
Abstract
Nitrones (e.g. α-phenyl-N-tert-butyl nitrone; PBN) are cerebroprotective in experimental stroke. Free radical trapping is their proposed mechanism. As PBN has low radical trapping potency, we tested Sgk1 induction as another possible mechanism. PBN was injected (100 mg/kg, i.p.) into adult male rats and mice. Sgk1 was quantified in cerebral tissue by microarray, quantitative RT-PCR and western analyses. Sgk1+/+ and Sgk1−/− mice were randomized to receive PBN or saline immediately following transient (60 min) occlusion of the middle cerebral artery. Neurological deficit was measured at 24 h and 48 h and infarct volume at 48 h post-occlusion. Following systemic PBN administration, rapid induction of Sgk1 was detected by microarray (at 4 h) and confirmed by RT-PCR and phosphorylation of the Sgk1-specific substrate NDRG1 (at 6 h). PBN-treated Sgk1+/+ mice had lower neurological deficit (p < 0.01) and infarct volume (p < 0.01) than saline-treated Sgk1+/+ mice. PBN-treated Sgk1−/− mice did not differ from saline-treated Sgk1−/− mice. Saline-treated Sgk1−/− and Sgk1+/+ mice did not differ. Brain Sgk3:Sgk1 mRNA ratio was 1.0:10.6 in Sgk1+/+ mice. Sgk3 was not augmented in Sgk1−/− mice. We conclude that acute systemic treatment with PBN induces Sgk1 in brain tissue. Sgk1 may play a part in PBN-dependent actions in acute brain ischemia.
Introduction
Ischemic stroke remains a major cause of mortality and morbidity worldwide, with few therapeutic options.1,2 The thrombolytic agent recombinant tissue plasminogen activator (rt-PA) is the only widely approved drug available.2,3 A substantial proportion of patients do not meet inclusion criteria for rt-PA, and the drug carries an increased risk of hemorrhagic transformation.2,3 Thus, additional therapeutic approaches, complementary to rt-PA, would have substantial clinical benefit.
Nitrone compounds such as α-phenyl-N-tert-butyl nitrone (PBN) and its congener 2,4-disulfophenyl-N-tert-butyl nitrone (NXY-059) are protective in rodent and primate models of stroke.4–9 PBN is a small, water-soluble molecule (Figure 1) with substantial brain penetration. Nitrone compounds “trap” and stabilize reactive free radical species that are a pathogenic factor in ischemia-induced cell injury.
1
As PBN (like NXY-059) is in fact a relatively weak free radical trapping agent,10–12 we hypothesized that some other mechanism might be involved in the protective action.
Chemical induction of Sgk1 in brain tissue following systemic treatment with PBN. (a, b) Systemic injection of PBN (100 mg/kg i.p.) induced brain Sgk1. (a) Microarray data show up-regulation of Sgk1 expression in rat brain 4 h after injection of PBN, relative to saline-injected rats (n = 5, 5, horizontal bars show the group mean). Inset: structure of PBN. (b) Quantitative RT-PCR data confirmed up-regulation of Sgk1 expression in rat brain (relative to the housekeeping gene 18S). (c,d) Systemic injection of PBN augmented brain Sgk1 activity in mice. (c) Representative blots of mouse brain lysates, immunoblotted using the p3-NDRG1 antibody raised against the phosphorylated decapeptide repeat in NDRG1 or total NDRG1. Wild-type mice were injected either with PBN (100 mg/kg i.p. “P”) or with saline vehicle (“V”) and brains sampled at 2, 4 or 6 h post-injection. (d) Semi-quantitative densitometry showing phosphorylation of the Sgk1-specific p3-Thr motif in NDRG1. Phosphorylation was augmented at 6 h post-injection of PBN, relative to vehicle-injected mice. Symbols show mean ± 1SD. *p < 0.05, **p < 0.01, ***p < 0.001.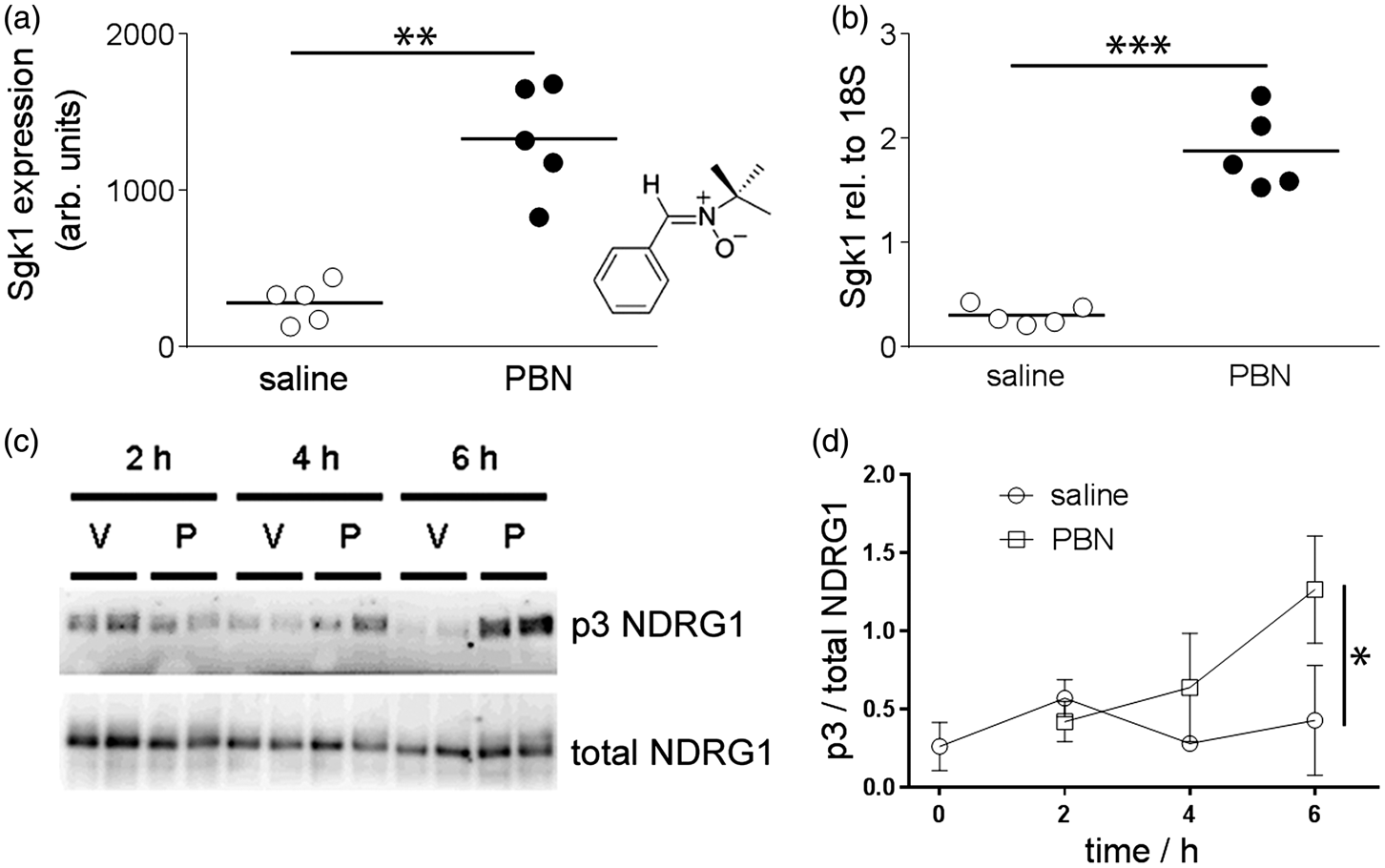
