
Abstract
Ischemic stroke is one of the most prevalent sources of disability in the world. The major brain tissue damage takes place upon the reperfusion of ischemic tissue. Energy failure due to alterations in mitochondrial metabolism and elevated production of reactive oxygen species (ROS) is one of the main causes of brain ischemia-reperfusion (IR) damage. Ischemia resulted in the accumulation of succinate in tissues, which favors the process of reverse electron transfer (RET) when a fraction of electrons derived from succinate is directed to mitochondrial complex I for the reduction of matrix NAD+.
We demonstrate that in intact brain mitochondria oxidizing succinate, complex I became damaged and was not able to contribute to the physiological respiration. This process is associated with a decline in ROS release and a dissociation of the enzyme's flavin. This previously undescribed phenomenon represents the major molecular mechanism of injury in stroke and induction of oxidative stress after reperfusion. We also demonstrate that the origin of ROS during RET is flavin of mitochondrial complex I. Our study highlights a novel target for neuroprotection against IR brain injury and provides a sensitive biochemical marker for this process.
Introduction
Ischemic stroke occurs as a result of a blockage of blood flow to the brain. It is one of the most prevalent sources of disability in modern times, almost pandemics in developed countries, with the number of people affected ∼1184 per 100,000 population. 1 It is a consensus that brain tissue damage occurs upon reperfusion of ischemic tissue. Mitochondrial bioenergetics failure and elevated production of reactive oxygen species (ROS) are considered among the major causes of brain ischemia-reperfusion (IR) tissue damage. The mitochondrial IR-induced damage is usually delayed; after seemingly complete recovery of aerobic ATP production in the first phase of reperfusion, mitochondria function fails later on, this process is termed “secondary energy failure.” The molecular mechanisms of this secondary failure are not understood (reviewed in the study by Kristian 2 ). Several putative mechanisms had been suggested for IR-induced brain mitochondria damage,3–5 including a decline in mitochondrial complex I function caused by IR.6,7 However, the reasons for complex I failure are also not known.
Mitochondrial complex I oxidizes tricarboxylic acid cycle-produced NADH by ubiquinone in the inner mitochondrial membrane. Electrons from NADH are accepted by non-covalently bound flavin mononucleotide (FMN) and further transferred to ubiquinone in the inner mitochondrial membrane,8,9 generating the protonmotive force 10 which fuels the production of ATP by mitochondrial ATPase. The electron transfer in complex I can be reversed so the enzyme can catalyze the reduction of matrix NAD+ by the ubiquinol at the expense of mitochondrial membrane potential.11,12 This reaction is termed “reverse electron transfer” (RET). Despite the fact that more than 55 years since the first observation of RET in the intact mitochondria have passed,11,13 not much is known about regulation and significance of this process in situ. However, it is known that the minimal requirements for this reaction to occur are the presence of the membrane potential and a high level of ubiquinone reduction, which in the brain can be provided by succinate oxidation. The accumulation of succinate well above its physiological levels is a common marker of brain ischemia. In rat brain, 5 min of ischemia results in ∼300% increase in succinate concentration, to the millimolar range.14,15 Upon reperfusion, succinate in post-ischemic brain returns to the control levels only after 30–40 min. 16 Succinate had also been shown to inhibit the oxidation of pyruvate and other NAD-linked respiratory substrates and cause an over-reduction of mitochondrial pyridine nucleotides. 17 Succinate had been proposed lately to be the primary oxidized substrate in brain homogenates of post-ischemic neonatal mouse brains (Dr. Vadim Ten's lab, submitted).
Recently, succinate-driven RET emerged as a potential area of great physiological importance.18–21 In this manuscript, we demonstrate that in intact brain mitochondria under conditions of RET, prolonged succinate oxidation damages complex I by inducing a dissociation of the enzyme's flavin. This previously undescribed phenomenon may represent the major molecular mechanism of IR-induced secondary energy failure in stroke. Our data also demonstrate that the origin of ROS during RET is flavin of mitochondrial complex I.
Materials and methods
Intact mitochondria isolation for in vitro studies
The animal protocol was approved by the Institutional Animal Care and Use Committee of Weill Cornell Medicine. All experiments were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals of the National Institutes of Health and the ARRIVE guidelines. To isolate intact mitochondria, a modified protocol combining differential centrifugation and digitonin treatment was used. 22 The forebrain hemispheres of C57BL/6 8–10 weeks male mice were excised and immediately immersed into ice-cold isolation medium (225 mM mannitol, 75 mM sucrose, 20 mM HEPES-Tris, 1mM EGTA, pH 7.4) supplemented with 1 mg/ml bovine serum albumin (BSA). Tissue was homogenized with 40 strokes by pestle “B” (tight) of a Dounce homogenizer in 10 ml of the isolation medium, diluted twofold and transferred into centrifuge tubes. The homogenate was centrifuged at 5900 g for 4 min in a refrigerated (+4℃) Beckman centrifuge. The supernatant was centrifuged at 12,000 g for 10 min, and the pellets were resuspended in the same buffer and 0.02% digitonin was added. The suspension was homogenized briefly with five strokes in a loosely fitted Potter homogenizer and centrifuged again at 12,000 g for 10 min, then gently resuspended in the isolation buffer without BSA and washed once by centrifuging at 12,000 g for 10 min. The final mitochondrial pellet was resuspended in 0.1 ml of washing buffer and stored on ice.
