
Abstract
Maternal environmental factors such as diet have consequences on later health of the offspring. We found that maternal high-fat diet (HFD) exposure renders adult offspring brain more susceptible to ischemic injury. The present study was further to investigate whether HFD consumption during rat pregnancy and lactation influences the cerebral vasculature in adult male offspring. Besides the endothelial damage observed in the transmission electron microscopy, the MCAs of offspring from fat-fed dams fed with control diet (HFD/C) also displayed increased wall thickness and media/lumen ratio, suggesting that cerebrovascular hypertrophy or hyperplasia occurs. Moreover, smaller lumen diameter and elevated myogenic tone of the MCAs over a range of intralumenal pressures indicate inward cerebrovascular remodeling in HFD/C rats, with a concomitant increase in vessel stiffness. More importantly, both wire and pressure myography demonstrated that maternal HFD intake also enhanced the MCAs contractility to ET-1, accompanied by increases in ET types A receptor (ETAR) but not B (ETBR) density in the arteries. Furthermore, ETAR antagonism but not ETBR antagonism restored maternal HFD-induced cerebrovascular dysfunction in adult offspring. Taken together, maternal diet can substantially influence adult offspring cerebrovascular health, through remodeling of both structure and function, at least partially in an ET-1 manner.
Introduction
Epidemiological and animal studies suggest that, besides the parental heredity and adult lifestyle, the specific environmental factors that a developing offspring experiences in early life, may also contribute to the development and health problems of adults. 1 Ischemic stroke is one of the most common diseases for mortality and morbidity of human being. It is known that challenges early in development can alter the brain in a manner that makes it more susceptible to brain injury as an adult. For example, brief mother–infant separation or neonatal immune challenge could influence the stroke survival and recovery in adult rodents.2–4 The environment that offspring experiences in early life, including intrauterine and early postnatal conditions, is highly influenced by maternal diet and metabolic status. Nowadays, the most common maternal dietary imbalance is the excessive intake of dietary fat in many countries. Our previous study demonstrated that maternal high-fat diet (HFD) consumption renders the brain more susceptible to the consequences of ischemic injury, supported by the evidence showing that adult rat offspring of HFD-fed dams during pregnancy and lactation develops aggravated infarct volume and function deficits following stroke. 5 Although the study showed that the programing of the central neurotrophin and neuroendocrine system may contribute to aggravated stroke outcomes in maternal HFD offspring, 5 the mechanisms by which maternal HFD compromises adult ischemia occurrence and outcomes remain to be fully elucidated.
The incidence and outcome of an ischemic stroke are directly affected by the structure and function of cerebral vasculature, especially the middle cerebral artery (MCA). Numerous studies have demonstrated hazardous influence of adult HFD consumption on the cerebral arteries in animals, such as changes in MCA lumens, wall thickness, cerebral vessel compliance, and blood–brain barrier (BBB) permeability.6–10 More directly, the MCAs of obese Zucker rats undergo structural remodeling and these rats have greater cerebral injury. 11 Although it remains to be established whether maternal HFD exposure would also program the cerebral vasculature in a manner that makes it more vulnerable and susceptible to ischemic attack as an adult, there is accumulating evidence supporting that perinatal overnutrition or HFD consumption induces health problems in adult offspring, such as obesity, insulin resistance, metabolic syndrome.12,13 Nevertheless, other studies also reported that offspring of HFD-fed dams can have normal glucose tolerance and body composition in adulthood.14,15 Moreover, Khan et al.13,16 demonstrated long-term detrimental influence of maternal HFD intake on the peripheral circulation in adult offspring. Therefore, it is important to address whether maternal HFD consumption in the early life can influence cerebrovascular health of adult offspring.
Vascular endothelium plays a critical role in regulating vascular tone, permeability, coagulation, and smooth muscle growth. Endothelin-1 (ET-1), a potent vasoconstrictor peptide, 17 exerts opposing vasoconstriction and vasodilation effects via ET types A receptor (ETAR) and B receptor (ETBR), respectively.18–20 Studies have demonstrated ET-1 as an early target in vascular disease process, such as hypertension and atherosclerosis.21–23 The presence of ET-1-mediated hyperactivity of microvasculature is implicated in several animal models of obesity, diabetes, and insulin resistance.24,25 Moreover, ET-1 levels, as well as ETAR expression, are elevated and may be related to accelerated myocardial and vascular hypertrophy in obesity patient and experimental model. 26 Furthermore, ETAR blockade attenuates vascular dysfunction and that ETBR antagonism exhibits differential effects depending on the dose of the antagonists and the disease state in type 2 diabetes rats. 21 More specifically, ET-1 has been shown to mediate cerebrovascular dysfunction in several adult animal models.27,28 For example, diabetes promotes the remodeling of MCA structure in an ET-1-dependent manner in type 2 diabetic rats, characterized by increased media/lumen (M/L) ratio.29,30 Nevertheless, the role of ET-1 signaling in maternal HFD-induced cerebrovascular remodeling remains unknown.
In the current study, we hypothesized that maternal HFD consumption would program cerebrovascular function and structure in adult offspring. To test the hypothesis, structural and functional changes of the MCAs were assessed in adult male offspring of fat-fed dams during pregnancy and lactation, since the MCAs contribute considerably to the total vascular resistance and the control of cerebral blood flow. 31 Additionally, comparisons were also made between maternal HFD offspring and animals fed with HFD after weaning. The further goal of current study was to evaluate the involvement of ET-1 system in maternal diet-induced cerebrovascular remodeling, with additional emphasis on the contribution of ET receptor in the processes by exploring the effect of selective receptor antagonism on the cerebral vasculature.
Material and methods
Animals
Female Sprague–Dawley rats (120–140 days), obtained from Experimental Animal Center of the Chinese Academy of Sciences (Shanghai, China), were housed under a 12 h:12-h light/dark cycle (lights on at 0700 h) at 22 ± 2℃ temperature and provided with food and water ad libitum. The dam rats were fed with either a standard control diet (5.3% fat (corn oil), 21.2% protein, 57.4% carbohydrate, 4.6% fiber; Medicience Ltd, Jiangsu, China) or HFD (25.7% fat, 19.5% protein, 41.3% carbohydrate, 3.5% fiber; estimated fats: palmitic acid 4.5%, stearic acid 1.99%, palmitoleic acid 0.12%, oleic acid 6.86%, linoleic acid 2.58%, α-linolenic acid 0.25%, arachidonic acid 0.19%; Medicience Ltd), for 10 days before mating and throughout pregnancy and lactation. The day of parturition was set as day 0, and litters were evenly culled to eight per mother on day 1, to standardize competition for food and maternal attention. Litters were kept with their dams until weaning on day 21. Thereafter, weaned male rats were housed three per cage and fed a control diet (C) or HFD until 180 day, to obtain four groups (control offspring fed with control diet (C/C) or HFD (C/HFD), offspring from fat-fed dams fed with control diet (HFD/C) or HFD (HFD/HFD)). Animal weight was recorded weekly post-weaning. Experiments were performed according to the Regulations of the Chinese Council on Animal Care, approved by the Institutional Animal Care and Use Committee of Wenzhou Medical University and reported following the ARRIVE guidelines. All surgical procedures were carried out under ketamine anesthesia (100 mg/kg i.p.; Pharmacia and Upjohn Ltd, Crawley, UK) and xylazine (10 mg/kg i.p.; Bayer, Leverkusen, Germany).
Metabolic parameters and ET-1 measurement
The adult offspring were implanted with indwelling cardiac catheter for collection of blood samples without interruption as described previously. 32 Blood samples were collected via the cardiac catheter and plasma ET-1 level was determined with enzyme-linked immunoassay kits (Purevalley Biotech., Beijing, China). Baseline cerebrovascular ET-1 levels were also measured in the MCA homogenate, using the immunoassay kits. Subset of rats were applied with temporary middle cerebral artery occlusion (MCAO), 5 and post-stroke plasma ET-1 levels were determined 2 h later. In brief, a silicon-coated 5–0 nylon monofilament (tip diameter: 0.31 ± 0.02 mm) was introduced into the external carotid artery and advanced up the internal carotid artery to the Circle of Willis (18–19 mm from the carotid bifurcation), where it blocked the origin of the MCA to decrease blood flow for 1.5 h. Cerebral blood flow reduction of at least 80% of baseline was confirmed as ischemic attack by Laser Doppler Flowmetry (Moor Instruments, UK).