Serum and glucocorticoid inducible kinase-1 (Sgk1; MW 50 KDa) is a cytoplasmic serine-threonine kinase associated with cell survival.13,14 Developmental studies suggest an important role for Sgk1 in vasculogenesis 15 and organismal lifespan. 16 A homolog of Akt, Sgk1 is expressed in brain tissue of rodents,13,17,18 primates 19 and humans. 20 In vitro, Sgk1 protects neuronal primary cultures from apoptotic stimuli, the mechanism being (at least in part) sequestration of the Forkhead-like transcription factor FOXO3a.21,22 Further evidence for the pro-survival role of Sgk1 in brain tissue comes from studies of behavioral learning, 23 brain tissue biochemistry20,21,24 and neuropathological examination of brains of older people. 20 Others have demonstrated Sgk1 expression and function in brain vascular tissue 25 and in oligodendrocytes. 26
We have previously reported differences in Sgk1 expression and activity in brains of older people with Alzheimer’s disease, relative to age-matched control brains. 20 Here we tested whether acute PBN treatment changes Sgk1 expression in rodent brain tissue and whether Sgk1 participates in the brain protective action of PBN in experimental focal ischaemia.
Materials and methods
Animals
Adult male Wistar rats (345–360 g) were obtained from Harlan Laboratories, UK. Sgk1−/− mice were produced as described 27 (see Supplementary Methods for further details). As homozygous Sgk1−/− offspring were rare on heterozygote matings, sister colonies of Sgk1−/− and Sgk1+/+ strains were established. Each litter was genotyped to confirm colony integrity (see Supplementary Figure S1). Animals were housed under controlled environmental conditions (temperature 21℃, 12-h light/dark cycles 07.00–19.00) within St George’s Biological Research Facility. They were provided ad libitum with water and standard chow. Young adult male mice aged 8–10 weeks were used for all experiments. For PBN injections, PBN (Sigma-Aldrich, Poole, UK) was dissolved in 0.9% saline at 20 mg/ml (2% w/v; equivalent to 110 mM) on the day of use. Animals were injected with PBN (100 mg/kg i.p.) or an equivalent volume of saline.
For generation of Sgk1−/− mice, all experimental protocols were approved by the Institutional Animal Use and Care Committees of Dartmouth Medical School, Lebanon, NH, and all procedures adhered to the American Physiological Society’s “Guiding Principles in the Care and Use of Animals”. 27 At the St George’s site, all procedures were approved by the St George’s University of London Ethical Review Board for Animal Use and were performed in accordance with the Animals (Scientific Procedures) Act 1986 and in compliance with EU Directive 63-2010. The data are reported in accord with ARRIVE guidelines. 28
Middle cerebral artery occlusion
Transient middle cerebral artery occlusion (MCAo; 60-min duration) 29 was carried out in young adult male Sgk1+/+ and Sgk1−/− mice using a filament method. General anesthesia was induced with isoflurane (2%) in 1:1 Oxygen:N2O and maintained with 1.5% isoflurane. Within a litter, male mice were randomized using a random number table to receive PBN or saline vehicle. Injections were administered (PBN or an equivalent volume of saline vehicle) 5 min after induction of MCAo, by an individual blind to treatment. In “sham” control animals, the common carotid artery was exposed but not ligated (n = 5). Neurological deficit was assessed by an individual blind to treatment on a 28-point score (including body symmetry, gait, ability to climb, circling behaviour and whisker responsiveness). 30 Neurological score was measured prior to MCAo and then at 24 h and 48 h post-occlusion. At 48 h post MCAo, animals were killed by schedule 1 approved method (cervical dislocation). Brains were rapidly removed and sliced at 1 mm in a matrix. Infarct volume was estimated using TTC vital stain (direct method) 29 by an individual blind to treatment and genotype (example in Figure S1). Animals subjected to sham surgery exhibited no behavioural deficit and no detectable lesion (n = 5; not shown).