Accessing respiration and ROS production in intact mitochondria
Mitochondrial respiration and extramitochondrial release of H2O2 were measured using a high-resolution respirometer (Oroboros) equipped with two-channel fluorescence optical setup to monitor simultaneously oxygen level and fluorescence in 2 ml of mitochondrial suspension. For H2O2 measurements, fluorescence was recorded using a green 525 nm LED as an excitation light source, a photodiode (S2386-8K, Hamamatsu Photonics, Japan) as a sensor, and an emission filter with cut off 580 nm (#34, Rosco Laboratories, USA), fixed in a 3D printed mount (Form2, Formlabs, USA). The photodiode was connected to the amperometric port of the Oxygraph and the fluorescence was recorded using the Datlab software. The calibration of Amplex UltraRed response was performed at the end of the run by adding several 93 pmol/ml aliquots of freshly made H2O2 to the chamber containing all the components of the assay.
Mitochondria (0.14–0.18 mg of protein) were added to 2 ml respiration buffer composed of 125 mM KCl, 0.2 mM EGTA, 20 mM HEPES-Tris, 4 mM KH2PO4, pH 7.4, 2 mM MgCl2, 2 mg/ml BSA, 10 µM Amplex UltraRed (Invitrogen), and 4 U/ml horseradish peroxidase at 37℃. The following substrates were used: 2 mM malate and 5 mM pyruvate or 5 mM succinate and 1 mM glutamate. To initiate State III respiration, 200 µM ADP was added to the mitochondrial suspension. Respiration was fully sensitive (inhibited) by 1 mM cyanide or 1 µM antimycin A.
To enable us to rapidly change oxygen concentration in the mitochondrial suspension, we have included a gas headspace (1 ml) above the measurement chamber. The headspace could be continuously purged with humidified gaseous nitrogen through the stopper at a rate of 10–60 ml/min. By varying the partial pressure of gaseous nitrogen in the headspace, we are able to control its concentration in the liquid sample via N2 exchange between the two phases. When required, the gas flow was turned off, and the stoppers moved to seal the oxygen chamber so that no headspace remained.
Complex I activity measurements in disrupted mitochondria
The NADH:HAR oxidoreductase activity of complex I 23 was determined spectrophotometrically using a plate reader (SpectraMax M5, Molecular Devices, USA). Oxidation of 150 µM NADH (ɛ340nm = 6.22 mM−1 × cm−1) by 1 mM HAR was followed at 340 nm in 0.2 ml of standard respiration buffer (pH = 7.5) supplemented with 25 µg/ml alamethicin (to disrupt mitochondria), 2 mM MgCl2, 1mM cyanide, and 0.01–0.025 mg protein/ml mitochondria.
Flavin fluorescence measurements
The flavin fluorescence in mitochondria oxidizing succinate was measured by Hitachi 7000 spectrofluorimeter set at 450 nm excitation and 525 nm emission. The reaction was carried out under exactly the same conditions as the respiratory assays, except that Amplex UltraRed and horseradish peroxidase were omitted from the medium. Special controls for photobleaching of the preparation during prolonged measurements were performed.
Mitochonfrial membrane potential measurements
Mitochondrial membrane potential was asessed indirectly using membrane-permeable fluorescent safranin O probe (2 µM), excitation 525 nm, emission 580 nm essentially as described previously. 24
All chemicals were purchased from Sigma-Aldrich (USA) unless stated otherwise. Protein content was determined by bicinchoninic acid assay with deoxycholate (0.1%) for solubilizing the mitochondrial membranes (ThermoFisher, USA). All activities were measured at 37℃ except NADH:HAR (25℃). All results are expressed as means ± SEM. Six different preparations of mitochondria were tested. When comparing different groups, a two-sample t-test was used. Other experimental details are described in the legends of the figures.
Results
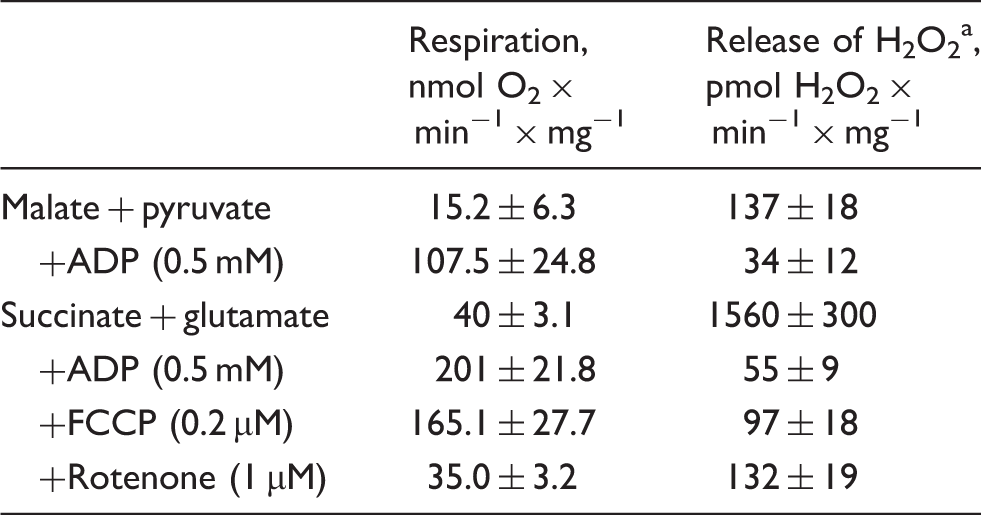
In order to assess the oxygen dependence of the hydrogen peroxide (H2O2) emission from intact brain mitochondria, first we carried out experiments in the closed respirometer chamber. Figure 1(a) shows representative traces of oxygen consumption and H2O2 release by intact brain mitochondria oxidizing NAD-dependent substrates, or succinate. Table 1 shows quantitative characteristics of the respiration and H2O2 emission. The rate of H2O2 emission upon coupled succinate oxidation was around 10-fold higher than in the presence of NAD-dependent substrates. As expected, the succinate-supported H2O2 production was suppressed by either adding ADP or an uncoupler, as well as by rotenone, confirming that ROS generation was fueled by RET. It should be noted that the addition of succinate rapidly induces reduction of the entire pool of matrix NAD(P).