On the day of experiment, rats were fasted overnight for 14 h, and venous blood was collected via the cardiac catheter (0 min), for basal plasma glucose (HK/G6P-DH enzymatic UV test; Roche Diagnostic systems, Shanghai, China) and insulin (DGR Instruments, Marburg, Germany) measurements. Rats subsequently received an orogastric gavage of 2 g·kg−1·4 ml−1 glucose (Sigma-Aldrich, Shanghai, China) for oral glucose tolerance test, and blood was collected at 5, 10, 15, 30, 45, 60, 90, and 120 min for glucose measurement.
Vascular morphology
The rats were anesthetized with an i.p. injection of sodium pentobarbitone (50 mg/kg, Rhone Merieux Ltd, Harlow, UK) and transcardially perfused with heparinized saline (5 U/ml) followed by 4% paraformaldehyde in 0.1 M PBS (pH = 7.4), at a pressure of 110–120 mmHg. Brain tissue containing the MCA root in long was dissected and fixed in paraffin, sectioned at 5 µm, and subjected to hematoxylin–eosin (HE) staining. Wall thickness, lumen diameter, and outer diameter of the MCA root were measured from captured images of HE-stained cross-sections using SPOT program (Diagnostic Instruments, Sterling Heights, USA), and M/L ratio of the MCA root was calculated.
Vascular ultrastructure
Ultrastructure appearance of cerebral artery was observed by a transmission electron microscopy. Three rats in each group were randomly selected and anesthetized with sodium pentobarbitone. The rats were perfused through the left ventricle at a pressure of 110–120 mmHg, with approximately 50 ml of heparinized saline (5 U/ml) followed by 100 ml of 4% paraformaldehyde and 2.5% glutaraldehyde in 0.1 mol/L cacodylic acid buffer (pH 7.3). The fixed brain was dehydrated through an ethanol series embedded in epoxy resin and cut into ultrathin sections (brain tissue containing MCAs in long, approximately 1.0 mm × 1.0 mm × 2.0 mm) under a microscope. The sections were mounted on copper grids, stained in uranyl acetate and citric acid lead, and observed under an H-7500 transmission electron microscope (Hitachi Ltd, Tokyo, Japan).
Pressure myography
Rats were euthanized with CO2 inhalation and decapitated at the age of 180 day. The first branch-free segment of the MCAs proximal to the Circle of Willis was dissected and placed in cold Krebs buffer. The vessel segment was mounted on two glass micropipettes (outer diameter, 125–150 μM) and secured with a 40-µm-thin wire in a pressure myograph (Living systems instrumentation, Burlington, USA). The pressure myograph was connected to a pressure servo-controller via a pressure transducer, allowing for pressure to be manipulated under 0-flow conditions. A calibrated video dimension analyzer was used to determine lumen diameter. 11 Briefly, after equilibration at 80 mmHg in 37℃ Krebs buffer (gassed with 21% O2, 5% CO2 and 74% N2), the myogenic response was determined by measuring lumen diameter as pressure was increased by 20 mmHg increments over a range of intralumenal pressures (40 to 140 mmHg). The measurements were also made in the absence of calcium to determine passive vessel structure. Complete loss of active tone was verified by confirming the lack of a response in lumen diameter on the addition of papaverine. Comparing the lumen diameters under active and passive conditions, the percentage of myogenic tone was calculated as follows: % myogenic tone = [1−(IDA/IDP)]×100, where IDA is inner diameter under active conditions, and IDP is inner diameter under passive conditions. 21 On the other hand, changes in lumen diameter were measured after 2 min of exposure to increasing concentrations of ET-1 (0.1–100 nM) to determine the impact of maternal HFD on the contractility of the MCAs to the vasoconstrictor, expressed as the percentage of max response to nimodipine. At the end of each experiment, the artery was perfused with Ca-free buffer that contained 1 μM nimodipine to determine the maximum vasodilatory capacity.
Vascular stiffness parameter (β) was calculated for the passive vessels using the pressure–diameter data obtained over the entire pressure range. A regression was performed for each vessel using the following equation: ln (p/pS) = β (d/dS−1). Where p is the internal pressure, pS is a reference pressure chosen in the physiological pressure range, d is the external diameter, and dS is the external diameter of the vessel at the reference pressure. The reference pressure was chosen to be 80 mmHg because it lies within the physiological pressure range and gives a good fit to the equation (mean R2 > 0.90).
Wire myography
Isometric tension exerted by the vessels was recorded via a force transducer using the Living Systems wire myograph. Approximately 2-mm-long MCA segments, dissected as described above, were mounted in the chamber and adjusted to a baseline tension of 1 g. The MCAs were exposed to ET-1 in the absence or presence of nitric oxide synthase inhibitor L-nitro-arginine methyl ester (L-NAME, 300 µmol/L) pre-incubated for 30 min. After stabilization, cumulative dose-response curves to ET-1 (0.1–100 nM) were generated and the force was expressed as the percentage of baseline. 21 Moreover, the response to ET-1 was also determined in subsets of offspring which applied with acute specific ET-1 receptor antagonism by i.p. injection of, either ETAR antagonist, atrasentan (5mg/kg, ApexBio, Shanghai, China), or ETBR antagonist, A-192621 (30 mg/kg, ApexBio). After 3 h, the MCAs were collected and the response to ET-1 was subsequently determined without L-NAME. Endothelium-dependent relaxation to acetylcholine (ACh, 1 nM-1 μM) was also determined after the MCAs were constricted to 60% over the baseline tension, using serotonin (5-HT).
Quantitative reverse transcriptase-polymerase chain reaction
Basal expression of ETAR and ETBR mRNA in the MCAs of non-stroked offspring was determined by real-time quantitative RT-PCR. Total RNA was extracted from the MCA strips using TRIZOL reagent (Sigma-Aldrich) in accordance with the manufacturer’s instructions. For the quantitative RT-PCR, the following primers were used: ETAR (Forward) 5′-CAGCCTGGCCCTTGGAGACCTTAT-3′, (Reverse) 5′-TTCTGTGCTGCTCGCCCTTGTATT-3′; ETBR (Forward) 5′-GATACGACAACTTCCGCTCCA-3′, (Reverse) 5′-GTCCACGATGAGGACAATGAG-3′. The Light-Cycler™ (Roche Biochemicals, Lewes, UK) was used for real-time quantitative analysis of ETAR and ETBR mRNA expression. cDNA was synthesized using reverse transcriptase (Sigma-Aldrich) after RNA quality was verified by a spectrometer. The cycling conditions consisted of an initial single cycle of 10 min at 95℃, followed by 40–45 cycles of 30 s denaturation at 95℃, 45 s of annealing at 56℃, and 30 s of extension at 72℃ and 1 min of final extension at 95℃. Reaction conditions for ETAR, ETBR mRNA, and 28S rRNA were optimised separately to give the best results for each primer and for the different quantities of target in samples. Preliminary experiments were taken to prepare the PCR products and then generate standard curves in real-time RT-PCR. ETAR or ETBR mRNA was quantified, against the respective standard curve, using the Light-Cycler™ software. The 28S rRNA was quantified as a reference gene against a separate standard curve of samples containing known concentrations of 28S rRNA product. PCR product for ET receptor mRNA was sequenced and analyzed using an ABI Stepone system (Applied Biosystems Inc., Foster City, USA). The values of ETAR and ETBR mRNA were expressed as a ratio of ET receptor mRNAs and 28S rRNA.
Western blot analysis
ETAR and ETBR protein levels were determined by immunoblotting. MCAs strips were homogenized in RIPA lysis buffer. Total protein content in the sample was determined by the Bradford method (Thermo Scientific, IL, USA). Vascular extracts were separated on 10% SDS-PAGE gel and transferred to a nitrocellulose membrane in Tris-glycine transfer buffer supplemented with 20% methanol. The immunoblots were blocked for 1 h in 5% bovine serum albumin diluted in 0.2 M Tris base, 1.4 M NaCl, 0.1% Tween 20, and 0.02% NaN3. The membrane was then incubated overnight with primary antibodies for ETAR and ETBR (Santa Cruz Biotechnology, Dallas, USA). The specificity of the bands was confirmed by using increasing concentrations of competing peptide for each antibody. The protein bands were visualized with ECL detection reagent and quantified by optical densitometry and ImageJ software. The densitometry values represented the pixel intensity, and were normalized to GAPDH to correct for loading.