Microarray analysis
Male Wistar rats were injected at time 0 min and 120 min with either saline or PBN (n = 5 per group). Two doses were used to allow for the plasma half-life of PBN (terminal half-life in rat ∼2 h). 31 At 240 min, animals were killed by cervical dislocation, brains rapidly removed, cerebella discarded and cerebral hemispheres frozen in liquid nitrogen. Whole hemispheres were homogenized in Trizol, total RNA extracted and cleaned using RNeasy columns (QIAGEN GmbH, Hilden, Germany). Individual samples were labelled, hybridized to U34A genechip microarrays, washed and scanned in accordance with the manufacturer’s protocols (Affymetrix UK Ltd., High Wycombe, UK). The Sgk sequences (probeset L01624, ID 81963) on this array were specific for rat Sgk1.
Quantitative RT-PCR
For rat brain lysates, total RNA was converted to cDNA using random primers and Life Technologies Superscript II. The cDNA was diluted (5 ng per µl) and Taqman reactions performed with 20 ng cDNA per well under standard conditions in 25 µl reaction volume. Taqman probesets for rat Sgk1 and 18S ribosomal RNA were obtained from Applied Biosystems, Paisley, UK (Rn00570285_m1 and #4310893E, respectively). For mouse brain lysates, total RNA was isolated from cerebra of young adult male mice from the Sgk1+/+ and Sgk1−/− colonies. Whole cerebral hemispheres were homogenized and passed through QIAshredder™ columns (QIAGEN GmbH, Hilden, Germany). RNA was extracted using an RNeasy® Mini kit (QIAGEN GmbH, Hilden, Germany). Total RNA (500 ng) was converted to cDNA using a Precision Nanoscript reverse transcription kit with random primers (Primerdesign Ltd, Southampton, UK). Amplification reactions were performed in a 20-µl reaction volume containing 250 ng cDNA for Sgk3 and Sgk1 assays, and 25 ng cDNA for the housekeeping gene Gapdh, using Taqman® Universal mastermix with uracil-N-glycosylase (Applied Biosystems, Paisley, UK). Taqman® expression assays for mouse Sgk3 (assay # Mm01227735_m1), Sgk1 (Mm00441387_g1) and gapdh (Mm99999915_g1) were purchased from Applied Biosystems-Life Technologies, Paisley, UK.
Antibodies
Polyclonal anti-NDRG1 was generated in sheep against full length human NDRG1 and antigen-affinity purified.32,33 A sheep antibody that recognizes NDRG1 phosphorylated at Thr346, Thr356 and Thr366 (p3-NDRG1) was raised against the nonapeptide RSRSHpTSEG, whose sequence is common to all three phosphorylation sites, and was antigen-affinity purified. 32 Rabbit polyclonal anti-Sgk1 (S-5188) was from Sigma-Aldrich (Poole, Dorset, UK), characterized in our previous report. 20 Rabbit polyclonal anti-GFAP (Z-0334) and mouse monoclonal anti-CD31 (clone JC70A) were from Dako (Ely, Cambs., UK).
Western blotting
Male Sgk1+/+ mice were injected at time 0 min and 120 min with either saline or PBN (100 mg/kg i.p.). At 2, 4 or 6 h (n = 2–4 per time-point) after the first injection, animals were killed by cervical dislocation, the brain rapidly removed from the skull and the cerebral hemispheres frozen in isopentane within liquid nitrogen. Samples were stored at −80℃ for subsequent lysis. For Western blots, tissues were lysed and proteins separated by SDS-PAGE as described previously.20,33 Lysis buffer contained (mM): 50 Tris-HCl pH 7.4, 150 NaCl, 50 NaF, 2 EDTA, 5 EGTA, 10% v/v glycerol, 1% v/v Triton X-100, supplemented just before use with 1 mM PMSF and 1% v/v protease inhibitory cocktail (P-2714, Sigma-Aldrich, Poole, UK). Lysates were run on SDS-PAGE gels and transferred to PVDF (0.45 µm; Millipore, UK). Membranes were blocked for 60 min with Tris-buffered saline with 0.1% v/v Tween-20 (TBS-T) containing virtually fat-free milk powder (5% w/v; TBS-TM). Identical blots were exposed to NDRG1 (1:6,000) or p3-NDRG1 antisera (1:3,000), diluted in TBS-TM and applied overnight at 4℃, the latter in the presence of 0.5 mg/L dephosphorylated immunogenic peptide, to reduce non-specific binding to non-phosphorylated NDRG1. 32 Rabbit anti-sheep IgG secondary antibody (1:10,000, Sigma-Aldrich, Poole, UK) was applied for 60 min at room temperature. Proteins were visualized by chemiluminescence (ECL+ kit, GE Healthcare, Amersham UK) and semi-quantitative densitometry analysis carried out using ImageJ software (http://rsb.info.nih.gov/ij/). The ratio of p3-NDRG1/total NDRG1 was determined for each sample.
Multiple labelling immunofluorescence
Coronal brain sections were cut on a cryostat (15 µm thickness) and fixed in 100% ethanol at −20℃ for 20 min then washed in PBS-T. Non-specific binding was blocked by incubation with 3 % w/v BSA (Jackson Immunochemicals) in PBS-T for 1 hour at room temperature. Sections were then incubated overnight with primary antibodies in a humidified chamber at 4℃.