11
Therefore, in the steady-state conditions of the succinate oxidase reaction in vitro, there is no RET as a classical reduction of matrix nucleotides showed by Britton Chance.
11
In the present paper, we use term RET-like conditions describing the process of oxidation of succinate by a fully competent respiratory chain of intact mitochondria in steady state.
H2O2 release during the forward and reverse electron transfer. Representative traces of respiration (top) and generation of H2O2 (bottom) by intact mitochondria from mouse brain. (a) The reaction was started by addition of mitochondria 0.16 mg of protein/ml to the medium composed of 125 mM KCl, 0.2 mM EGTA, 20 mM HEPES-Tris (pH 7.4), 4 mM KH2PO4, 2 mM MgCl2, 2 mg/ml BSA, 10 μM Amplex UltraRed and 4 U/ml horseradish peroxidase at 37℃. Substrates were either 5 mM pyruvate and 2 mM malate (right) or 5 mM succinate and 1 mM glutamate (left). Black traces correspond to oxygen concentration (top) and Amplex UltraRed fluorescence (bottom). The rate of the change of these signals per minute is shown in red. (b) Release of H2O2 during coupled oxidation of 5 mM succinate and 1 mM glutamate as a function of oxygen concentration. 100% rate corresponds to 1400 pmol H2O2 × min−1 × mg protein−1. Oxygen concentration as measuring directly by the Oroboros respirometer was rapidly varied by continuously purging the headspace with nitrogen as described in Materials and Methods section. Respiration activities and H2O2 release by coupled mouse brain mitochondria. Measured simultaneously in 125 mM KCl, 0.2 mM EGTA, 20 mM HEPES-Tris, 4 mM KH2PO4, pH 7.4, 2 mM MgCl2, 2 mg/ml BSA, 10 µM Amplex UltraRed, and 4 U/ml horseradish peroxidase at 37℃, 0.14–0.19 mg of protein/ml mitochondrial protein. Substrates were either 5 mM malate and 2 mM pyruvate or 5 mM succinate and 1 mM glutamate. Values are given as mean ± SEM. Three different preparations of mitochondria were used, all conditions were repeated in triplicates.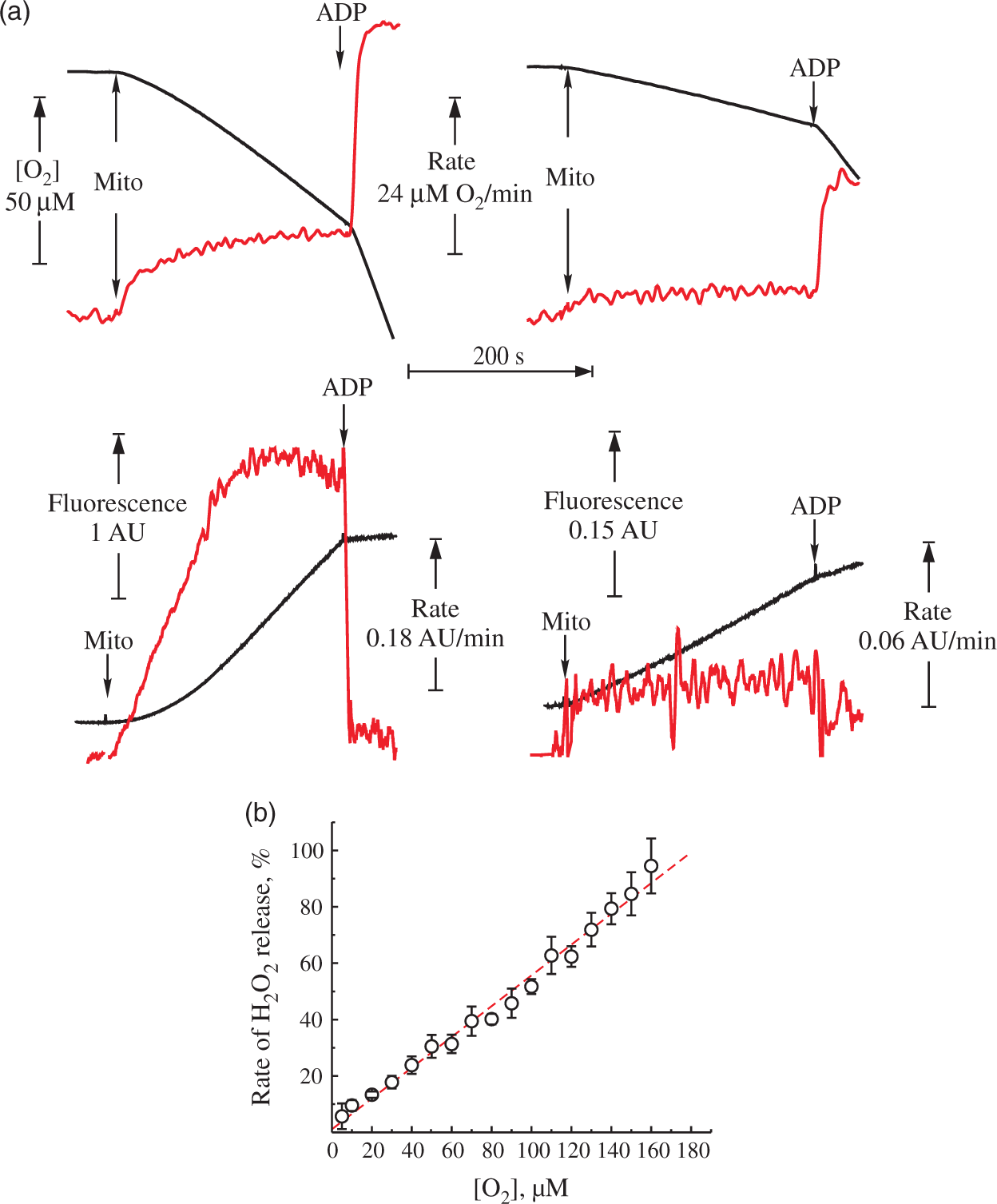
We estimated the effect of the oxygen concentration on H2O2 release during the oxidation of succinate by purging the headspace in our system with nitrogen to quickly reduce the partial pressure of O2 in the solution. RET-associated H2O2 release during succinate oxidation was almost linearly dependent on the oxygen concentration in the medium (Figure 1(b)).