ET receptor antagonism
The subsets of HFD/C and C/C rats were applied with either an ETAR antagonist, atrasentan (5mg/kg/day), administered in the drinking water, or ETBR antagonist, A-192621 (30 mg/kg/day), administered by oral gavage. Daily water consumption was measured for the atrasentan treatment. The doses were based on the recommendations of the manufacturer and previous publications. 21 The chronic treatment was given from week 12 through week 16. A corresponding control group was included and given the vehicle. The lumen diameter and myogenic tone of the MCAs were assessed by pressure myograph at the age of 180 day, as described above.
Statistical analysis
All quantitative data were represented as mean with standard deviation. Statistical comparisons in offspring body weight were made by repeated-measures ANOVA with Bonferroni correction for multiple comparisons. The glucose levels were assessed by ANOVA followed by Dunnett’s t-test to assess statistically significant differences within the same strain at different time points, as well as between the different strains at the same time point. The statistical significance of ET-1 and ET receptor levels were evaluated by a one-way ANOVA followed by Dunnett’s t-test. A multiple-measure ANOVA was performed for the dose-response curve to ET-1, comparing different groups of rats with a post hoc Tukey test. A two-way repeated measures ANOVA with a Bonferroni post-test was used to compare lumen diameter and myogenic tone between groups. P < 0.05 was considered statistically significant.
Results
Offspring body weight and glucose tolerance
Repeated-measures ANOVA revealed that maternal HFD exposure had no effect on growth rate during the experiment (Figure 1(a)). While in 10 weeks HFD feeding after weaning, C/HFD (n = 12) and HFD/HFD (n = 10) rats showed increased body weight, compared with C/C animals (n = 10, P < 0.05). After fasting overnight, blood glucose for the C/C (n = 6), HFD/C (n = 8), C/HFD (n = 8), and HFD/HFD (n = 7) rats were 4.38 ± 1.45, 4.47 ± 1.81, 4.68 ± 1.75, and 4.87 ±1.66 mmol/L, respectively; while insulin for each group were 1.18 ± 0.56, 1.82 ± 1.02, 2.22 ± 1.32, and 2.34 ± 1.37 mmol/L, respectively. No differences in fasting glucose or insulin concentrations were observed between groups (P > 0.05). Nevertheless, oral glucose tolerance was significantly impaired in HFD/C offspring, compared with C/C rats (P < 0.05, Figure 1(b)).
Growth rate (A) and glucose metabolism (B) in groups. (a) Body weight remained unchanged in HFD/C rats during the experiment, compared with C/C offspring. (b) Oral glucose tolerance test (orogastric gavage of 2 g/kg/4 ml glucose) was significantly impaired in HFD/C rats. *P < 0.05 C/HFD group versus HFD/C group at the same time period. #P < 0.05 HFD/C group versus C/C group. Results are shown as mean ± SD. Rat weight: C/C (n = 10), HFD/C (n = 12), C/HFD (n = 12), and HFD/HFD (n = 10). Glucose tolerance test: C/C (n = 6), HFD/C (n = 8), C/HFD (n = 8), and HFD/HFD (n = 7).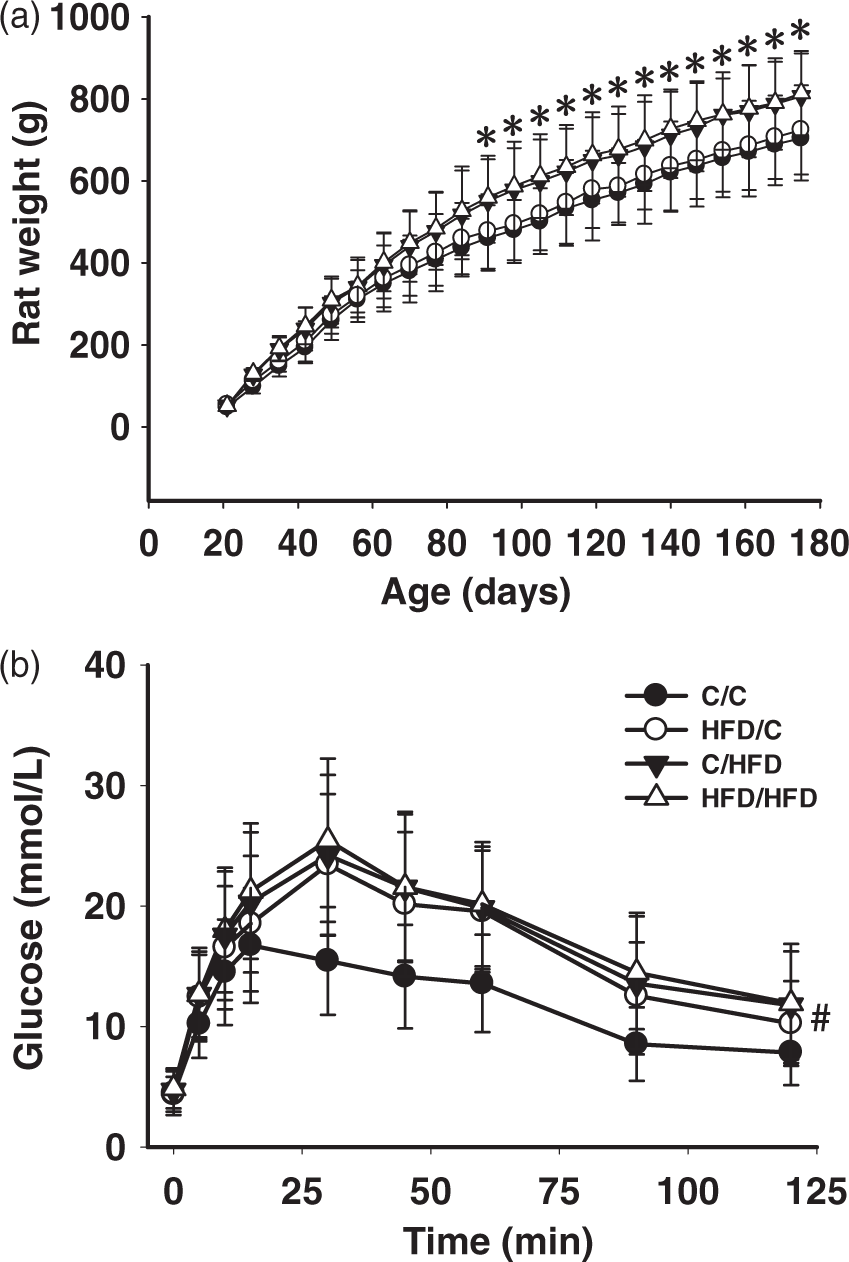
Cerebrovascular structure
Representative morphological examples of the MCAs from groups are shown in Figure 2(a) to (d). Summary analysis (Figure 2(e) and (f)) showed that, compared with C/C offspring (n = 5), HFD/C rats (n = 7) displayed increased wall thickness and M/L ratio, although total vessel size was not different (P < 0.05). Moreover, C/HFD (n = 6) and HFD/HFD rats (n = 6) showed similar raised wall thickness and M/L ratio with HFD/C offspring (P > 0.05). Additionally, external diameters for the C/C, HFD/C, C/HFD, and HFD/HFD rats were 310.15 ± 32.64, 320.46 ± 33.11, 327.45 ± 33.64, and 330.71 ± 34.13 µm, respectively; and no difference was observed between groups (P > 0.05).