Primary antibodies were diluted in 3% w/v BSA in PBS-T: Sgk1 (diluted 1:200), GFAP (1:200), CD31 (1:30). Sections were incubated with appropriate secondary antibodies conjugated to Alexa488 or Alexa546, diluted 1:200 in 3% BSA in PBS-T at room temperature for 1 h. After nuclear labeling with DAPI (30 min, 0.3 µM in PBS-T), sections were mounted and photographed with a Zeiss LSM 510Meta confocal microscope. Red fluorescence was viewed with 543 nm excitation and 545–575 nm emission bandwidth. Green fluorescence was viewed with 488 nm excitation and 505–530 nm emission bandwidth. DAPI was viewed with 364 nm excitation and 385–470 nm emission bandwidth. Neighboring sections were processed identically in parallel, but with omission of primary antibodies.
Human tissue
Immunohistochemical labelling for Sgk1 was examined in human brain tissue from individuals with acute ischemic lesions (death within 2–6 days, n = 6, 4 M/2 F, age 31–71 y). All human tissue was from the Oxford Brain Collection, John Radcliffe Hospital, Headington, Oxford. All tissue samples were donated following written informed consent by donors or their next of kin. This study had approval of Local Research Ethics Committees and the UK National Research Ethics Service.
For immunohistochemistry, paraffin wax-embedded sections (6 µm) were de-waxed and processed for standard immunohistochemical staining. 20 After exposure to H2O2 (3 % v/v) to abolish endogenous peroxidase activity, sections underwent high-pressure heat-induced antigen retrieval (30 s, 120℃) in Tris-citrate-EDTA buffer, pH 7.8. Non-specific binding was blocked with bovine serum albumin (BSA, Jackson Immunochemicals; 3% w/v) in phosphate-buffered saline containing 0.1% v/v Triton-X100 (PBT). Sgk1 antibody (1:20,000) or MBP (1:1000) in PBT containing 3% BSA were applied to tissue sections overnight (4℃). We have previously characterized the Sgk1 antibody by immune-depletion. 20 Peroxidase-conjugated secondary reagent (Envision kit®, Dako-Cytomation, Ely, UK) was applied for 60 min at room temperature. Antibody labeling was visualized using diaminobenzidine (DAB) chromagen, enhanced with copper II sulphate, and nuclei counterstained with Mayer’s haematoxylin. Sections were examined on a Zeiss Axioplan-2 microscope driven by Axiovision software (version 4.7).
Power calculation and statistical analysis
Based on our own and others’ previous data, we conservatively assumed a σ/mean ratio of 0.25 for studies of mRNA or protein abundance in native tissue34–36 and 0.35 for behavioural tests.29,37 From a standard power calculation, to detect differences between groups of at least 50% (α = 0.05, β = 0.80), we estimated a required group size of at least four/group for biochemical assays and eight/group for behavioural tests.
Differences in continuous variables (mRNA, protein abundance, infarct volume) were analyzed using Student’s t-test or ANOVA as appropriate. A p-value less than 0.05 was considered significant. For differences in categorical variables (neurological scores), an equivalent non-parametric test (Kruskal–Wallis) was used. The experimental unit was considered to be a single animal. Post-surgical deaths (n = 4 in total) were excluded from statistical analyses.
Results
Four hours after systemic administration of PBN or saline vehicle to adult rats, microarray analysis revealed altered expression by a factor of at least 2.0 in 11 genes (not shown) in brain tissue of PBN-treated rats, relative to saline-treated animals. The greatest change was in Sgk1 (4.7-fold up-regulation; p = 0.0015, Figure 1). Subsequent quantitation with RT-PCR confirmed significant up-regulation of Sgk1 in brain tissue of PBN-treated rats (Figure 1; 6.1-fold increase in Sgk1, normalised with respect to the housekeeper 18 S; p = 0.00046).
As an assay of Sgk1 enzymatic activity, the degree of phosphorylation of an Sgk1-specific triple phospho-Thr motif in the cytoplasmic protein NDRG1 was quantified (p3-NDRG1).20,32 Over a 6-h time course, the fraction of p3-NDRG1, relative to total NDRG1, in mouse brain lysates was greater in PBN-injected mice than in saline-injected mice (p = 0.049 relative to drug treatment, p = 0.088 relative to time, two-way ANOVA; Figure 1). At 6 h post injection, the p3-NDRG1/total NDRG1 fraction was 2.97 fold greater in PBN-injected than in saline-injected mice (p = 0.014). In brain lysates from Sgk1−/− mice, the degree of p3-NDRG1 phosphorylation was very low (not shown) indicating that the assay is Sgk1-specific.32,38 These data indicate an increase in Sgk1 expression and enzymatic activity in cortical tissue 4–6 h after a systemic injection of PBN.
To test whether Sgk1 induction is required for PBN-dependent brain protection, Sgk1+/+ and Sgk1−/− mice were subjected to transient MCAo (60 min). PBN improved behavioural outcome, relative to saline, in Sgk1+/+ mice at 48 h post-MCAo but not in Sgk1−/− mice (p = 0.007, p = 0.497 respectively; Figure 2). PBN reduced lesion size at 48 h, relative to saline injection, in Sgk1+/+ mice (0.16-fold reduction; Figure 2) but not in Sgk1−/− mice, (p = 0.0025 for drug effect, p = 0.72 for genotype; Figure 2). Saline-treated Sgk1−/− mice were not significantly different from saline-treated Sgk1+/+ mice in terms of behavioural outcome (p = 0.889) or infarct size (p = 0.217, Figure 2). Four post-surgical mortalities were excluded from these analyses (1 Sgk1+/+ saline-treated, 2 Sgk1+/+ PBN-treated, 1 Sgk1−/− saline-treated; 8 % overall mortality). All four animals failed to recover consciousness following general anaesthesia (determined by paw pinch and whisker stimulation) and were humanely terminated within the first few hours after MCAo surgery. These findings support the cerebro-protective action of PBN in wild-type mice and suggest that this action may be lost in mice lacking Sgk1.