As shown in Figure 2(a), during the time course of succinate oxidase reaction in the closed chamber when the oxygen gradually decreases due to the respiratory activity, the rate of H2O2 release also declines. We would like to emphasize that the resting respiration rate during the oxidation of succinate stayed almost constant at the wide range of oxygen concentration and dropped sharply only at 20–25 µM [O2]. In order to evaluate the rate of H2O2 emission from mitochondria at constant concentration of oxygen (around 190 µM [O2]), we carried our similar experiments in the open respirometer chamber. Unexpectedly, we also observed a decline in H2O2 release rates during RET in the open chamber (Figure 2(d)), while the oxygen concentration in the medium did not change (Figure 2(b)).
Consumption of oxygen and H2O2 release in closed (a), (c) and open (b), (d) respirometer chamber. Oxygen concentration (top) and Amplex UltraRed fluorescence (bottom) were measured simultaneously in 125 mM KCl, 0.2 mM EGTA, 20 mM HEPES-Tris, 4 mM KH2PO4, pH 7.4, 2 mM MgCl2, 2 mg/ml BSA, 0.25 mg/ml of mouse brain mitochondria, 5 mM succinate, 1 mM glutamate 10 µM Amplex UltraRed and 4 U/ml horseradish peroxidase. Black traces correspond to oxygen concentration (top) and raw Amplex UltraRed fluorescence (bottom). The rates of the change of these signals per minute are shown in red.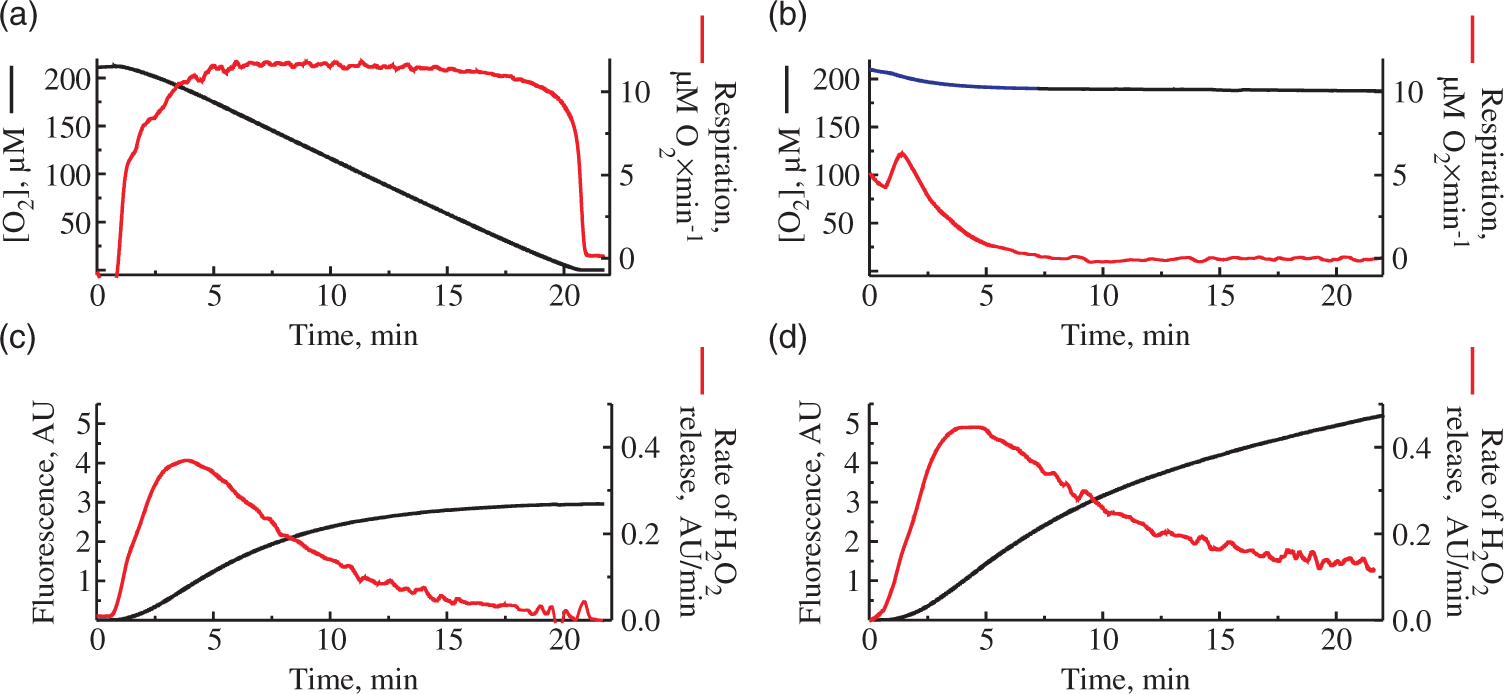
To further investigate this phenomenon, we tested the effect of amount of mitochondria in the medium to the observed decline in the H2O2 emission rate. Representative traces of the H2O2 release rate decline in time at different concentrations of mitochondrial protein in the assay are shown in Figure 3(a). The half-time of this decline was around 4.5 min (Figure 3(b)) and was not dependent on the concentration of mitochondria in the range of 0.1–0.5 mg of protein/ml. Therefore, the decline was not due to the accumulation of any reaction product in the medium. The inhibitory action of accumulated H2O2 under our conditions is not likely, because, upon oxidizing NAD-linked substrates, there is virtually no accumulation of H2O2 in the suspension of mitochondria, whereas during RET-catalyzed H2O2 generation, the accumulation is minimal peaking at about 30 nM of H2O2.