The effect of maternal HFD intake on the MCA morphology. Representative images of the MCA root from C/C (n = 5), HFD/C (n = 7), C/HFD (n = 6), and HFD/HFD (n = 6) rats are shown in a–d, respectively. (e) and (f) summary showing the increased wall thickness and media/lumen of the MCAs in HFD/C rats. *P < 0.05 versus C/C group. Values are mean ± SD. Scale Bar: 50 µm.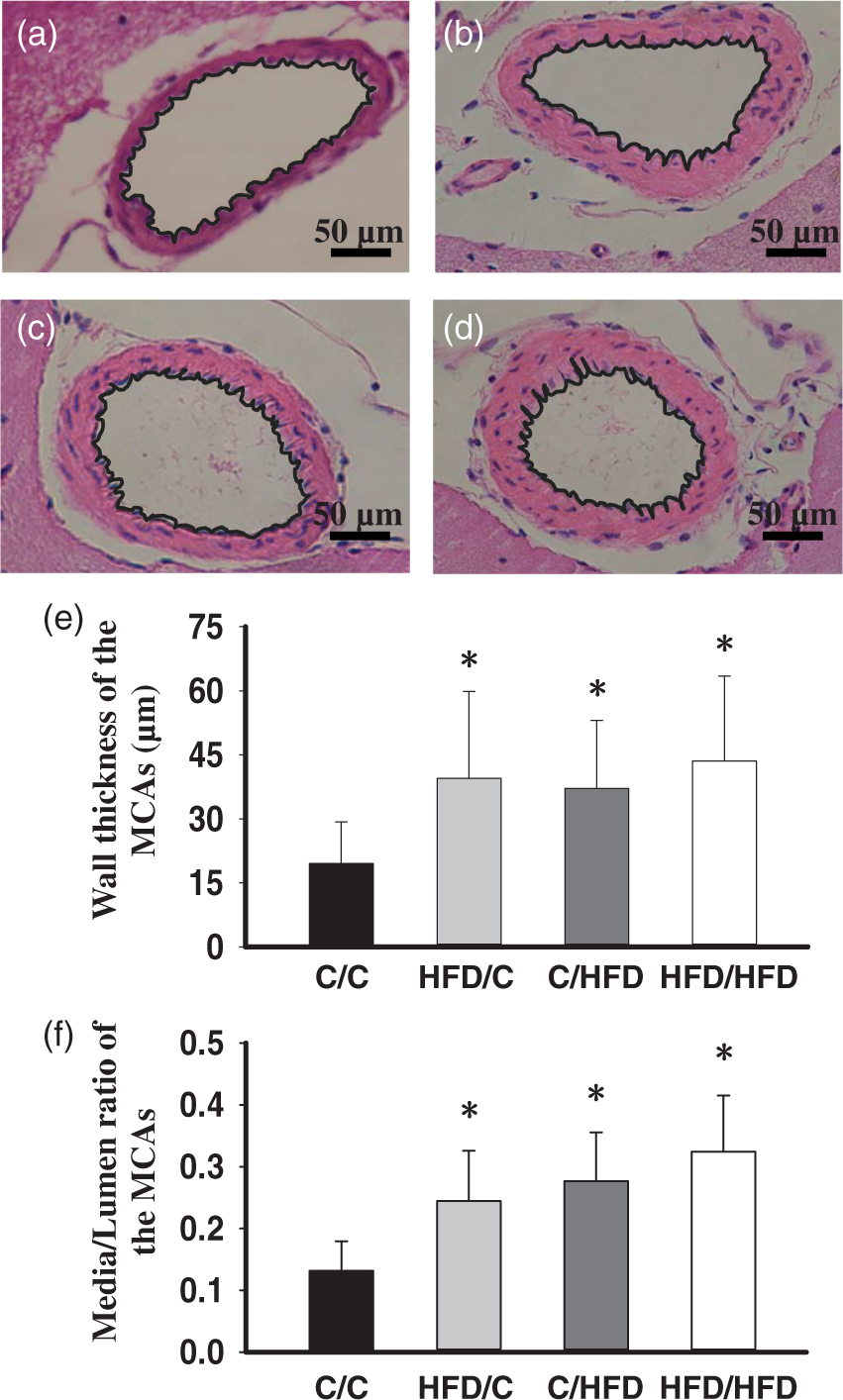
Representative ultrastructural examples of the MCAs are shown in Figure 3. No evidence of damage was found in the endothelial cell of the MCAs in C/C rats (Figure 3(a)), showing an intact basement membrane and tight junction. While ultrastructural damage such as vacuoles in the cytoplasm and endothelial cell denudation, cavity between endothelial cell and base membrane, as well as disintegrated basal lamina were observed in HFD/C offspring (Figure 3(b)). As shown in Figure 3(c), endothelial cellular swelling, junctional intercellular enlarged, and endothelial denudation were similarly found in the C/HFD rats. Obvious alterations in BBB ultrastructure were also observed in the HFD/HFD group, including decreased luminal area, endothelial denudation and swollen endothelial nucleus, as well as an abnormally thin basal lamina.
The effect of maternal HFD consumption on ultrastructure appearance of the MCAs. (a) C/C group. The normal base membrane (white arrow) and endothelial cell (EC) were observed. (b) Showing the endothelial damage and endothelial denudation (triangle) in HFD/C rats. (c) C/HFD group. Endothelial cellular swelling, disintegrated basal lamina (dark arrow), and endothelial denudation (triangle) were observed. (d) Showing obvious endothelial denudation, swollen endothelial nucleus, and enlarged intercellular junction (dark arrowhead) in HFD/HFD offspring. n = 3 for each group. Scale Bar: 0.5 µm.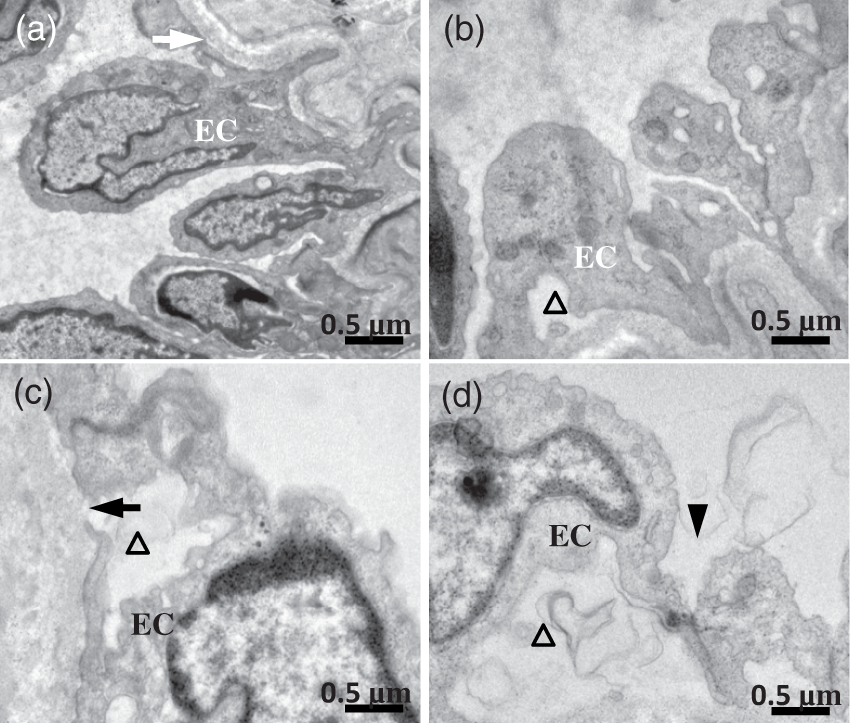
Pressure myography
The lumen diameter and myogenic tone of the MCAs were assessed by pressure myograph to determine whether these important contributors to cerebral blood flow were influenced by maternal diet. It should be noted that the maximal diameter of MCAs after incubation of nimodipine (without calcium) was not significantly different between the groups (P > 0.05). Moreover, repeated ANOVA revealed the lumen diameter of MCAs from the HFD/C rats (n = 8) was significantly decreased over the entire range of pressures measured, compared with these of the C/C group (n = 7, Figure 4(a), P < 0.05). C/HFD rats (n = 7) demonstrated a smaller lumen diameter over 40 and 140 mmHg, compared with C/C rats (P < 0.01). Moreover, the myogenic tone of the MCAs in HFD/C rats was significantly increased from 60 to 120 mmHg, compared with those from C/C rats (Figure 4(b), P < 0.05). Moreover, over the entire range of measured pressure, no differences in myogenic tone were observed between the HFD/C and C/HFD groups (P > 0.05). These results suggest that the MCAs of the maternal HFD offspring undergo an inward remodeling. Additionally, HFD/C offspring had a significant increase in the stiffness of the MCAs, with a larger β-coefficient (P < 0.05, Figure 4(c)). Similarly, the stiffness parameter of MCAs was also raised in C/HFD and HFD/HFD offspring, compared with that of C/C rats (P < 0.05). Furthermore, maternal HFD intake enhanced the contractile response of the MACs to ET-1 in offspring (Figure 4(d)), as indicated by increased change in lumen diameter. Nevertheless, there were no differences in the response of the MCAs to ET-1 between HFD/C and HFD/HFD rats (P > 0.05).