Effects of transient focal ischemia and acute PBN treatment in Sgk1+/+ and Sgk1−/− mice.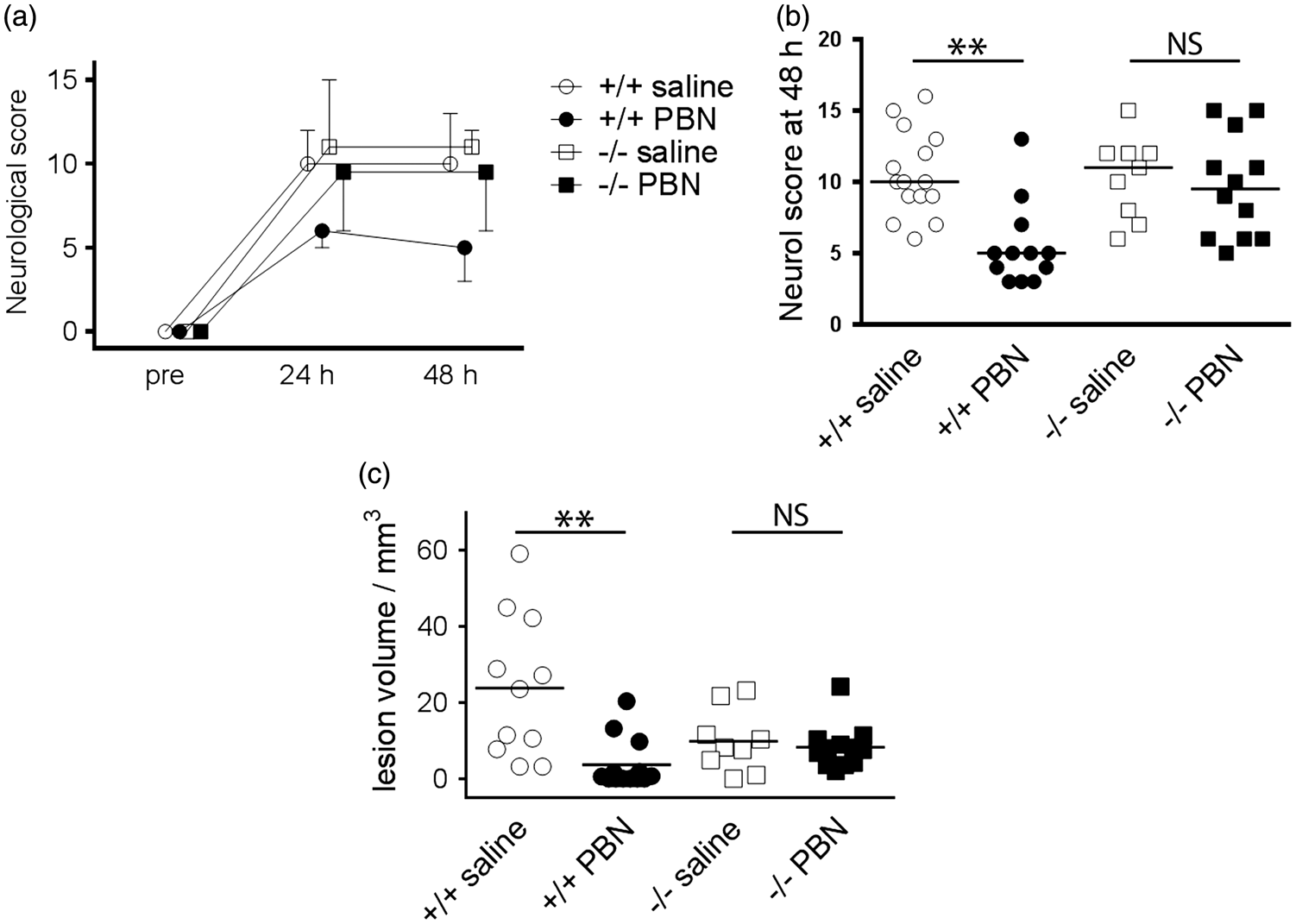
Immunohistochemical labelling of Sgk1 in uninjured mouse cortical tissue showed widespread cellular labelling, with a primarily nuclear location (Figure 3(a)). Cortical neurones were strongly positive for Sgk1, as in our previous studies in human cortical tissue.