25
In agreement with that, preincubating intact mitochondria with 45–90 µM H2O2 for 5–10 min before the addition of substrates did not change the parameters for the decline in H2O2 rates. The addition of 50 U/ml MnSOD to the medium did not change the dynamics for the decline in H2O2 emission (data not presented). Therefore, it is unlikely that in our conditions, ROS produced during RET had pronounced damaging effect on mitochondrial enzymes involved in RET. We found that the only condition that decreased the rate of decline was temperature. At 25℃ in the open chamber, the half-time of the decline in H2O2 release rate was increased to 10–12 min (not shown).
H2O2 release rate decline at different protein concentration and safranin response. (a) Three different protein concentrations were used to monitor H2O2 release in RET-like conditions. Curves 1–3 correspond to 0.5, 0.25, and 0.125 mg of protein/ml protein in the measuring assay. Dashed lines show fitting of exponential decay function. (b) Half-life of exponential decay of H2O2 release rate at difference protein concentrations (n = 3–4 for each data point). (c) Safranin O fluorescence after addition of 5 mM succinate and 1 mM glutamate to 0.15 mg of protein/ml. Assay conditions are as shown in Figure 2(b), except 2 µM safranin substituted Amplex UltraRed and horseradish peroxidase in (c). Concentration of FCCP was 0.1 µM.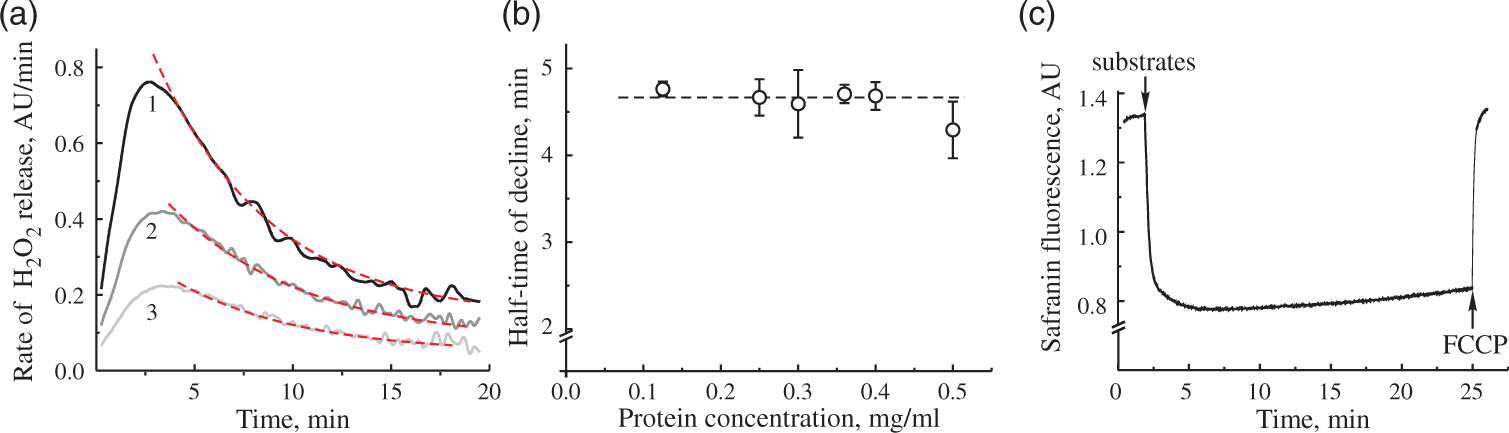
Several factors directly affect the H2O2 release rate during coupled oxidation of succinate: absolute activity of respiratory chain enzymes and the magnitude of membrane potential across the inner mitochondrial membrane. Therefore, first we tested the dependence of H2O2 release rate during RET on the potential dependent fluorescence of safranin O. 26 As shown in Figure 3(c), no significant changes in the dye fluorescence were observed during RET, indicating that membrane potential was mainly preserved during this time interval changing only by less than 8%.