The influence of maternal HFD consumption on the lumen diameter (a), myogenic tone (b), stiffness (c) and contractility, (d) of the MCAs, using the pressure myograph. Compared with C/C rats, HFD/C offspring displayed smaller lumen diameter, raised myogenic tone and increased stiffness. Moreover, maternal HFD intake increased the contractile response of the MCAs to ET-1, expressed as the percentage of max response to nimodipine. *P < 0.05 HFD/C group versus C/C group on the same intralumenal pressure. #P < 0.05 versus C/C rats. Results are shown as mean ± SD. ET-1 contractility study: C/C (n = 5), HFD/C (n = 6), C/HFD (n = 6), and HFD/HFD (n = 5); for other studies: C/C (n = 7), HFD/C (n = 8), C/HFD (n = 7), and HFD/HFD (n = 7).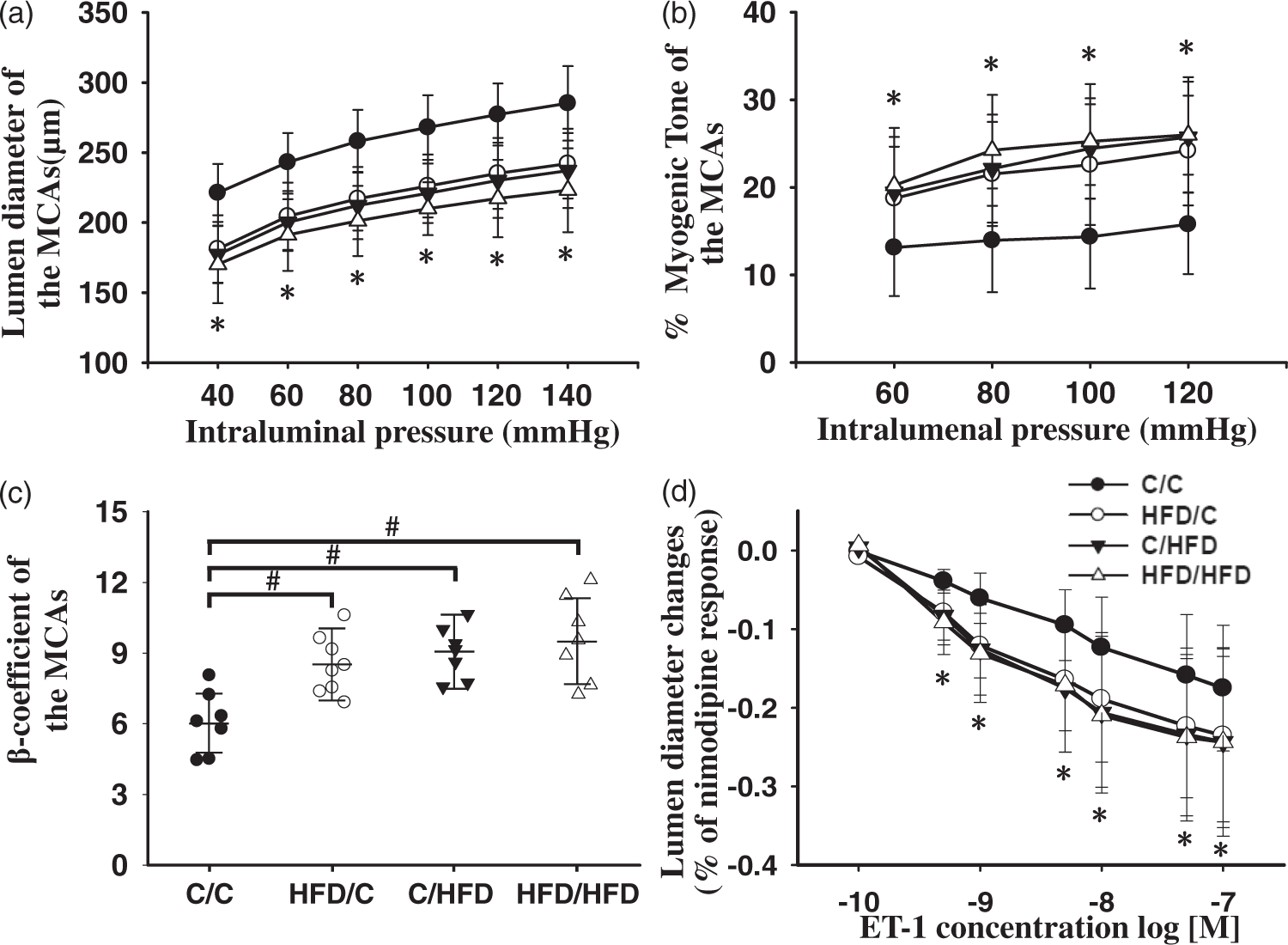
ET-1 levels and ET receptor expression
Unlike the unchanged basal plasma ET-1 level following increasing dietary fat intake during perinatal or adult period (in fmol/ml, C/C = 1.38 ± 0.47, HFD/C = 1.47 ± 0.68, C/HFD = 1.68 ± 0.54, HFD/HFD = 1.77 ± 0.56, P > 0.05), maternal HFD consumption significantly increased ET-1 concentration in the MCAs tissue (Figure 5(a)). While plasma ET-1 levels were significantly upregulated following MCAO procedure in all groups, post-stroke ET-1 level in the HFD/C rats was significantly increased (n = 8, P < 0.05 vs. C/C, Figure 5(b)). Furthermore, to determine whether increased ET-1-mediated constriction is accompanied by changes in ET receptor density in the MCAs, ET receptor mRNA and protein levels were measured by RT-PCR and immunoblotting, respectively (Figure 5(c) to (g)). There was a significant increase in ETAR mRNA and protein in HFD/C offspring (n = 8, P < 0.05), but parameters for ETBR remained unchanged. Moreover, compared with the C/C (n = 7), C/HFD (n = 8), and HFD/HFD (n = 7) offspring also showed increased ETAR mRNA and protein (P < 0.05).
Basal ET-1 and ET receptor expression in the MCAs, and post-stroke plasma ET-1 levels. Maternal HFD consumption increased baseline MCA ET-1 levels (a) and plasma ET-1 levels in 2 h after the MCAO (b). The analyses indicated an increase in ETAR mRNA (c) and protein (f) but not ETBR mRNA (d) and protein (g) in HFD/C offspring. (e) Representative immunoblot showing ETA and ETB receptor protein levels. The mRNA values are expressed as a ratio of ET receptor subtype mRNA and 28S rRNA. Densitometry values reported are normalized to GAPDH levels to account for differences in loading. *P < 0.05 versus C/C group. Results are shown as mean ± SD. MCA ET-1 and receptors: C/C (n = 7), HFD/C (n = 8), C/HFD (n = 8), and HFD/HFD (n = 7), plasma ET-1 determination: C/C (n = 6), HFD/C (n = 8), C/HFD (n = 7), and HFD/HFD (n = 7).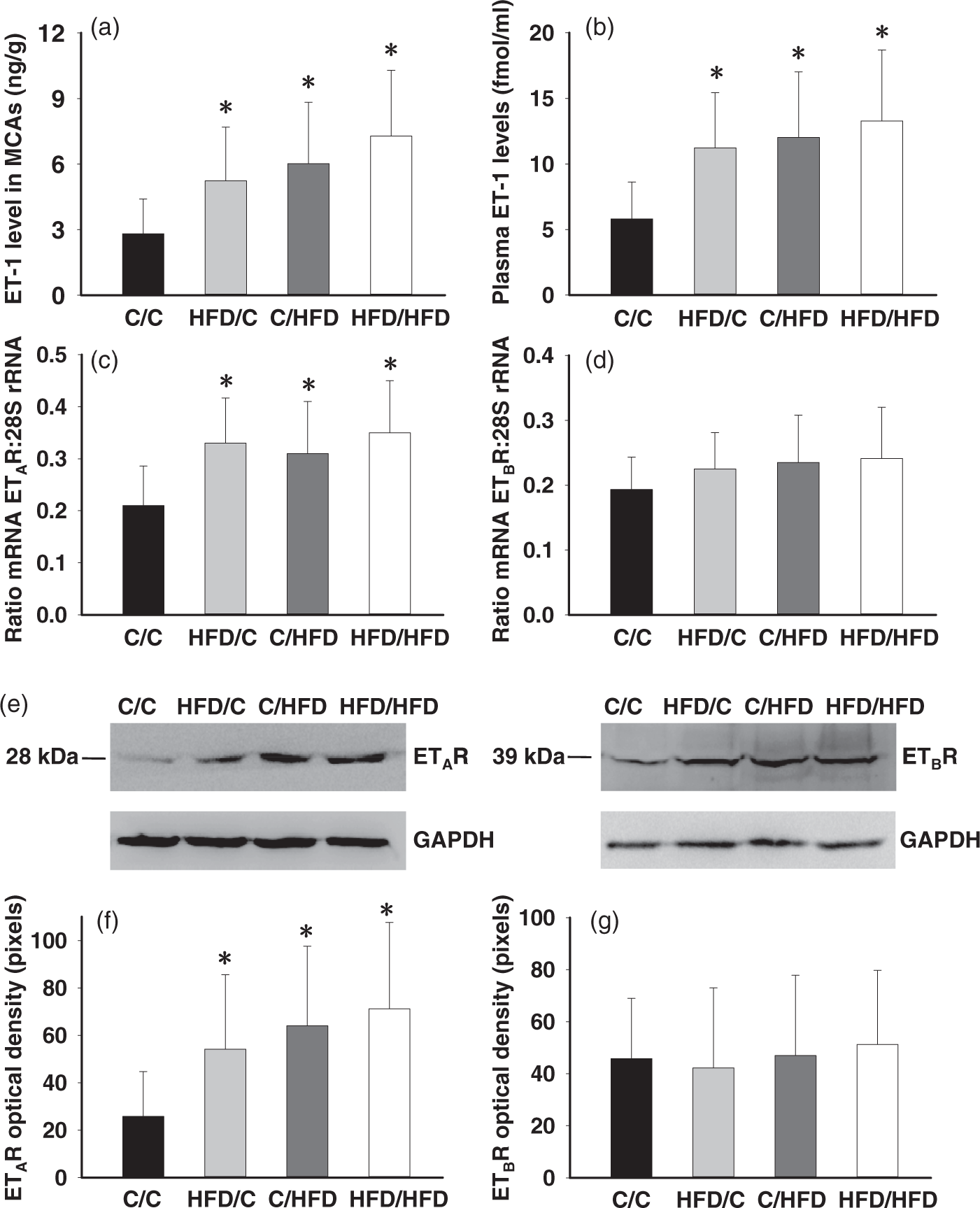
Wire myography