20
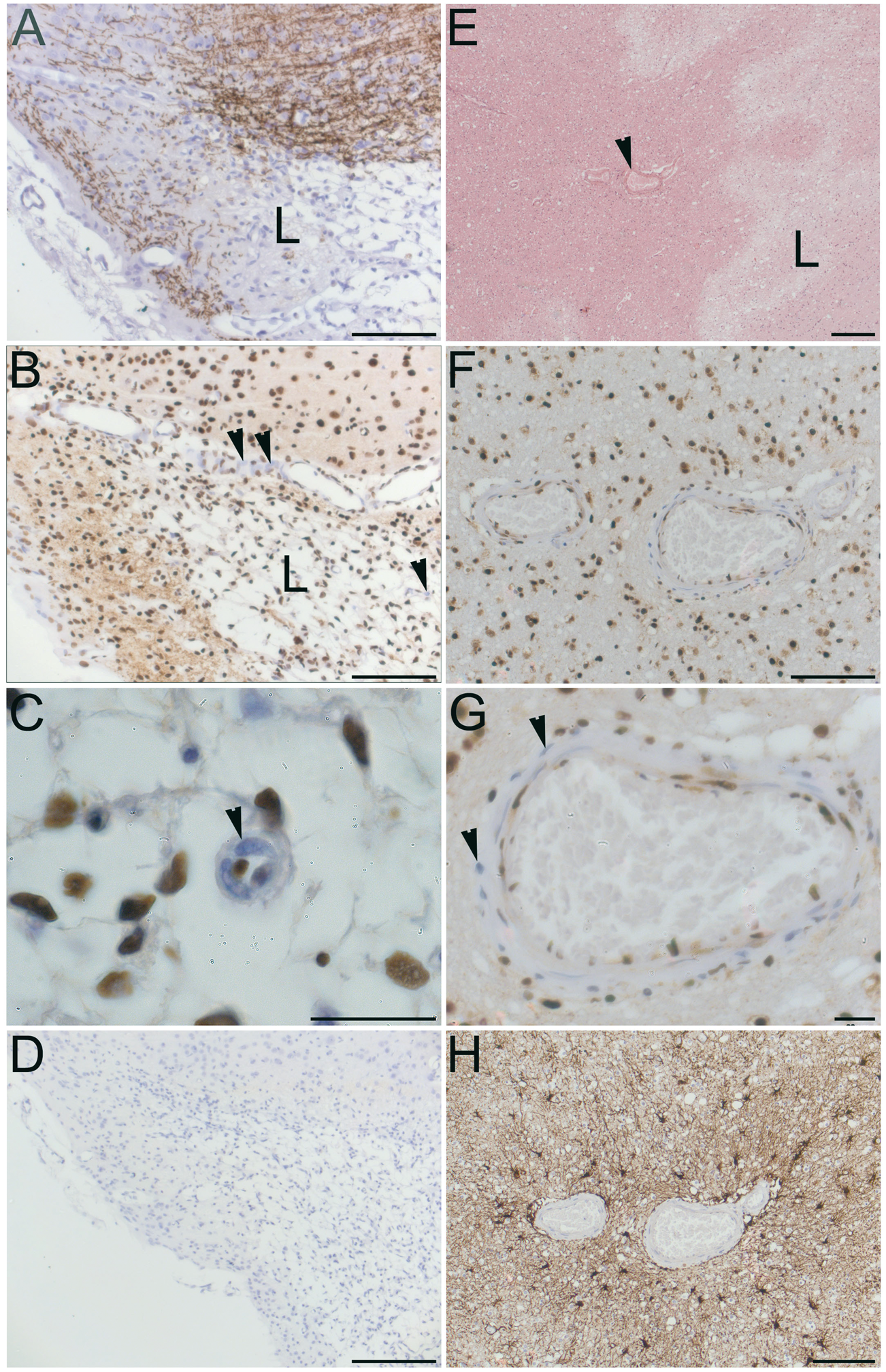
Sgk1 labelling showed little overlap with markers for astrocytes (GFAP) or for endothelial cells (CD31) (Figure 3(a) to (d)). In mice examined at 48 h post-MCAo, Sgk1 immunolabelling was sparse within lesional tissue of the ipsilateral cerebral cortex (Figure 3(e)). Cellular Sgk1 labelling at an equivalent location within the contralateral hemisphere remained robust (not shown). In small arteries adjacent to the MCAo-induced lesion, Sgk1-negative vascular cells were evident (Figure 4(a) to (d)). A similar finding was observed in human brain tissue adjacent to an acute ischemic lesion. Post mortem tissue was sampled from people who died shortly after a focal ischemic stroke (up to six days post-event, n = 6). In small arteries within the peri-lesional area, Sgk1-negative cells were evident within the vessel wall (example in Figure 4(e) and (f)).
Immunohistochemical labelling of Sgk1+/+ mouse cerebral cortex.(a) Robust cellular labelling for Sgk1 (green) is evident in cerebral cortical tissue from an unlesioned mouse. Cells within the cortical pyramidal layer are clearly labelled. There is relatively little overlap with the astroglial marker, GFAP (red). (b) As a negative control, a neighbouring section treated identically except for omission of primary antibodies, shows little non-specific labelling. (c, d) Higher magnification images confirm little overlap of Sgk1 labelling (green) with the astrocyte marker GFAP (panel C, red) or with an endothelial cell marker, CD31 (d, red). (e) Cortical tissue in a mouse at 48 h after MCAo. Ipsilateral cortical tissue within the ischaemic lesion. Astrocytic cells labelled with GFAP (green) are evident within the lesional area. Sgk1 labelling (red) is sparse. In all panels, nuclear chromatin is labelled with DAPI (blue). Scale bars 20 µm (a–b, e) or 10 µm (c–d). Sgk1-negative vascular cells adjacent to ischaemic lesions in rodent and human brain. (a–d) cerebral cortex, two days post-MCAo. (a) Cortical tissue immunolabelled for myelin basic protein (MBP, brown). Loss of MBP clearly delineates the ischaemic lesion (L). (b) Neighboring section immunolabelled for Sgk1 (brown). Nuclear chromatin is counterstained with hematoxylin (blue). Nuclear Sgk1immunoreactivity is widespread. Sgk1 is absent from some vascular cells (arrowheads) within and adjacent to the ischemic lesion (L). (c) High magnification view of a small penetrating artery, clearly showing unlabeled vascular cells (example marked with arrowhead). (d) A negative control section treated identically but without primary antibody.