Potentially, the H2O2 release rate during RET also depends on the activity of complexes I and II. Therefore, we have assessed the respiratory activities of mitochondria before and after incubation under RET conditions. As shown in Figure 4(a), succinate oxidase activity in both resting and ADP-stimulated state was unaffected by 20-min incubation. Contrary to that, ADP-stimulated respiration on malate and pyruvate was decreased by around 75% (Figure 4(b)). This strongly indicates that prolonged RET affected the NADH:ubiquinone reductase activity of complex I. Consequently, it is highly likely that an impairment of complex I function was responsible for the decline in H2O2 emission rate during the prolonged oxidation of succinate (Figure 3(a)).
Effect of incubation of intact brain mitochondria in RET-like conditions on the succinate- and NADH-dependent respiration ((a) and (b), respectively). Mitochondria 0.15–0.2 mg of protein/ml were incubated in the presence of 5 mM succinate and 1 mM glutamate (as shown in Figure 2(b)). After 2 min (gray) and 20 min (white), 1 µM rotenone was added for the registration of succinate oxidase only (a). Alternatively, 25 nM atpenin, 5 mM pyruvate, and 2 mM malate were added for the NAD+-dependent respiration (b) After the additions, respirometer chamber was closed and resting respiration (state II) was measured. ADP-stimulated (state III) respiration was measured after addition of 0.5 mM ADP.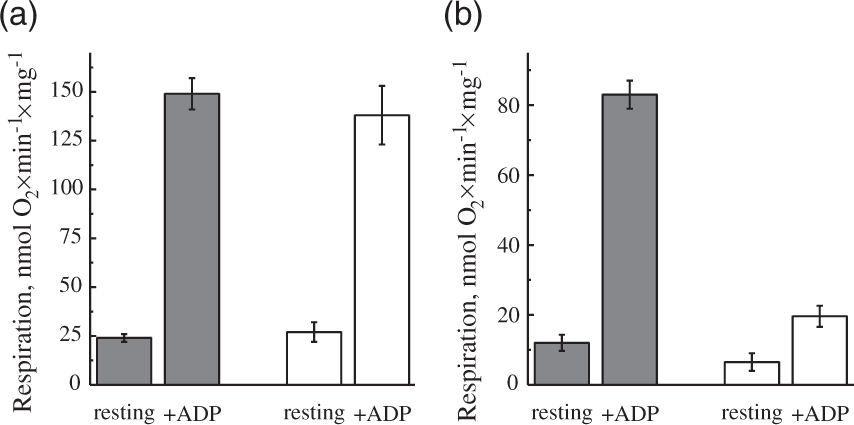
The loss of NADH:ubiquinone reductase activity of mitochondrial complex I can be caused by several factors. To elucidate the potential mechanism, we have assessed the changes in the activity of complex I during the time course of the decline with an artificial electron acceptor HAR (NADH:HAR reductase) spectrophotometrically in permeabilized mitochondria (Figure 5). We found that NADH:HAR activity decreased in parallel with the H2O2 release rate by intact mitochondria. Moreover, the addition of 10 µM exogenous FMN had resulted in a partial recovery of NADH:HAR activity. These data unequivocally indicate that the decline in the rate of H2O2 generation was due to the loss of complex I flavin caused by prolonged succinate oxidation under RET conditions.
Effect of incubation of intact brain mitochondria in RET-like conditions on NADH:HAR reductase activity. Mitochondria (0.3 mg of protein/ml) were incubated in the presence of 5 mM succinate and 1 mM glutamate as shown in Figure 2(b) and H2O2 release rate was monitored after reaching the maximum (squares). Small aliquots were taken in time and NADH:HAR reductase activity was assayed as described in Materials and Methods section with or without 10 µM FMN (black and red circles, respectively). One hundred percent rate of H2O2 release corresponds to 1530 ± 150 pmol H2O2×min−1×mg protein−1.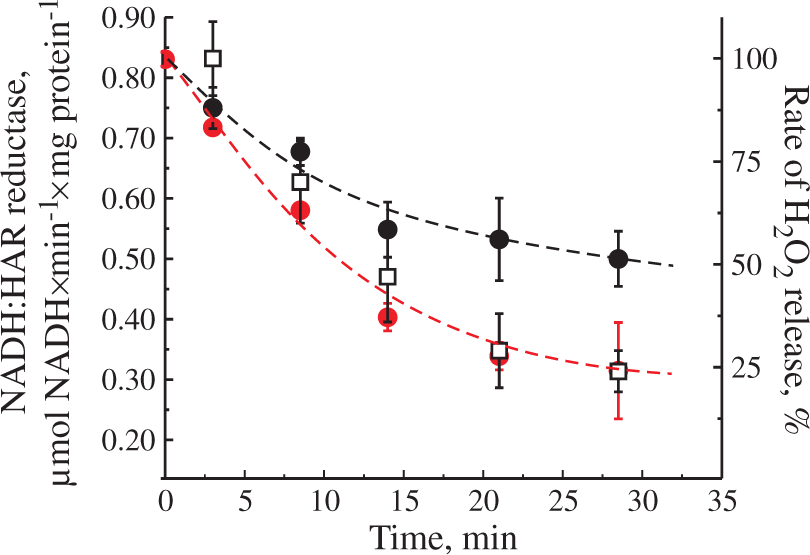
To further support this conclusion, we have assessed flavin fluorescence in intact mitochondria exactly under the same conditions as those employed to measure H2O2 emission (Figure 6(a)). A fast decline in the fluorescence intensity after addition of substrates indicating a reduction of flavin was observed for the first 2 min. The decline was followed by a slow increase in fluorescence. This could be caused by either an increase in flavin's fluorescence intensity upon its dissociation from the protein or by a fast autoxidation of the reduced flavin in the matrix. The dynamics of the flavin fluorescence intensity (Figure 6(a)) followed exactly the inverted pattern of the decline in H2O2 emission rate (Figure 6(b)). This indicates that in steady-state conditions, the rate of H2O2 emission upon RET was directly proportional to the amount of complex I-bound flavin.
