 1a channels show pH of half-maximal activation (pH0.5) at 6.2, whereas ASIC 1a/ASIC 2a channels show pH0.5 at 4.8 (Bassilana et al., 1997), and ASIC 2a channels show pH0.5 at 4.35 (Champigny et al., 1998). Association with ASIC 2a also slows the inactivation kinetics of ASIC 1a and increases its selectivity for Na+ over Ca2+ (Bassilana et al., 1997). This might be significant in the context of ischemia, in which intracellular Ca2+ overload has been implicated as a primary mechanism of neuronal injury (Kristian and Siesjo, 1998; Tymianski and Tator, 1996). However, little is known about channel composition and regulation in in vivo systems. Phosphorylation, interaction with other molecules, and disruption of normal ion gradients might alter channel properties. Hence, considerations as to the effect of up-regulating the ASIC 2a subunit remain speculative until the ASIC system has been more completely characterized. It will be informative to compare in normal and ischemic brains subunit expression and to assess channel composition with antibodies specific to other ASICs.
Whether expression of ASIC 2a provides a defense against the damaging effects of ischemic acidosis remains unresolved. Acidosis is a prominent event in ischemia, but one whose impact on cell survival is complicated and controversial (Tombaugh and Sapolsky, 1993).
Severe acidosis (pH 5.5 to 6.0) after prolonged ischemia causes pan necrosis, but mild acidosis after transient ischemia appears not to contribute to selective neuronal necrosis. Mild acidosis may, in fact, be neuroprotective (Simon et al., 1993; Tombaugh and Sapolsky, 1993).
Extracellular pH of 6.5 to 7.0 suppresses NMDA channel currents, thereby attenuating the injurious effects of glutamate (Tang et al., 1990). However, pH below 6.5 impairs glial glutamate uptake and abolishes the protective effect of acidosis (Swanson et al., 1995). The new effect of acidosis described in the current study provides another possible point from which to explore the impact of acidosis on the outcome of ischemia.