(e–f) Acute ischemic lesion in human cerebral cortex within the left MCA territory, four days post-stroke (male, aged 45 y). (e) In a haematoxylin-eosin-stained section, the ischemic lesion (L) is seen as pale, less eosinophilic than surrounding peri-lesional tissue. A small penetrating artery in the peri-lesional area is marked (arrowhead). (f) Neighboring section immunolabelled for Sgk1 (brown). Again, nuclei are strongly positive for Sgk1immunoreactivity. Sgk1 is sparse or absent from some cells within the wall of a small artery (landmark vessel from panel E). (g) Sgk1-negative cells are clearly seen in a higher magnification image (arrowheads). (h) As a control for the secondary antibody specificity, a neighbouring section immunolabelled with a different primary antibody (the astrocyte marker GFAP) shows a different pattern of immunoreactivity. Scale bars: 20 µm (c, g), 1 mm (panel E), 100 µm (a, b, d, f, h).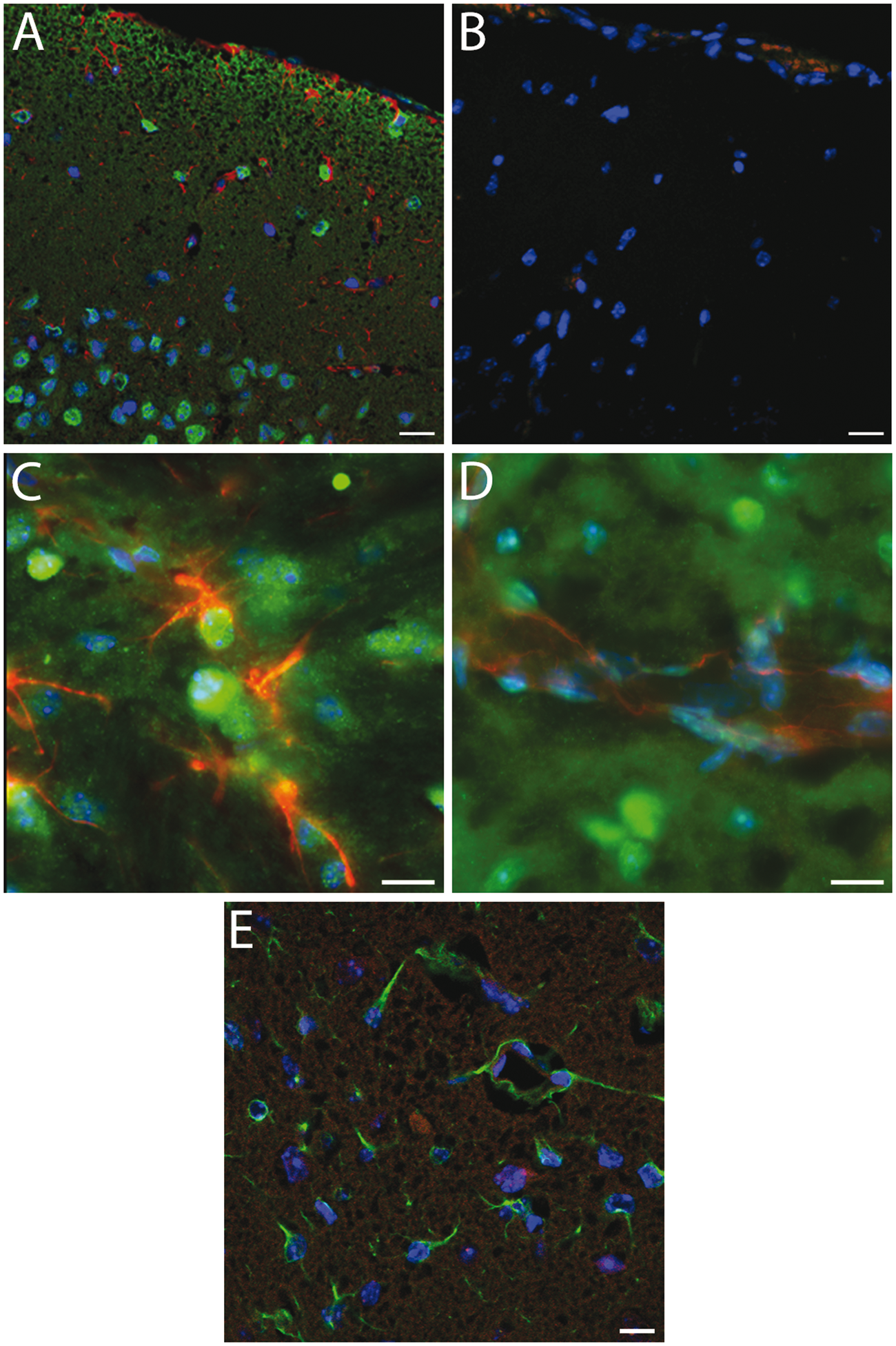
As previous studies showed significant expression of the Sgk1 homologue Sgk3 in brain tissue,18,39 it seemed plausible that Sgk3 might compensate for Sgk1 in Sgk1−/− mice. Quantitative RT-PCR revealed no difference in Sgk3 mRNA levels in Sgk1−/− animals relative to Sgk1+/+ mice. The Sgk3/Gapdh ratio was 7.6 ± 1.5 × 10−4 in Sgk1+/+ mice, 11.1 ± 3.7 × 10−4 in Sgk1−/− mice (mean ± SD, n = 9, 6, respectively; p = 0.33, Student t-test. See Supplementary Figure S2). The Sgk1/Sgk3 mRNA expression ratio in Sgk1+/+ mouse brain was 10.6 ± 4.06 (n = 4, Figure S2). In Sgk1−/− animals as expected, Sgk1 mRNA was undetectable (n = 4). These findings suggest that Sgk3 expression does not compensate for Sgk1 in Sgk1 null mice.
Discussion
In agreement with our results, other laboratories have observed brain protection by PBN in transient focal ischemia,6,8,9 as reviewed elsewhere. 5 Here we observed rapid augmentation of Sgk1 in rodent brain tissue, following acute systemic administration of PBN. We therefore examined whether Sgk1 participates in PBN-mediated neuroprotection in vivo.