Flavin fluorescence recovery (a) and dynamics of H2O2 release (b) in intact brain mitochondria during oxidation of succinate in RET-like conditions. Parameters of the assay are shown in Figure 2, except 0.4 mg of protein/ml mitochondria was used. AmplexUltra Red and horseradish peroxidase were absent from the medium for flavin fluorescence measurements. Black traces correspond to Amplex UltraRed fluorescence and the rate of H2O2 release per min per mg of protein is shown in red. Note that fluorescence response (averaged from three measurements) is shown in inverted scale.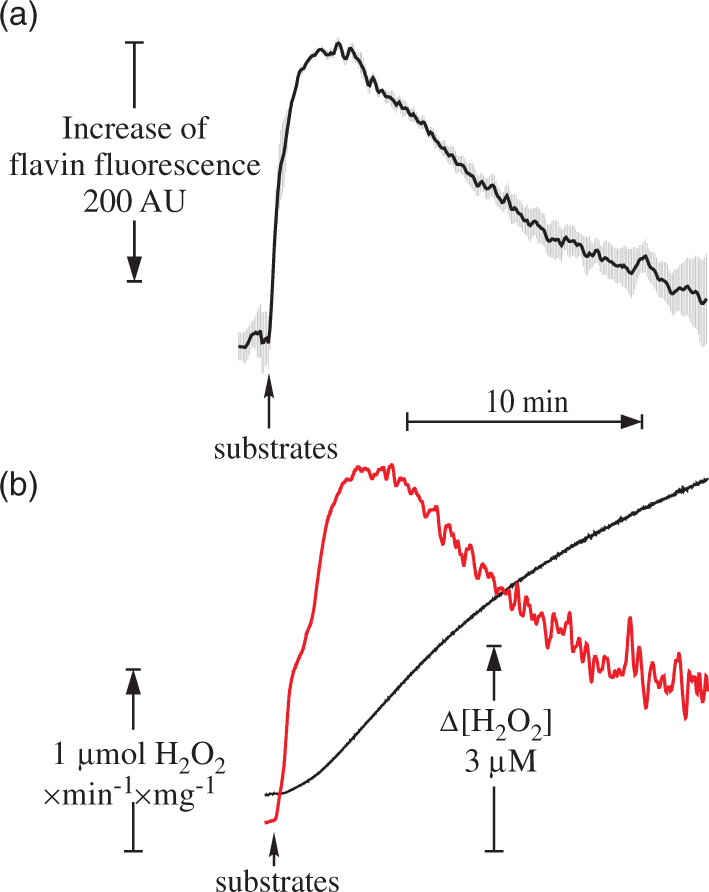
Discussion
In this study, we have shown that prolonged oxidation of succinate by brain mitochondria under RET conditions stimulates dissociation of flavin from mitochondrial complex I and results in the loss of activity of the enzyme. In turn, this impairs the oxidation of NAD-linked (physiological) substrates as well as the generation of ROS by mitochondria.
The process of RET, first observed more than half a century ago,11,13 has recently gained significant attention in ischemia/reperfusion studies. First, after a period of oxygen deprivation succinate level is elevated in highly metabolizing tissues such as the brain14,15,27 and heart.18,28 Potentially, given the availability of substrate and energization of the inner mitochondrial membrane, RET can take place after oxygen is resupplied to the tissue. Second, succinate-dependent respiration is characterized by the highest rates of ROS production by mitochondria.18,19,29–32 In our study, RET also provided the greatest rate in H2O2 release equivalent to around 3.5% of total oxygen consumption, which is very close to what observed for the mitochondria from adult rat brain. 26 Therefore, in postischemic tissue, RET is active and may render injury to the tissue found in several models of tissue ischemia.18,33
Mitochondrial complex I is the most sensitive respiratory enzyme to ischemia in the brain.6,7,19,34 The mechanism of complex I damage during this process is not fully understood. Surprisingly, incomplete ischemia results in greater complex I damage in a rat model of brain oxygen deprivation. 35 Most likely, succinate accumulated in the ischemic core may diffuse to the surrounding tissues and have devastating consequences to the penumbra area, where both substrates (oxygen and succinate) are available. This may explain greater decrease on mitochondrial complex I damage in the models of incomplete brain ischemia.