The MCAs of HFD/C offspring (n = 8) displayed hypersensitivity to ET-1 compared with C/C rats (n = 6), with or without NO synthase inhibitor L-NAME (Figure 6(a) and (b)). Compared with C/C offspring, C/HFD (n = 8) and HFD/HFD rats (n = 7) also displayed increased contractile response to ET-1 (P < 0.01), in the presence or absence of L-NAME. Further analysis revealed that L-NAME markedly enhanced the contractile response of the MCAs to ET-1 in all groups, but the increments were not different between groups (P > 0.05). Moreover, acute ETAR but not ETBR antagonism prevented the hypersensitivity of the MCAs to ET-1 in HFD/C offspring (Figure 6(c) and (d)). Whereas upon treatment with ETAR antagonist, control rats showed decreased contractile responses to ET-1 (0.5–10 nM). Additionally, in C/C rats on A-192621, vasoconstriction to ET-1 was augmented over the applied concentration range, suggesting an inhibition of endothelial ETB-mediated NO release that normally balances ET-1 constriction. On the other hand, following preconstriction with 5-HT, ACh caused concentration-dependent relaxation, which was unchanged with increasing dietary fat content during pregnancy and lactation (Figure 6(e), P > 0.05). Moreover, no difference in endothelium-dependent vasodilation to Ach was observed between HFD/C (n = 7) and C/HFD (n = 6) offspring (P > 0.05).
The reactivity of the MCAs to ET-1 and ET receptor antagonism, as well as ACh. Maternal HFD intake induced hyperreactivity of the MCAs to ET-1, in the absence (a) or presence (b) of L-NAME. Moreover, acute ETAR (atrasentan, c) but not ETBR antagonist (A-192621, d) prevented hypersensitivity of the MCAs to ET-1 in HFD/C offspring. (e) relaxation to ACh was not altered in HFD/C rats compared with controls. *P < 0.05 versus C/C across ET-1 concentration range. #P < 0.05 HFD/C + vehicle versus HFD/C + atrasentan. Results are shown as mean ± SD. MCA reactivity: C/C (n = 6), HFD/C (n = 8), C/HFD (n = 8), and HFD/HFD (n = 7), antagonist studies: C/C + vehicle (n = 5), C/C + antagonist (n = 6), HFD/C + vehicle (n = 6), HFD/C + antagonist (n = 7), Ach relaxation: C/C (n = 5), HFD/C (n = 7), C/HFD (n = 6), and HFD/HFD (n = 6).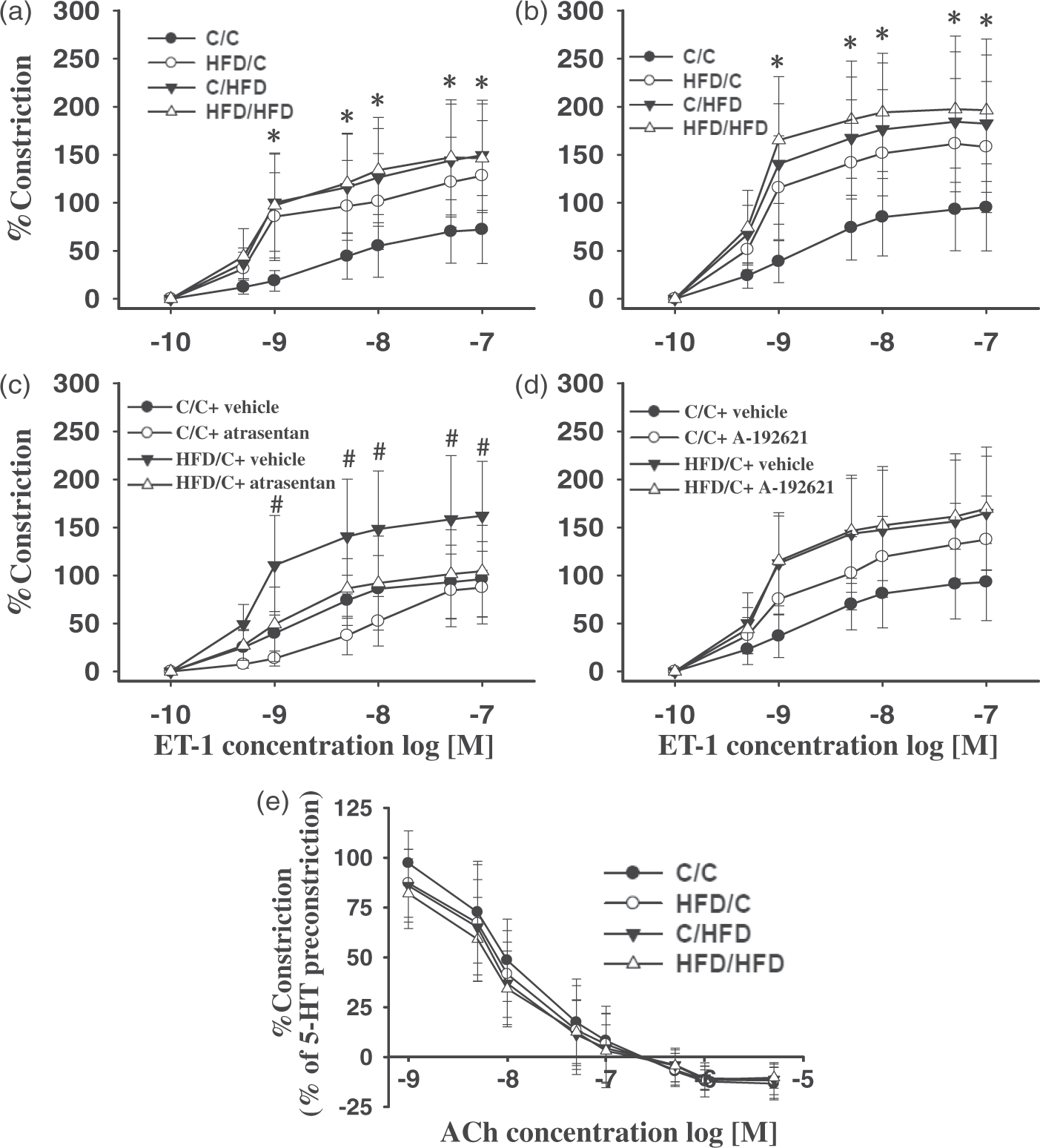
Selective ET receptor antagonism
Repeated ANOVA revealed that ETAR antagonist atrasentan significantly increased the lumen diameter of the MCAs from HFD/C rats over the range of 60 to 140 mmHg (Figure 7(a)). However, A-192621-treated HFD/C offspring (n = 7) displayed similar lumen diameter of MCAs with that of the vehicle-treated HFD/C offspring (n = 5, Figure 7(b), P >0.05). Moreover, the myogenic tone was significantly decreased in MCAs from HFD/C rats treated with atrasentan from 80 to 120 mmHg, compared with those of vehicle-treated HFD/C rats (Figure 7(c), P < 0.05). Nevertheless, ETBR antagonist did not alter the myogenic tone of the MCAs over the entire range of measured pressure in HFD/C rats (n = 7, P > 0.05). These results suggest that chronic ETAR but not ETBR antagonism restores maternal HFD-induced cerebrovascular remodeling. Nevertheless, it should be noted that chronic antagonism of either ETAR or ETBR has no effect on the lumen diameter or myogenic tone of the MCAs in C/C rats.