We found that the well-established brain protective effects of PBN (in terms of lesion size and behavioural deficit) were absent in Sgk1−/− mice. This suggested that Sgk1 may play a role in the brain protective action of PBN. From the established cell survival actions of Sgk1, we hypothesized a worse outcome for Sgk1−/− than for Sgk1+/+ mice following focal ischaemia. Contrary to this prediction, saline-treated Sgk1−/− mice did not have significantly worse behavioral outcome and greater lesion size relative to saline-treated Sgk1+/+ mice. In terms of lesion size (Figure 2(c)), there was an unexpected trend for smaller ischaemic lesions in the Sgk1−/−animals (relative to Sgk1+/+). Similar paradoxical findings are reported from another laboratory, where acute treatment with Sgk1 inhibitor drugs (GSK650394 and EMD638683) was examined. 40 Animals injected with Sgk1 antagonists (directly into the brain, i.c.v.) had smaller infarct volume after transient MCAo than did vehicle-injected mice. 40
We observed widespread immunohistochemical labelling for Sgk1 in mouse cerebral cortex, with pronounced nuclear labelling (Figure 3). Cortical neurones were positive for Sgk1, while astrocytes and vascular endothelial cells showed relatively little labelling. This pattern is in agreement with our previous findings in human brain tissue. 20 Our immunohistochemistry data suggest that Sgk1 is lost from the ischemic lesion tissue (Figure 3(e)).
We examined Sgk1 expression in the peri-lesional region adjacent to acute ischemic lesions of rodents and human stroke patients. Sgk1-negative vascular cells are evident in small arteries adjacent to the ischemic lesion (Figure 4).
We hypothesized that up-regulation of the homologue Sgk3 might compensate for Sgk1 in Sgk1−/− animals.13,15 Although Sgk3 is expressed on a different chromosome from Sgk1, 39 Sgk3 has similar biochemical attributes and tissue distribution to Sgk1.13,39 (The third member of this family, Sgk2, is not expressed in brain tissue39,41). We found that Sgk3 mRNA expression in Sgk1+/+ mouse brain was approximately one-tenth (0.094) that of Sgk1. Brain Sgk3 mRNA abundance was not different in Sgk1−/− mice, relative to Sgk1+/+ animals, suggesting that compensatory Sgk3 upregulation had not occurred in Sgk1−/− animals.
The proposed mechanism of action of PBN-mediated brain protection is free radical trapping. Data derived from several experimental approaches demonstrate that PBN is in fact a weak radical trapping agent.10–12 Based on the present data, we suggest that chemical induction of Sgk1 may be an alternative mechanism by which PBN exerts its effects in animal models of focal cerebral ischemia. We speculate that PBN drives rapid Sgk1 induction in brain tissue (either directly or through another rapid chemical messenger). Sgk1 is a short-lived protein, with cytoplasmic half-life ∼30 min (though the brain-specific isoform Sgk1.1 has a longer half-life of 180 min).17,36,42 Our data suggest that Sgk1 is depleted within the ischemic lesional area at 48 h post-ischaemia (Figure 3). PBN-dependent Sgk1 induction within ischaemic brain tissue may promote cell survival (as previous experimental studies indicate)20–22,24 resulting in less damage to functional networks, observed as milder behavioural impairment. Others have suggested alternative mechanisms of nitrone action, 5 including endothelial protection, 43 blood–brain barrier augmentation 44 and reduced leukocyte adhesion. 45
Transient augmentation of brain Sgk1 could be beneficial in stroke and other acute neurological states. In human populations, the SGK1 gene was associated with risk of ischemic stroke (OR 1.29) in two independent Scandinavian cohorts. 46 The association survived adjustment for hypertension and diabetes mellitus, indicating a cerebrovascular action. 46 PBN is a small, water-soluble molecule with substantial brain penetration. The adverse effects are mild over a wide dose-range in experimental species, including rats, mice, gerbils and pigs. 47 Potential limitations to therapeutic Sgk1 augmentation (even if only transient) are the important cellular actions of Sgk1 in other tissues, including large arteries 25 and renal tissue. 13
Our study has several limitations. First, we lack mechanistic evidence linking PBN to Sgk1 expression. This is an area for future experimental study. Second, we administered the drug immediately (within 5 min) following onset of brain ischaemia. While many previous experimental studies have followed this dosing timescale, in order to maximise the likelihood of detecting a bio-effect, it clearly does not reflect a realistic therapeutic scenario in clinical stroke. Third, the germline Sgk1−/− mice we used may be subject to developmental effects of Sgk1 deletion. 15 While inducible null animals would allow adult-onset gene deletion, such animals were not available to us.
In conclusion, our results suggest that rapid chemical induction of cerebral Sgk1 may follow systemic treatment with PBN. This mechanism may play a part in PBN-mediated brain protection.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by an Action Medical Research UK project to AHH, RJY-M and JTM (grant number SP-4394). ANF-T was supported by National Institutes of Health (Grant number DK 41481). GFT was supported by National Institutes of Health (Grant number DK 58898). The Oxford Brain Bank is supported by the UK Medical Research Council (MRC G1000691) and Brains for Dementia Research (BDR) (Alzheimer Society and Alzheimer Research UK), Autistica UK and the NIHR Oxford Biomedical Research Centre. The funders had no involvement in the design or execution of the study.
Acknowledgements
We are grateful to Timothy J Bull, Richard Linedale and Robert J Hall and our colleagues in the Biological Research Facility at St George’s for expert advice and assistance. We thank Professor Margaret Esiri and Ms Carolyn Sloan for access to human tissue samples. We gratefully acknowledge tissue donors and their families, and the staff of the Oxford Brain Bank.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
CMcC, PA, ARG, CNK, ANFT, GFT, RJYM, JTM and AHH made a substantial contribution to the concept and design. CMcC, ASO, ARG and AHH made a substantial contribution to the acquisition of data. CMcC, PA, AS, ASO, ARG, CNK, ACP, JTM and AHH made a substantial contribution to the analysis and interpretation of data. AHH drafted the article. CMcC, PA, AS, ARG, ACP, ANFT, GFT, RJYM, JTM and AHH revised it critically for important intellectual content. All authors approved the final submitted version.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.