In our initial experiments, we observed a linear dependence of H2O2 release by intact brain mitochondria during RET which is in accordance with the earlier studies on the intact brain in vivo. 36 Superoxide anion generation by submitochondrial particles oxidizing succinate 37 also showed a linear dependence of H2O2 production upon oxygen concentration. This is in apparent disagreement with recent studies of liver mitochondria 38 and may be due to the different enzymatic composition of the preparations from different tissues.
The unexpected observation in our further study is the gradual decline in H2O2 release rate by intact brain mitochondria during oxidation of succinate at constant oxygen level. The rate of decline is not dependent on the concentration of mitochondria (0.1–0.5 mg of protein/ml) and half-time of the process is around 4.5 min. We assessed activities of complexes I and II before and after oxidation of succinate and found that only complex I activity is strongly diminished after mitochondria were exposed to RET-like conditions. We found that this decline is due to the dissociation of the flavin from mitochondrial complex I since NADH:HAR reductase activity of the enzyme declines in simultaneously with ROS production. Artificial acceptor HAR takes electrons directly from the flavin of complex I at the hydrophilic domain and this process does not involve downstream components of the enzyme such as iron-sulfur clusters and ubiquinone-binding site.23,39 Moreover, supplementation of exogenous FMN to the assay medium partially recovered NADH:HAR reductase activity of enzyme supporting the critical role of flavin in the process of RET-induced inactivation.
Under RET conditions, electrons from succinate are transferred to ubiquinone and then directed to the oxygen via downstream complex III and cytochrome c oxidase and partially upstream to complex I. The above process provides respiratory activity and builds up a potential across the mitochondrial membrane, and the latter one is likely to be responsible for the generation of ROS upstream of ubiquinone binding site of complex I. In the steady state, coupled succinate oxidation would maintain high NAD(P)H/NAD(P)+ ratio in the matrix and increases reduction of the tightly bound FMN of complex I. Non-covalently bound FMN resides on the bottom of a deep cavity of the catalytic subunit NDUFV1 8 and is accessible to oxygen, therefore, reduced flavin favors ROS formation. FMN has been proposed to be the main site of ROS-production in complex I in several studies.40–44 However, the FeS clusters of complex I45,46 and semiquinone47,48 had also been discussed as the source of ROS origin (see Andreyev et al. 49 for review). Our experiments strongly support the idea that exactly FMN, and not the FeS clusters, is the major site of H2O2 generation in intact brain mitochondria in conditions of RET during succinate oxidation.
Another important finding of our studies is that FMN quickly dissociates from the enzyme in intact brain mitochondria when it is reduced in the RET-like conditions. Reductive dissociation of FMN from the three-subunit FP-fragment of complex I, 50 as well as from the intact bovine and isolated prokaryotic enzyme51,52 have been demonstrated in vitro. The conditions required for dissociation of FMN in these studies (high pH or the presence of the detergents) are hardly related to any conceivable physiological situation. The significant change in angle of a butterfly-like structure of the isoalloxazine ring 53 and a general loosening of the complex I molecule structure upon reduction 54 may contribute to the strong change in FMN affinity to its binding site. It should be stressed that release of complex I reduced flavin in the mitochondrial matrix may result not only in complex I impairment but also may lay at the basis of oxidative stress after IR. Fully reduced flavin reacts avidly (within seconds) with oxygen, and H2O2 is the predominant product. 55 After the release, the reduced flavin can be readily oxidized by molecular oxygen resulting in an increase of local ROS generation in the matrix. Oxidized FMN molecule can either be metabolized or reinserted back to complex I. At present, the process of non-covalent inclusion of FMN to complex I and the general mechanisms by which mitochondria can provide their flavin cofactors remain unsolved. 56
Our observation suggests that the main cause of decrease in complex I activity after ischemia is the reversible release of flavin from the enzyme. This is first direct evidence of the mechanism of complex I damage during oxidation of succinate in postischemia-like conditions. Our in vitro findings were corroborated by in vivo data from transient cerebral ischemia by middle cerebral artery occlusion where 35 min ischemia significantly decreased the content of mitochondrial membrane non-covalently bound FMN (Kahl et al., 2017, Stroke, submitted). To our knowledge, the only report describing the loss of mitochondrial flavin during cardiac ischemia in dogs was published more than 35 years ago. 57 Also of note, a high proportion of acute stroke patients manifested riboflavin deficiency following reperfusion; however, the clinical significance of these findings is not yet known. 58 Further studies are expected to shed light on the mechanism of complex I inactivation in brain ischemia and is currently underway in our laboratory.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by MRC grant MR/L007339/1 (to A.G.) and by NIH grant NS-100850 (V.T.).
Acknowledgements
We are grateful to Anna Bunin for help in the preparation of this manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors' contributions
Anna Stepanova: acquisition of data, analysis, and interpretation of data, critical revision of the article; Anja Kahl: in vivo studies and data analysis; Csaba Konrad: fluorescent module setting up and programming; Anatoly Starkov: acquisition and analysis of data, critical revision of the article; Vadim Ten: analysis of data and critical revision of the article; Alexander Galkin: supervisor, conception, acquisition of data, analysis, and interpretation of data, drafting the article, critical revision of the article, and final approval.