The effects of chronic ET receptor antagonism on maternal HFD-induced remodeling of the MCAs. (a) and (b) showing ETAR antagonist atrasentan but not ETBR antagonist A-192621 prevented maternal HFD-induced decline in lumen diameter, respectively. (c) and (d) showing atrasentan but not A-192621 significantly attenuated increased myogenic tone in maternal HFD offspring, respectively. * P < 0.05 atrasentan-treated HFD/C rats versus vehicle-treated HFD/C group on the same intralumenal pressure. Results are shown as mean ± SD; C/C + vehicle (n = 5), C/C + antagonist (n = 7), HFD/C + vehicle (n = 5), HFD/C + antagonist (n = 7).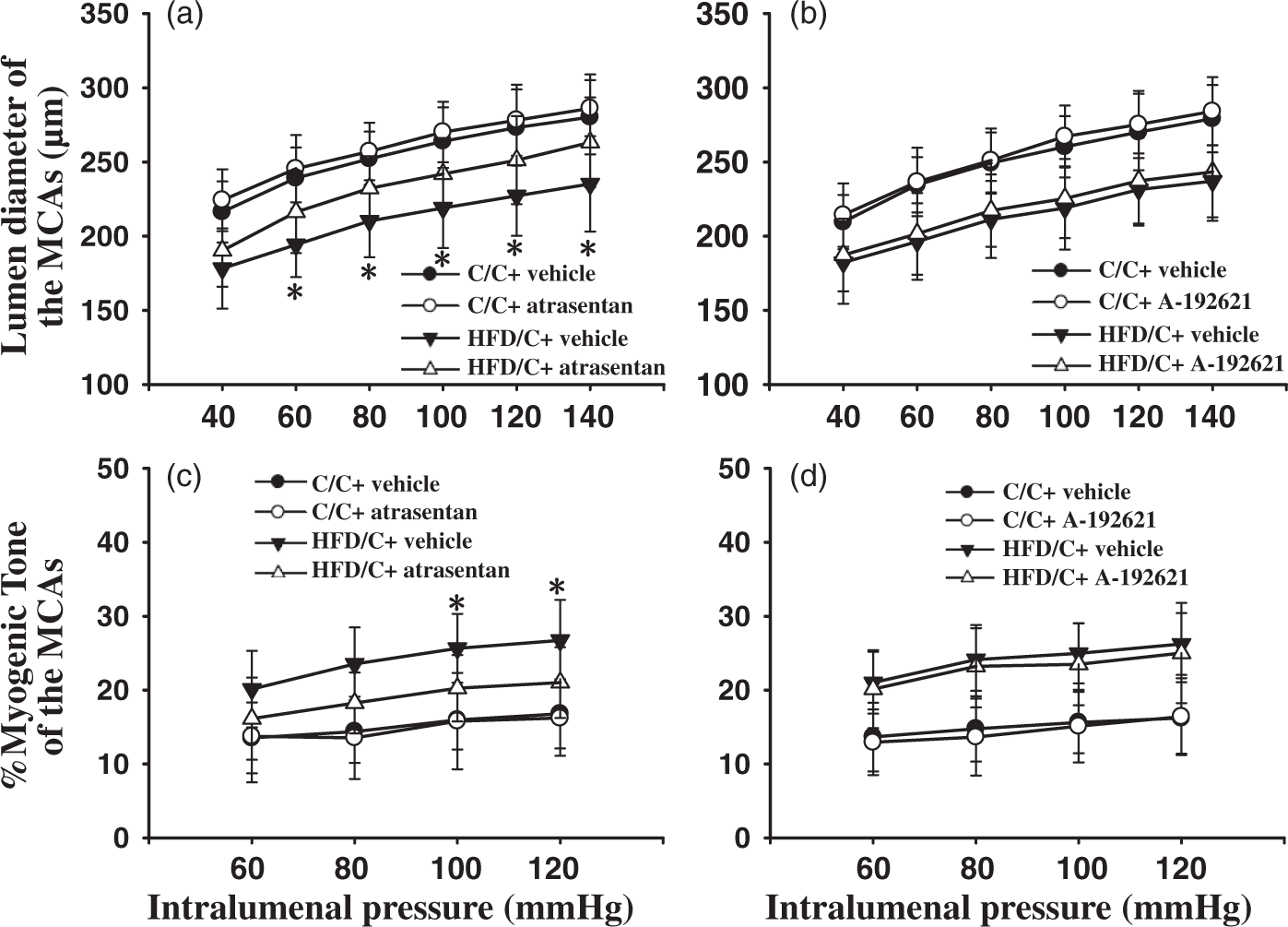
Discussion
Nutritional status or diet intake during early life has important influence on development and later health of the offspring, changing their responses to environmental challenges and thus their predisposition to disease. The present study examined the cerebrovascular structure and function changes in a rat model of maternal fat dietary regimen, initiating just before pregnancy until the end of lactation. We found that, similar to increasing HFD consumption in adulthood, 7 maternal dietary fat intake also remodel cerebrovascular structure and function in adult male offspring. To the best of our knowledge, this is the first study to document the long-term influence of maternal diet on the cerebral vasculature in adult offspring. The results are consistent with other data showing that a fat-rich diet during rat pregnancy and suckling induces hypertension and cardiovascular dysfunction in adult offspring.13,16,33 Moreover, maternal HFD exposure also confers susceptibility to mental health and behavioral disorders in offspring, such as depression, anxiety, impairments in social behavior, and cognitive deficit.32,34–37 Taken together, maternal diet has profound effects on many aspects of physiology and behavior in offspring.
The importance of early-life challenges in modulating the susceptibility to brain injury in adult offspring has been demonstrated in several animal models, such as brief mother–infant separation, neonatal immune challenge.2–4 Consistently, maternal HFD intake also renders the brain more susceptible to the consequences of ischemic injury in adult offspring. 5 The present study expands the result by demonstrating that maternal HFD consumption would substantially influence adult offspring cerebrovascular health, providing an important mechanism for maternal HFD-induced aggravated stroke outcome. Moreover, because of the cerebral circulation remodeling, maternal HFD exposure would also give rise to an offspring phenotype predisposed to the development of stroke and other cerebrovascular diseases in adulthood. The results are consistent with other data showing that adult fat feeding programs cerebral vessel and increases the ischemic damage, 7 which is also confirmed in our studies. It is further supported by the evidence showing that developmental programing of adulthood cardiovascular and metabolic disorders are observed in animals exposed to maternal diet rich in saturated fat or cholesterol.38,39 Nevertheless, it should be mentioned that, consistent with previous research,13,40 maternal HFD intake does not affect offspring growth rate in the present study. Since obesity has been reported to play an important role in diet-induced cerebral vessel remodeling in adults, 7 these data indicate that fat-rich feeding during perinatal or adulthood causes cerebrovascular remodeling through, at least partially, differential underlying mechanisms. It is worth noting that maternal food restriction has also been shown to modulate cerebrovascular structure and contractility in adult rat offspring, including increased artery stiffness and wall thickness, depressed pressure-evoked myogenic reactivity, via both glucocorticoid-dependent and glucocorticoid-independent mechanisms. 41
The MCAs contribute considerably to the total vascular resistance and the control of cerebral blood flow, 31 and most commonly occlude in humans when ischemic attack occurs. The cerebral blood perfusion is significantly impacted by changes in MCA stiffness such as vessel lumen diameter and myogenic tone. 42 Although maximal outer diameter of MCAs remains unchanged in the present study, smaller lumen diameter and elevated myogenic tone of the MCAs indicates that, even when near-maximally vasodilated, cerebral blood flow will be impaired in maternal HFD offspring. Moreover, a marked increase in wall thickness and M/L ratio of the MCAs suggests some hypertrophy or hyperplasia of the vessel wall occurs in the HFD/C rats. Nevertheless, it needs to be mentioned that true lumen diameters may not recorded since wall thickness and M/L ratio measurements were not taken from maximally dilated vessels. Additionally, obvious ultrastructural damages such as endothelial cell denudation are also observed in HFD/C offspring. All these changes in cerebral circulation substantially contribute to the increased risk for development of cerebrovascular disease and aggravated ischemic stroke outcomes. Consistently, changes in the mechanical and architectural properties of the cerebral vasculature are also observed in populations consuming fat-rich diet, specifically an inward remodeling of the MCAs. A recent study also showed that fat-rich diet beginning in childhood causes inward vessel remodeling with a concomitant increase in vessel stiffness, as well as a marked increase in vessel wall thickness. 7 However, besides a smaller vessel lumen diameter, a small reduction in outer diameter and wall thickness is observed in the MCAs of obese Zucker rats. 11 Overall, it appears that the maternal HFD-induced cerebrovascular remodeling would decrease cerebral blood flow and then increase the vulnerability and susceptibility to stroke, as well as other cerebrovascular diseases. Although oral glucose tolerance test is observed to be impaired in HFD/C offspring, the role of glucose metabolism dysregulation in maternal HFD-induced cerebrovascular dysfunction remains to be fully established. It is worth noting that the female but not male adult offspring of HFD-fed dams display raised blood pressure 13 ;therefore, the maternal HFD-induced cerebrovascular remodeling may occur independent of changes in blood pressure, at least in male animals.
Vascular endothelium plays a critical role in both function and structure of the cerebral vasculature. Vascular dysfunction, as a hyperreactivity to constrictor agents and/or a decreased relaxation to vasodilators, is associated with the occurrence and development of cerebrovascular diseases. Although endothelial damage is observed in the HFD/C rats, the endothelium-dependent relaxation to ACh is not affected, indicating the endothelial function may be not impaired in maternal HFD offspring. Some studies have shown ET-1 system dysfunction contributes to cerebrovascular diseases in several animal models, including diabetes, chronic alcoholic encephalopathy, moderate strength cold air exposure, exercise, and hypertension.27,28,43–46 More specifically, ET-1 is important in diet-induced vascular dysfunction in animal and patient.24,25 In the current study, maternal HFD rats display raised tissue MCA ET-1 levels, which may be associated with the increased wall thickness and M/L ratio.29,30 Moreover, ET-1 has been reported to stimulate cell growth via ETAR, ultimately promote accelerated myocardial and vascular hypertrophy. 47 These results suggest the ET-1 dysfunction in the artery tissue may contribute to the maternal HFD-induced cerebrovascular structure remodeling. Besides the structural changes, maternal HFD intake also increases the contractility of the MCAs to ET-1 under both isometric and isobaric conditions, using wire myograph and pressure myograph technique. Consistently, Ergul and investigators reported that animals fed with HFD in early life display hypersensitivity to constrictor including ET-1 following MCAO and show greater cerebral injury and poor outcomes. 48 Nevertheless, Deutsch et al. 7 demonstrated that childhood HFD consumption-induced changes in the adult cerebral vasculature are largely structural and do not relate to alterations in signaling pathways or the smooth muscle contractile machinery. The conflicting data presented above may result from the differences in animal species, experimental model, dietary intervention duration, contraction agents, and vascular bed studied. Additionally, increased post-stroke plasma ET-1 levels in this study suggest that ET-1 system may also contribute to increased ischemic brain injury susceptibility. It is further supported by the evidence showing ET-1 overexpression leads to increased ischemic brain damage and acute ETAR antagonism could reverse the effects.49–51 Thus, it appears that ET system is modulated in maternal HFD exposure model and may contribute to the cerebrovascular dysfunction, but specific receptor distribution, receptor affinity, signaling cascades mediating contraction have not been studied. Although maternal diet-induced programing of ET-1 system may be associated with the changed plasma gonadal hormone level, 52 stress hormone 53 and glucose metabolism, 21 the underlying mechanisms remain to be fully established.
The present study also examined the individual and relative roles of the ET receptors governing cerebrovascular dysfunction induced by maternal HFD intake. We observed that hypersensitivity of maternal HFD rats to ET-1 is accompanied by increases in ETAR density, indicating that the increased receptor expression at least partially contributes to the hyperreactivity of the MCAs. Atrasentan, a selective ETAR antagonist, prevents this hyperactive response. Whereas ETBR density remains unchanged in the MCA tissue and ETBR antagonism does not alter vascular responses of maternal HFD rats to ET-1, it causes enhanced constriction in control rats. Moreover, chronic ETAR but not ETBR antagonism prevents cerebrovascular dysfunction, including changes in lumen diameter and myogenic tone of the MCAs. These results suggest that ETAR blockade restores maternal HFD-induced cerebrovascular dysfunction but ETBR antagonism does not influence the remodeling. It is further supported by the evidence showing that ETAR antagonism attenuates mesenteric vascular dysfunction in type 2 diabetes rat and that ETBR blockade exhibits differential effects depending on the disease state. 21 Moreover, another study also reported that administration of ETAR antagonist at reperfusion prevents the decreased dilatory ability of basilar arteries in stroke animals. 54 These results provide evidence for a pivotal and differential role of ET receptor subtypes in the regulation of maternal HFD-induced cerebrovascular dysfunction. Nevertheless, the heterogeneity of ETBR function in those located on the endothelium versus those on vascular smooth muscle cells has been demonstrated in several studies. 55 Endothelial ETBR are believed to confer protective effects by causing relaxation to ET-1, where smooth muscle ETBR are vasoconstrictive in nature. It should be noted that either receptor density or contractile response of the ETBR was done with total vessel or its homogenate and cannot differentiate vascular versus endothelial ETBR. Further studies with endothelium-denuded vessels would help clarify the contribution of endothelial versus muscular ETBR.
Many studies have suggested that fetal predictive adaptive responses to a maternal imbalanced diet favor adulthood survival on a similar diet. 56 For example, Khan et al. 16 reported that the defect in endothelial function and the fall in heart rate in offspring of rats exposed prenatally and during suckling to the fat-rich diet and maintained from weaning on standard chow is absent when the offspring are fed the fat-rich diet. 16 Nevertheless, we demonstrated that cerebrovascular defects, including the endothelial dysfunction, observed in normally fed adult rats exposed to a maternal HFD, are still evident even the offspring are maintained on their habituated, suggesting that adaptive mechanisms may not program cerebrovascular protection against the adulthood dietary insult. In fact, no differences are observed in major cerebrovascular parameters between HFD/C and HFD/HFD groups. Actually, although Khan et al. reported predictive adaptive responses prevent endothelial dysfunction and reduce heart rate in offspring of fat-fed dams if rat offspring are raised on the same diet, but not raised blood pressure and adiposity. Therefore, the fetus or neonate may be partially capable of mounting a predictive adaptive response to an anticipated postnatal diet, but the influence of the maternal diet on cerebrovascular development may be irreversible, which requires more further investigations.
Conclusions
Overall, the present experiments support the hypothesis that a fat-rich diet during pregnancy and lactation fundamentally alters the middle cerebral arteries of adult offspring. The present study demonstrated, for the first time, that maternal diet and metabolic status in early life can drastically affect the structure and function of the cerebral vasculature, as revealed by pressure myography, wire myography, and electron microscopy. These maternal HFD-induced cerebrovascular remodeling would ultimately not only affect the adult sensitivity to ischemic injury, but also give rise to an offspring phenotype predisposed to the development of cerebrovascular disease in adult. Moreover, cerebrovascular ET-1 system may play an important role in maternal HFD-induced cerebral circulation dysfunction in offspring. Nevertheless, more studies need to be conducted to characterize the mechanisms by which maternal HFD consumption influences the cerebrovascular health.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: this study was funded by grants from the National Natural Science Foundation of China (81673144, 81301014), Foundation of Wenzhou Science and Technology Bureau (Y20160025).
Acknowledgements
The authors greatly appreciate the contribution from YiFan Cheng who performed the myography experiments and Hong Wang who performed the electron microscopy experiments.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
LCC, WXY, SB and LYS conceived and designed the research. LCC, WXY, ZYL and NXT performed experiments, ZWL and LYS conducted the statistical analyses, LCC, WXY and LYS drafted and revised the manuscript.